

Abstract
Objective
To determine the frequency and clinical relevance of immunoglobulin (Ig)G, IgA, and IgM N-methyl-d-aspartate receptor (NMDAR) antibodies in several diseases, and whether the IgG antibodies occur in disorders other than anti-NMDAR encephalitis.
Methods
Evaluation of IgG, IgA, and IgM NMDAR antibodies in serum of 300 patients with anti-NMDAR encephalitis, stroke, dementia, schizophrenia, or seronegative autoimmune encephalitis. Antibodies and their effect on cultured neurons were examined with cell-based assays and brain and live neuronal immunostaining. Retrospective analysis of the clinical diagnoses of a cohort of 1,147 patients with IgG NMDAR antibodies identified since 2005.
Results
Among the 300 patients studied, IgG NMDAR antibodies were only identified in those with anti-NMDAR encephalitis and all reacted with brain and live neurons. By cell-based assay, IgA or IgM antibodies were detected in 22 of 300 patients (7%) with different diseases, but only 10 (3%) reacted with brain and 7 (2%) with live neurons. In cultured neurons, IgG but not IgA or IgM antibodies caused a decrease of synaptic and extrasynaptic NMDAR. Among the cohort of 1,147 patients with IgG NMDAR antibodies, 1,015 (88.5%) had anti-NMDAR encephalitis, 45 (3.9%) a limited form of the disease, 41 (3.6%) autoimmune post–herpes simplex encephalitis, 37 (3.2%) overlapping syndromes (anti-NMDAR encephalitis and demyelinating disease), and 9 (0.8%) atypical encephalitic syndromes; none had schizophrenia.
Conclusions
IgG NMDAR antibodies are highly specific for anti-NMDAR encephalitis and cause a decrease of the levels of NMDAR. In contrast, IgA or IgM antibodies occur infrequently and nonspecifically in other diseases and do not alter the receptor levels.
The identification of antibodies against the N-methyl-d-aspartate receptor (NMDAR) and other synaptic proteins in neuropsychiatric diseases previously considered idiopathic has disclosed immunopathogenic mechanisms with important clinical implications.1 These findings resulted from (1) a “clinic-to-lab” approach centered in the study of patients with syndromes of unclear etiology in order to identify the underlying mechanisms (e.g., autoantibodies) and treat them accordingly, and (2) the use of rigorous criteria for antibody diagnosis, including cell-based assay (CBA), immunostaining of brain sections, and live cultured neurons.2 When these criteria were applied, the diagnostic value and syndrome specificity of the antibodies were enhanced. Subsequently, several studies have challenged the specificity of NMDAR antibodies.3–6 Different from a clinic-to-lab approach, these studies screen for antibodies irrespective of the patients' disease, often using a single test (CBA). This single-test approach, either with fixed3,5–7 or live cell CBA,8 using serum only and without confirmatory studies, has produced an extraordinary variability of findings among investigators and has suggested the presence of NMDAR antibodies in patients with a wide range of diseases3,5–8 and in healthy persons.5,9 Additional confusion comes from studies that do not clarify the clinical features, or mix the findings of the different antibodies (immunoglobulin [Ig]G, IgA, IgM).3,5–7 Here, we took a similar approach and examined the presence of serum IgG, IgA, and IgM NMDAR antibodies using CBA, but also including confirmatory tests in 300 patients with different diseases. In addition, we reviewed our experience with 14,475 patients investigated for IgG NMDAR antibodies since 2005. The findings are important because they show remarkable differences concerning syndrome association, diagnostic utility, and pathogenicity among the 3 indicated classes of antibodies.
Methods
Patients and samples
Given that most of the studies examining IgG, IgA, or IgM antibodies in diseases other than anti-NMDAR encephalitis only used patients' serum, we used a similar approach on sera of 300 randomly selected patients, including 60 with anti-NMDAR encephalitis,10 90 dementia, 50 stroke, 50 schizophrenia, and 50 possible autoimmune encephalitis without autoantibodies, defined as reported10 (supplemental material, links.lww.com/WNL/A364). For patients with encephalitis or stroke, serum was obtained at the acute stage, and for patients with chronic disease (dementia, schizophrenia), it was obtained at the time the diagnosis was established using standard clinical tests (dementia) or DSM-IV-TR (schizophrenia). Among patients with anti-NMDAR encephalitis, we randomly selected 40 sera from patients with IgG antibodies in serum and CSF, and 20 sera from patients with IgG antibodies only in CSF.
NMDAR antibody determination, Ig classes, and antibody effects on cultured neurons
Considering that the CBA for NMDAR with fixed HEK cells is widely used in clinical and commercial labs, and in the published studies on IgA and IgM antibodies, we designed an algorithm of antibody testing starting with this technique. All 300 serum samples were examined specifically for IgG, IgA, and IgM NMDAR antibodies in CBAs (representing 900 CBAs). In parallel, all 300 samples were examined for brain immunostaining as a confirmatory test, using an IgG biomarker that potentially identifies IgA or IgM antibodies (secondary antihuman IgG antibodies with potential cross-reactivity with IgA or IgM) (figure 1). Subsequently, all samples positive in any of these assays (CBA or brain immunostaining) and 50 randomly selected samples negative in both assays were examined in a second round of brain immunostaining with IgA- or IgM-specific secondary antibodies. Next, all samples positive by CBA or brain tissue immunohistochemistry were examined for cell surface reactivity with live hippocampal neurons. Finally, a representative sample positive for IgG (used as positive control) and all samples positive for IgA or IgM NMDAR antibodies (confirmed with at least 2 independent techniques) were investigated for pathogenic effects on cell surface NMDARs (total and synaptic).
Figure 1. Algorithm of antibody testing using confirmatory studies.

AE = autoimmune encephalitis; CBA = cell-based assay; Ig = immunoglobulin; NMDAR = N-methyl-d-aspartate receptor; NMDARE =NMDAR encephalitis.
Details of each technique have been reported and are described in supplemental information (links.lww.com/WNL/A364), including an in-house CBA upon which a current commercial test was developed with mild modifications (a validation study showed similar specificity for anti-NMDAR encephalitis11), brain tissue immunohistochemistry, antibody binding to live cultured neurons,2 epitope region analysis,12,13 and antibody effects on the levels of total and synaptic NMDAR.14,15 Investigators assessing the results were blinded to clinical information.
NMDAR antibody–positive patients identified in our laboratory
We reviewed the diagnoses of all patients with IgG NMDAR antibodies identified in Hospital Clínic, University of Barcelona, and the University of Pennsylvania since the initial study that led to the discovery of anti-NMDAR encephalitis.16 During the first 3 years, all patient samples were investigated with 3 techniques, including brain immunostaining (in which the antibodies of patients with anti-NMDAR encephalitis produce a highly characteristic pattern), CBA (that establishes the identity of the antigen), and live cultured neurons.2,17 This experience, including that with the first 100 consecutive patients, was reported2 and showed that immunolabeling of live neurons was always positive when the antibodies were demonstrated with both CBA and brain immunostaining. Therefore, we have since used both CBA and brain immunostaining as obligatory criteria to define serum or CSF positive for NMDAR antibodies. Cases with discordant results (∼7% sera; <1% CSF) are retested and, if needed, resolved with live neuronal staining.
Since 2005 until May 2017, 14,475 patients representing a wide variety of diseases (among others paraneoplastic, autoimmune encephalitis, multiple sclerosis and other demyelinating disorders, prion diseases, neurodegenerative diseases, peripheral neuropathies, viral encephalitis) have been examined for neuronal antibodies. Of these 14,475 patients, 8,361 and 100 healthy controls were simultaneously studied with CBA and brain tissue immunostaining irrespective of the disease or reason for consultation (approach used by J.D.). The other 6,114 were investigated in a sequential manner, including first brain immunohistochemistry, and if this was positive, the identity of the antigen was confirmed with CBA (approach used by F.G.). Two hundred sixteen patients with anti-NMDAR encephalitis were clinically studied by the authors.
Standard protocol approvals, registrations, and patient consents
Patients' serum and CSF samples are deposited in the collection of biological samples named “Neuro-immunology” registered in the Biobank of Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) or the neuroimmune tissue bank at the University of Pennsylvania. Written informed consent for the storage and use of samples for research purposes was obtained from patients. The study was approved by the ethics committee of the Hospital Clínic of Barcelona and the institutional review board of the University of Pennsylvania.
Results
Among the 60 patients with anti-NMDAR encephalitis, serum CBA confirmed that 40 had IgG antibodies against the NMDAR, and 10 of them had additional IgA or IgM antibodies. The other 20 samples were from patients known to have IgG NMDAR antibodies only in CSF (serum IgG negative) and 1 had IgM antibodies in serum. Among the 240 patients with diseases other than anti-NMDAR encephalitis, 11 had IgA, IgM, or both classes of antibodies, but none had IgG NMDAR (table 1 and figure 2, A–F). Because commercial antibody tests often use high serum concentration (dilution 1:10) instead of our standard dilution (1:40), we repeated all studies at this dilution in 50 randomly selected samples for CBA IgG, IgA, and IgM (n = 150 assays), and all showed similar results to those found with 1:40 dilutions (data not shown).
Table 1.
Determination of serum IgG, IgA, and IgM antibodies to NMDAR in 5 disorders
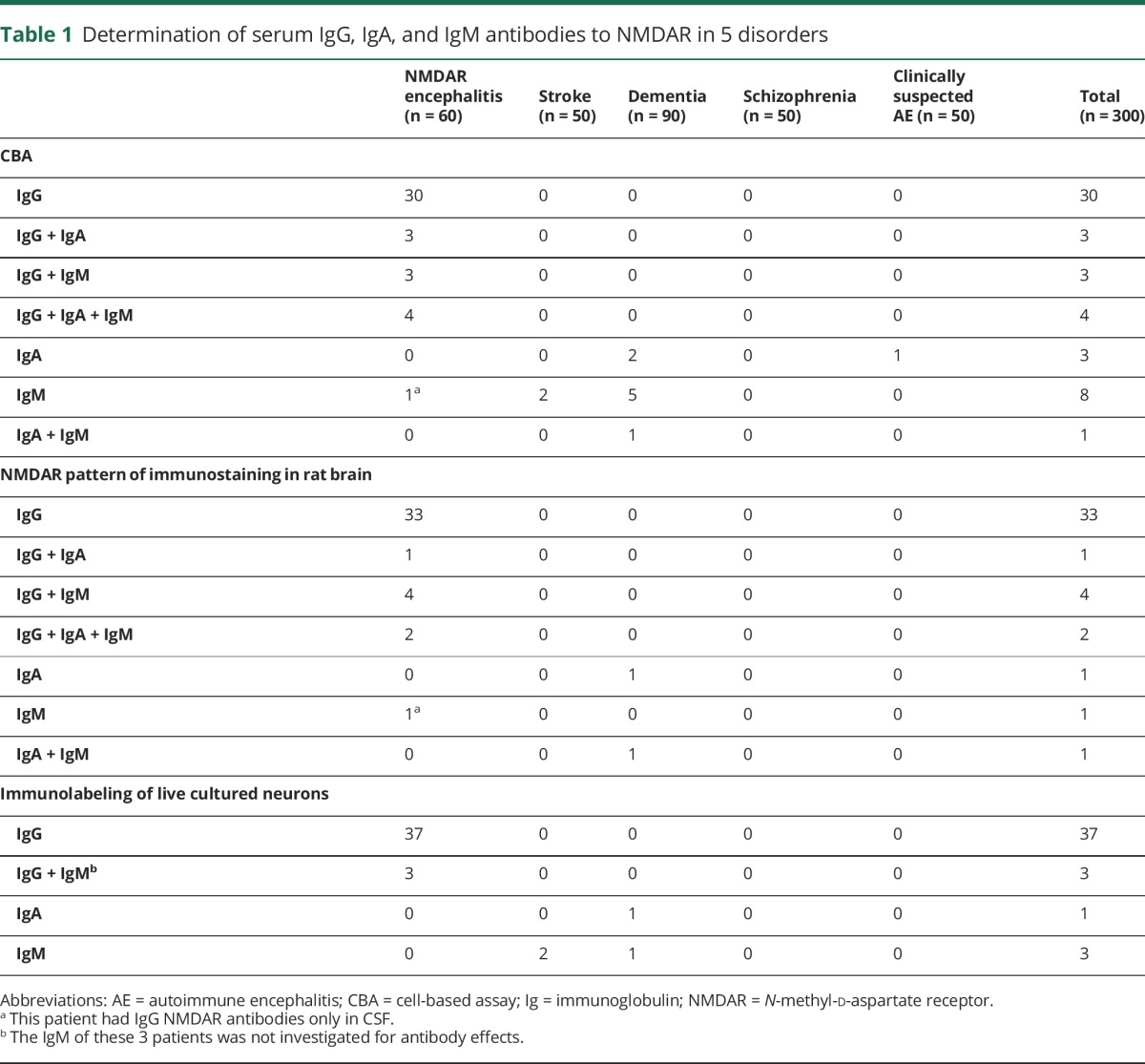
Figure 2. IgG, IgA, and IgM antibodies against the NMDAR demonstrated by CBA and brain tissue immunostaining.

CBA for NMDAR antibodies showing the reactivity of serum of 3 different patients with IgG (A), IgA (B), and IgM (C) antibodies. Patients' antibodies are shown with green fluorescence and a commercial antibody against a noncompeting NMDAR epitope with red fluorescence. (D–F) Lack of reactivity of a control sample (without NMDAR antibodies) using a similar CBA. (G–I) Brain reactivity of the 3 patients' samples (same as in A–C) using the corresponding secondary antibodies against IgG (G), IgA (H), and IgM (I). The lack of reactivity of the control sample using the indicated secondary antibodies is shown in panels J–L. In panels A–F, the cell nuclei are shown with DAPI; scale bar = 10 μm. In panels G–L, the slides have been mildly counterstained with hematoxylin; scale bar = 500 μm. CBA = cell-based assay; DAPI = 4′,6-diamidino-2-phenylindole; Ig = immunoglobulin; NMDAR = N-methyl-d-aspartate receptor.
Parallel studies using brain immunostaining with samples from all 300 patients showed that 40 (corresponding to the same positive by CBA) had IgG antibodies that produced the characteristic pattern of NMDAR reactivity. However, among the 22 IgA or IgM CBA positive samples, only 10 showed IgA or IgM brain immunostaining (figure 2, G–L). None of the 50 randomly selected cases that were IgA or IgM CBA negative showed brain immunostaining. Overall, these data show that using 2 different techniques (CBA and tissue immunostaining), IgG NMDAR antibodies are disease-specific (anti-NMDAR encephalitis) whereas IgA or IgM antibodies are not.
Next, we examined whether any class of NMDAR antibodies detected by CBA or brain immunostaining reacted with live neurons. All 40 sera with IgG NMDAR antibodies showed cell surface reactivity, whereas only 3 sera with isolated IgM antibodies and 1 with isolated IgA antibodies showed cell surface reactivity (figure e-1, links.lww.com/WNL/A362). These data indicate that only 4 of 240 patients (2%) with diseases other than anti-NMDAR encephalitis had IgM or IgA antibodies that reacted with live neurons.
To determine whether IgA or IgM antibodies altered the levels of NMDAR, cultured live neurons were exposed to 4 representative samples containing only IgA or IgM antibodies (all 4 samples were from patients without anti-NMDAR encephalitis). After 24-hour exposure to patients' serum, those with IgG antibodies produced a specific and significant decrease of the number of clusters of cell surface (total) and synaptic NMDAR. In contrast, none of the 4 samples with IgA or IgM antibodies altered the levels of total or synaptic NMDAR (figure 3). Moreover, when the reactivity with previously described GluN1 subunit mutants was compared between 3 samples of patients with only IgA or IgM antibodies and the IgG antibodies of patients with anti-NMDAR encephalitis, the epitope repertoire was different; the IgA and IgM antibodies recognized the “G369I” mutant whereas the IgG antibodies did not. This is in line with previously reported studies.12,13
Figure 3. Effects of patients' antibodies on the levels of cell surface NMDAR.

(A) Dendrites from live neurons exposed to serum IgG antibodies (NMDAR IgG+ S) show a decrease of total cell surface clusters of NMDAR (green fluorescence in top row) compared to neurons exposed to control serum (NC S), and 4 different sera with IgM (NMDAR IgM+ S1; NMDAR IgM+ S2) or IgA (NMDAR IgA+ S3; NMDAR IgA+ S4) antibodies. None of the patients' samples affected the levels of PSD95 (red fluorescence in middle row). The serum with IgG antibodies (NMDAR IgG+ S), but none of the 4 sera with IgA or IgM antibodies, also caused a decrease of synaptic clusters of NMDAR (yellow fluorescence in lower row). Synaptic NMDARs are defined by the colocalization of green (NMDAR) and red (PSD95) fluorescence. Scale bars in all panels = 5 μm. (B) Quantification of the above studies demonstrating that only the serum with IgG NMDAR antibodies caused a significant decrease of total surface and synaptic NMDAR. Data obtained from 3 independent experiments (16–18 dendrites per experiment, for each condition) showing the median with interquartile range. Statistical significance was analyzed with the Kruskal-Wallis test followed by the Dunn post hoc test for nonnormally distributed data. A value of p < 0.05 was considered significant in post hoc testing after correction for multiple comparisons (Dunn test); ***p < 0.001. Ig = immunoglobulin; NC = normal control; NMDAR = N-methyl-d-aspartate receptor; PSD95 = postsynaptic density protein-95.
Lastly, we reviewed the clinical diagnoses of all patients with IgG NMDAR antibodies identified in our centers (Hospital Clínic, University of Barcelona, and University Pennsylvania) since 2005. Of 14,475 patients whose serum or CSF was examined for neuronal antibodies, 1,147 had IgG NMDAR antibodies; of these, 971 were identified among referrals to J.D., mainly focused on CNS inflammatory diseases, and 176 among referrals to F.G., focused on a variety of diseases. None of the 100 healthy controls had NMDAR antibodies. The clinical features and follow-up on many of the IgG NMDAR–positive patients have been included in previous reports.18,19 Here, we focused on whether there were patients with IgG antibodies identical to those found in anti-NMDAR encephalitis2 but who did not have this disorder. Table 2 shows that patients with IgG NMDAR antibodies segregated in 5 groups: (1) 1,015 (88.5%) developed a typical syndrome of anti-NMDAR encephalitis (as reported in reference 20); (2) 45 (3.9%) showed monosymptomatic or highly limited symptoms, mostly psychiatric,21 which otherwise are typical of anti-NMDAR encephalitis; (3) 41 (3.6%) developed autoimmune encephalitis post–herpes simplex encephalitis; (4) 37 (3.2%) showed overlapping syndromes (anti-NMDAR encephalitis and demyelinating disorders); and (5) 9 (0.8%) developed encephalitic syndromes atypical for anti-NMDAR encephalitis. The median age of these 9 patients was 33 years, 6 were female, and all had NMDAR antibodies in the CSF. Two patients have been reported,22,23 and the others are described in table e-1 (links.lww.com/WNL/A363).
Table 2.
Summary of 1,147 patients with IgG NMDAR antibodiesa

Discussion
The main findings of this study show that, (1) IgG NMDAR antibodies in serum or CSF are highly specific for the syndrome of anti-NMDAR encephalitis; only 0.8% of the patients had atypical symptoms, which always occurred in the context of CNS inflammation; (2) IgA or IgM NMDAR antibodies occur infrequently and nonspecifically in patients with or without autoimmune encephalitis (e.g., stroke, dementia), and the epitope repertoire is different from that of IgG antibodies of patients with anti-NMDAR encephalitis; and (3) in cultured live neurons, IgA or IgM antibodies did not alter the levels of NMDAR, whereas IgG antibodies caused a significant decrease of synaptic and extrasynaptic NMDAR.
The robust association of IgG NMDAR antibodies with the syndrome of anti-NMDAR encephalitis replicates our initial studies.2,24,25 This disease occurs with a characteristic spectrum of symptoms and less frequently with limited or monosymptomatic manifestations.20,26 Only 9 of 1,147 patients (0.8%) with IgG NMDAR antibodies showed symptoms that were atypical for anti-NMDAR encephalitis. These 9 patients had clinical, MRI, or CSF findings suggestive of an inflammatory CNS disorder without other diseases that could be potentially linked to the antibodies. In addition, 41 patients had NMDAR antibodies in the context of autoimmune encephalitis post–herpes simplex encephalitis. Data of some of these patients have been published,18,27,28 and the remainder will be reported as part of an ongoing prospective study. Long-term follow-up (>2 years) of 312 patients (277 in references 20, 35 unpublished) showed that none developed schizophrenia despite that low antibody titers are often detectable for months or years.13
Our results are different from those of reports suggesting the presence of NMDAR antibodies in 10% to 22% of patients with a wide variety of diseases.3,5,6,9,29 The reasons for these discrepancies are unclear, but several possibilities can be considered, such as patient selection, the lack of cross-validation or confirmatory studies, the use of different types of CBAs, and the investigators' experience interpreting the CBAs. It is unlikely that patient selection has a major role given that the indicated reports found NMDAR antibodies in the same proportion of patients across all diseases studied as in healthy participants.
In the current study, the focus of one of the investigators (J.D.) on inflammatory and autoimmune CNS diseases represents a referral bias, but the cases referred to F.G. include a wider spectrum of diseases of the central and peripheral nervous system (listed in methods) and, in this setting, the detection of IgG NMDAR antibodies remained tightly associated with anti-NMDAR encephalitis.
The need to use cross-validation studies for serum IgG NMDAR cannot be overemphasized.30 In a study of 312 patients with schizophrenia in which the serum CBA result was ambiguous in a few cases, all were negative when verification assays were applied.31 Using the same commercial CBA, other authors found that approximately 1% of patients with a wide variety of diseases and healthy participants had IgG NMDAR.3 Later, the same authors suggested that any person between the ages of 40 and 80 years has a 10% to 20% chance of being seropositive for any class of NMDAR antibodies.5 This extraordinary variability of results among investigators using the same commercial test is most likely influenced by differences in the CBA interpretation. It has been suggested that CBA with live cells may improve disease sensitivity but in a study using this approach, 23% of patients considered to be NMDAR antibody positive did not have anti-NMDAR encephalitis or even an autoimmune disease.8
In the current study, we used an in-house CBA (the same test that led to the discovery of anti-NMDAR encephalitis)2,17 instead of the commercially available CBA11 or a live cell CBA,8,32 and this may be perceived as a limitation. The focus of our study is not the revalidation of our CBA or that of others' but to raise concern about the diagnostic disease specificity of any CBA when only serum is used without confirmatory assays. Patients are tested worldwide in a variety of laboratories, many using the commercial CBA, and a few using in-house live cell CBA. Data reported from those laboratories,3,5–8,31,32 as well as our current and previous findings,13 support our conclusions about the problems derived from using any CBA with serum only without supportive tests.
Experience using CBA is important but one is not fully aware of the limitations of this test (particularly at low serum dilution such as 1/10 or 1/40) until confirmatory studies are run in parallel. When we tried to confirm the serum CBA results with immunostaining of brain sections and live neurons, the initially considered IgA- or IgM-positive cases dropped from 22/300 (7%) to 7/300 (2%), or from 11/240 (5%) to 4/240 (2%) when patients with anti-NMDAR encephalitis were excluded. Most of the previous reports on IgA and IgM antibodies did not systematically use supportive investigations and focused on patients without anti-NMDAR encephalitis,3,6,9,29 likely explaining the difference with our results (10%–20% vs 2%). A study showed that some patients with progressive cognitive decline developed IgA NMDAR antibodies. Cultured neurons exposed to the IgA of some of these patients showed a remarkable decrease of NMDAR and other synaptic proteins.33 We did not find this effect in the 2 cases we studied with IgA antibodies; the reason for this discrepancy is unknown.
We found that IgG from patients with anti-NMDAR encephalitis, but not IgA or IgM antibodies from patients with other diseases, caused a significant reduction of the levels of synaptic and extrasynaptic NMDARs. These findings are different from those of a recent study using round-shaped cells considered to be human cortical neurons derived from pluripotential stem cells, showing that after a 30-minute incubation with IgG, IgA, or IgM antibodies from patients with any disease (followed by a 20-minute incubation without antibodies), there was decreased availability of NMDAR on the body cell membrane.5 These early structural effects are surprising and may represent antibody-independent effects or a yet unknown antibody. In our experience, using confocal microscopy, bona fide cultured neurons, and IgG from patients with anti-NMDAR encephalitis (or a human recombinant IgG NMDAR),34 the antibody-induced reduction of the level of NMDAR becomes detectable after >2-hour exposure, reaching a maximal decrease at 12-hour exposure.15 We obtained similar results at the nanoscale level, using superresolution stochastic optical reconstruction microscopy, where the earliest detectable effect (clustering of receptors) is noted 2 hours after antibody exposure, preceding the internalization of NMDAR (data not published).
The current findings and previous reports show that serum IgA and IgM NMDAR antibodies do not associate with any specific syndrome3–6,9 regardless of the technique or laboratory used, remaining to be shown whether they have any clinical value. Despite this, some investigators have suggested that these antibodies may aggravate coexisting diseases (schizophrenia, stroke, neurodegeneration) and place approximately 10% of healthy persons at risk of developing neurologic symptoms if the blood-brain barrier is disrupted.5,9 To confirm these hypotheses, it is critical to, first, characterize the antibodies beyond CBA (e.g., brain and live neurons immunostaining); second, demonstrate that the antibodies reach the CNS in patients; and third, show that they consistently associate with a specific set of symptoms or comorbid disease. There is precedence for antibodies that in experimental models suggested pathogenicity but did not prove out on clinical grounds (e.g., GluR3 in Rasmussen encephalitis,35,36 or GluN2 in lupus37). Similar caution should be taken in the interpretation of the findings of a recent study in which 19% of patients with schizophrenia and 3% of healthy participants were found to have IgG NMDAR antibodies in serum. Although the IgG of those with schizophrenia caused a decrease of levels of NMDAR in cultures of neurons, the clinical implications are still unclear. No distinctive symptoms were identified in patients with antibodies, and in contrast to patients with anti-NMDAR encephalitis, those with schizophrenia did not have antibodies in CSF, had consistently very low levels of serum antibodies (not visible in peroxidase brain immunostaining), and the epitope regions appeared to be different.38
As for the diagnosis of anti-NMDAR encephalitis, there are reliable criteria that assist in identifying the disease on clinical grounds10 and highly specific antibody tests to confirm the clinical diagnosis.2,13 If serum is used with a single technique (CBA), the risk of false-positive results for the diagnosis of anti-NMDAR encephalitis (e.g., misinterpretation of the test, or antibody detection without disease association)8,31,39 and false-negative results (e.g., because of antibodies only present in CSF)13,40,41 increase regardless of the CBA or laboratory used. Previous studies and many case reports have shown the superiority of CSF for the diagnosis of this disorder.13 Analysis of CSF provides highly reproducible findings regardless of the CBA13 or laboratory used, and confirmatory tests are rarely needed. If serum is positive but the CSF is negative, the serum finding is probably a false result or the patient does not have anti-NMDAR encephalitis.39
Acknowledgment
The authors thank Dr. Anna Planas (Centro Superior de Investigaciones Científicas, CSIC-IDIBAPS, Universitat de Barcelona) and Drs. Xabier Urra and Raquel Sánchez del Valle (Hospital Clínic, Universitat de Barcelona) for providing samples and clinical information of patients.
Glossary
- CBA
cell-based assay
- DSM-IV-TR
Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition, Text Revision)
- Ig
immunoglobulin
- NMDAR
N-methyl-d-aspartate receptor
Footnotes
CME Course: NPub.org/cmelist
Author contributions
Design/conceptualization of the study: M.H., F.G., J.D. Data collection: M.H., E.M.-H., H.A., M.S., A.S., M.R.R. Analysis and interpretation of the data: M.H., M.R.R., F.G., J.D. Statistical analysis: H.A. Figure development: M.H., M.P.-P., J.D. Drafting of the manuscript: M.H., F.G., J.D.
Study funding
This study was supported in part by Instituto Carlos III/FEDER (CD14/001555, E.M.-H.; CM14/00081, H.A.; FIS PI15/00377, F.G.; FIS PI14/00203, J.D.); Mutua Madrileña Award (AP162572016, T.A.); Fondation de l'Université de Lausanne et Centre Hospitalier Universitaire Vaudois (UNIL/CHUV), Lausanne, Switzerland (M.S.); NIH (RO1NS077851, J.D.); AGAUR Generalitat de Catalunya (SGR93, J.D.); and Fundació CELLEX (J.D.).
Disclosure
M. Hara, E. Martinez-Hernandez, H. Ariño, T. Armangué, M. Spatola, M. Petit-Pedrol, and A. Saiz report no disclosures relevant to the manuscript. M. Rosenfeld receives royalties from Athena Diagnostics for the use of Ma2 as an autoantibody test and from Euroimmun for the use of NMDA as an autoantibody test. F. Graus received a licensing fee from Euroimmun for the use of IgLON5 as an autoantibody test. J. Dalmau receives royalties from Athena Diagnostics for the use of Ma2 as an autoantibody test and from Euroimmun for the use of NMDA as an autoantibody test. Dr. Dalmau received a licensing fee from Euroimmun for the use of GABAB receptor, GABAA receptor, DPPX, and IgLON5 as autoantibody tests; he has received an unrestricted research grant from Euroimmun. Go to Neurology.org/N for full disclosures.
References
- 1.Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med 2018;378:840–851. [DOI] [PubMed] [Google Scholar]
- 2.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahm L, Ott C, Steiner J, et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol 2014;76:82–94. [DOI] [PubMed] [Google Scholar]
- 4.Castillo-Gomez E, Kastner A, Steiner J, et al. The brain as immunoprecipitator of serum autoantibodies against N-methyl-D-aspartate receptor subunit NR1. Ann Neurol 2016;79:144–151. [DOI] [PubMed] [Google Scholar]
- 5.Castillo-Gomez E, Oliveira B, Tapken D, et al. All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatry 2017;22:1776–1784. [DOI] [PubMed] [Google Scholar]
- 6.Hammer C, Stepniak B, Schneider A, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry 2014;19:1143–1149. [DOI] [PubMed] [Google Scholar]
- 7.Steiner J, Walter M, Glanz W, et al. Increased prevalence of diverse N-methyl-D-aspartate glutamate receptor antibodies in patients with an initial diagnosis of schizophrenia: specific relevance of IgG NR1a antibodies for distinction from N-methyl-D-aspartate glutamate receptor encephalitis. JAMA Psychiatry 2013;70:271–278. [DOI] [PubMed] [Google Scholar]
- 8.Zandi MS, Paterson RW, Ellul MA, et al. Clinical relevance of serum antibodies to extracellular N-methyl-D-aspartate receptor epitopes. J Neurol Neurosurg Psychiatry 2015;86:708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zerche M, Weissenborn K, Ott C, et al. Preexisting serum autoantibodies against the NMDAR subunit NR1 modulate evolution of lesion size in acute ischemic stroke. Stroke 2015;46:1180–1186. [DOI] [PubMed] [Google Scholar]
- 10.Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wandinger KP, Saschenbrecker S, Stoecker W, Dalmau J. Anti-NMDA-receptor encephalitis: a severe, multistage, treatable disorder presenting with psychosis. J Neuroimmunol 2011;231:86–91. [DOI] [PubMed] [Google Scholar]
- 12.Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci 2012;32:11082–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 2014;76:108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vitaliani R, Mason W, Ances B, Zwerdling T, Jiang Z, Dalmau J. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol 2005;58:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armangue T, Titulaer MJ, Malaga I, et al. Pediatric anti-N-methyl-D-aspartate receptor encephalitis: clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;162:850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titulaer MJ, McCracken L, Gabilondo I, et al. Late-onset anti-NMDA receptor encephalitis. Neurology 2013;81:1058–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-D-aspartate receptor encephalitis. JAMA Neurol 2013;70:1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labate A, Quattrone A, Dalmau J, Gambardella A. Anti-N-methyl-D-aspartate-glutamic-receptor encephalitis presenting as paroxysmal exercise-induced foot weakness. Mov Disord 2013;28:820–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iizuka T, Kaneko J, Tominaga N, et al. Association of progressive cerebellar atrophy with long-term outcome in patients with anti-N-methyl-D-aspartate receptor encephalitis. JAMA Neurol 2016;73:706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iizuka T, Sakai F, Ide T, et al. Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology 2008;70:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Florance NR, Davis RL, Lam C, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol 2009;66:11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho ACC, Mohammad SS, Pillai SC, et al. High sensitivity and specificity in proposed clinical diagnostic criteria for anti-N-methyl-D-aspartate receptor encephalitis. Dev Med Child Neurol 2017;59:1256–1260. [DOI] [PubMed] [Google Scholar]
- 27.Armangue T, Leypoldt F, Malaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014;75:317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armangue T, Moris G, Cantarin-Extremera V, et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology 2015;85:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doss S, Wandinger KP, Hyman BT, et al. High prevalence of NMDA receptor IgA/IgM antibodies in different dementia types. Ann Clin Transl Neurol 2014;1:822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Witte LD, Hoffmann C, van Mierlo HC, et al. Absence of N-methyl-D-aspartate receptor IgG autoantibodies in schizophrenia: the importance of cross-validation studies. JAMA Psychiatry 2015;72:731–733. [DOI] [PubMed] [Google Scholar]
- 31.Chen CH, Cheng MC, Liu CM, Liu CC, Lin KH, Hwu HG. Seroprevalence survey of selective anti-neuronal autoantibodies in patients with first-episode schizophrenia and chronic schizophrenia. Schizophr Res 2017;190:28–31. [DOI] [PubMed] [Google Scholar]
- 32.McCracken L, Zhang J, Greene M, et al. Improving the antibody-based evaluation of autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm 2017;4:e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pruss H, Holtje M, Maier N, et al. IgA NMDA receptor antibodies are markers of synaptic immunity in slow cognitive impairment. Neurology 2012;78:1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malviya M, Barman S, Golombeck KS, et al. NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann Clin Transl Neurol 2017;4:768–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science 1994;265:648–651. [DOI] [PubMed] [Google Scholar]
- 36.Watson R, Jiang Y, Bermudez I, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology 2004;63:43–50. [DOI] [PubMed] [Google Scholar]
- 37.Steup-Beekman G, Steens S, van Buchem M, Huizinga T. Anti-NMDA receptor autoantibodies in patients with systemic lupus erythematosus and their first-degree relatives. Lupus 2007;16:329–334. [DOI] [PubMed] [Google Scholar]
- 38.Jezequel J, Johansson EM, Dupuis JP, et al. Dynamic disorganization of synaptic NMDA receptors triggered by autoantibodies from psychotic patients. Nat Commun 2017;8:1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armangue T, Santamaria J, Dalmau J. When a serum test overrides the clinical assessment. Neurology 2015;84:1379–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viaccoz A, Desestret V, Ducray F, et al. Clinical specificities of adult male patients with NMDA receptor antibodies encephalitis. Neurology 2014;82:556–563. [DOI] [PubMed] [Google Scholar]
- 41.Murdie D, Cooney G, Ferguson J. Seronegative anti-N-methyl-D-aspartate receptor encephalitis. Biol Psychiatry 2016;79:e67–e68. [DOI] [PubMed] [Google Scholar]
- 42.Titulaer MJ, Hoftberger R, Iizuka T, et al. Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 2014;75:411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]