

Abstract
Objective
To assess resting cerebral blood flow (CBF) in the whole-brain and cerebral white matter (WM) and gray matter (GM) of adults with Fabry disease (FD), using arterial spin labeling (ASL) MRI, and to investigate CBF correlations with WM hyperintensity (WMH) volume and the circulating biomarker lyso-Gb3.
Methods
This cross-sectional, case-control study included 25 patients with genetically confirmed FD and 18 age-matched healthy controls. We quantified resting CBF using Quantitative Signal Targeting With Alternating Radiofrequency Labeling of Arterial Regions (QUASAR) ASL MRI. We measured WMH volume using semiautomated software. We measured CBF in regions of interest in whole-brain, WM, and deep GM, and assessed correlations with WMH volume and plasma lyso-Gb3.
Results
The mean age (% male) for FD and healthy controls was 42.2 years (44%) and 37.1 years (50%). Mean whole-brain CBF was 27.56 mL/100 mL/min (95% confidence interval [CI] 23.78–31.34) for FD vs 22.39 mL/100 mL/min (95% CI 20.08–24.70) for healthy controls, p = 0.03. In WM, CBF was higher in FD (22.42 mL/100 mL/min [95% CI 17.72–27.12] vs 16.25 mL/100 mL/min [95% CI 14.03–18.48], p = 0.05). In deep GM, CBF was similar between groups (40.41 mL/100 mL/min [95% CI 36.85–43.97] for FD vs 37.46 mL/100 mL/min [95% CI 32.57–42.35], p = 0.38). In patients with FD with WMH (n = 20), whole-brain CBF correlated with WMH volume (r = 0.59, p = 0.006), not with plasma lyso-Gb3.
Conclusion
In FD, resting CBF is increased in WM but not deep GM. In FD, CBF correlates with WMH, suggesting that cerebral perfusion changes might contribute to, or result from, WM injury.
Fabry disease (FD) is an X-linked disease due to deficiency of lysosomal α-galactosidase A, causing widespread cellular accumulation of glycosphingolipids, including globotriaosylceramide (Gb3).1 Clinical features include cutaneous capillary lesions (angiokeratomata), painful peripheral neuropathy (acroparesthesia), hypertrophic cardiomyopathy, chronic renal impairment, and cerebrovascular disease. Blood vessel (including endothelial) dysfunction, due to prominent vascular lipid deposition, is likely to contribute to clinical CNS features of FD. Indeed, ischemic stroke occurs in up to 20% of patients with FD,2,3 while cognitive and mood impairments are also common.4,5 Characteristic MRI features of FD include white matter (WM) hyperintensities (WMH),6 lacunar infarcts,7 and microbleeds,8,9 suggesting that small vessel disease (SVD) contributes to its cerebrovascular consequences.
FD vasculopathy might relate to cerebrovascular disease by altering cerebral blood flow (CBF), which is quantifiable as a potential biomarker to detect and monitor the disease. Previous studies on CBF in FD, mainly using PET in hemizygous males,10–12 have been conflicting.13 Arterial spin labeling (ASL) MRI allows noninvasive measurement of CBF without ionizing radiation.14 We used Quantitative Signal Targeting With Alternating Radiofrequency Labeling of Arterial Regions (QUASAR) ASL MRI, a reproducible and accurate technique,15 to test the following hypotheses: (1) CBF is altered in male and female patients with genetically confirmed FD, compared to matched healthy controls; (2) CBF is altered to different extents in WM and deep gray matter (GM); and (3) in the whole group, CBF is correlated with WMH volume (a marker of SVD), and, in the FD group, with circulating lyso-Gb3 (a putative biomarker of disease severity).
Methods
Study population
Inclusion criteria were genetically confirmed FD referred to the National Hospital for Neurology and Neurosurgery, above the age of 18, and with no contraindication to MRI scanning. Healthy adult volunteers, matched as closely as possible for age and sex (as a group), were also recruited for comparison. Patients with other known CNS diseases were excluded. Eligible participants were recruited consecutively from April 2012 to July 2013; each participant underwent detailed clinical assessment, blood testing, and MRI brain scanning during a single visit.
Standard protocol approvals and patient consents
The Joint Research Ethics Committee of the UCL Institute of Neurology and National Hospital for Neurology and Neurosurgery, London, UK, approved the study. We obtained written informed consent from each participant.
MRI acquisition and postprocessing
We used a 3.0T MRI scanner with a 32-channel receive coil (Phillips Healthcare Systems, Best, the Netherlands). The center axial section was orientated along the subcallosal line. We assessed CBF using the QUASAR sequence: a multiple time point, pulsed ASL sequence. The scan parameters were repetition time (TR) 4,000 ms, echo time (TE) 23 ms, 13 inversion times (40–3,640 ms), ΔT1 300 ms, field of view 240 × 240, voxel size 3.75 × 3.75 × 6 mm3, 7 axial slices, slice gap 2 mm, flip angle = 35/11.7°, sensitivity encoding = 2.5, 84 averages (48 with vascular crusher gradients of 4 cm/s, 24 with no vascular crusher gradients, 12 with low flip angle), scan time 6 minutes 7 seconds.
We performed postprocessing using a Windows 7 PC (Microsoft, Redmond, WA) running IDL 6.1 (ITT Visual Information Solutions, Boulder, CO), which produced maps of CBF and tissue R1 (1/T1; longitudinal relaxation rate). To help with the placement of regions of interest (ROI), we acquired a fast spin echo multiecho scan with the following measures: TR 5,230 ms, TE1 16 ms, ∆TE 16 ms, 7 echoes, field of view 240 × 180 × 152 mm3, voxel size 1 × 1 × 2 mm3, 90 axial slices, acquisition time 6 minutes 48 seconds. The fourth echo, corresponding to a TE of 64 ms, was also used for lesion identification as a T2-weighted scan. We acquired a T1-weighted 3D turbo gradient echo sequence with the following measures: TR 6.9 ms, TE 3.1 ms, inversion time 824 ms, field of view 256 × 256 × 180, voxel size 1 × 1 × 1 mm3, 180 sagittal slices, scan time 6 minutes 31 seconds.
ROI identification
We registered proton density (PD) T2-weighted images to the R1 map calculated from the QUASAR sequence using an affine transformation with the NiftyReg toolkit (University College London; sourceforge.net/projects/niftyreg). We placed ROIs, each 6 × 6 voxels, onto the R1 map using the registered PD T2 images as a guide by a single trained radiology trainee (P.P.). An experienced vascular neuroradiology consultant (I.D.) verified all ROI placements. A total of 23 ROIs were placed in both right and left cerebral hemispheres, using Jim 6.0 (Xinapse systems [xinapse.com]): 15 in WM and 8 in deep GM. The WM regions were frontal lobe (above the ventricles), frontal lobe (at the level of the ventricles), frontal lobe (below the ventricles), parietal lobe, occipital lobe, temporal lobe anterior pole, genu of the corpus callosum, splenium of the corpus callosum, and central pons. The GM regions were anterior thalamus, thalamic pulvinar, caudate head, and putamen. Examples of the ROI placements are shown in figure 1.
Figure 1. Representative regions of interest, identified on the R1 images.

Regions of interest are shown as red boxes. (A) Anterior temporal lobe and central pons. (B) Occipital lobes. (C) Frontal lobes below the level of the ventricles. (D) Corpus callosum genu and splenium, putamen, anterior thalamus, and thalamic pulvinar. (E) Frontal lobes above the ventricles. (F) Frontal lobes above the ventricles, and parietal lobes.
Clinical measures
Participants were all assessed on the same day as their MRI. We recorded demographic data using a standardized case report form.
Whole-brain WM lesion quantification
WM lesions were assessed for volume on the PD T2 scans as bright lesions >2 mm, using the phase-sensitive inversion recovery images for anatomical referencing. Hyperintense lesions (>2 mm) around the anterior commissure (anterior perforating substance) were again excluded as larger perivascular spaces are commonly found in this area. By this definition, WM lesions on all participant MRI examinations were segmented using JIM version 5.0 (Xinapse Systems, Northants). A semiautomated method for ROI placement was used: ROIs were automatically assigned over lesions and refined manually by consensus between a trained observer and experienced vascular neuroradiology consultant. The mean total ROI volume was calculated automatically.
Plasma globotriaosylsphingosine (lyso-Gb3) was measured using a rapid multiplexed assay developed at UCL Institute of Child Health (Dr. Kevin Mills) with a reference range 0–1.8 ng/mL.
Statistical analysis
We performed all analyses in Stata 12.1 (Stata Corporation, College Station, TX). Since our values were not normally distributed and unpaired, we used nonparametric statistical tests. We calculated mean CBF of whole-brain by obtaining the mean of all available ROI for each patient. We compared whole-brain mean CBF between patients and controls using the nonparametric rank sum test. Mean WM and deep GM mean CBF was also calculated for each patient from all available ROIs, and then compared between the patients and controls using nonparametric rank-sum test. Median CBF and interquartile range was calculated for CBF from the different regions of the brain. Results were presented as means with 95% confidence intervals (CIs); statistical significance was taken to be p ≤ 0.05. Spearman rho correlation coefficients were calculated for measuring the relationship between CBF and clinical measures in FD, including WMH volume and lyso Gb-3. We excluded patients with FD with zero WMH from correlation analyses.
Results
General demographics
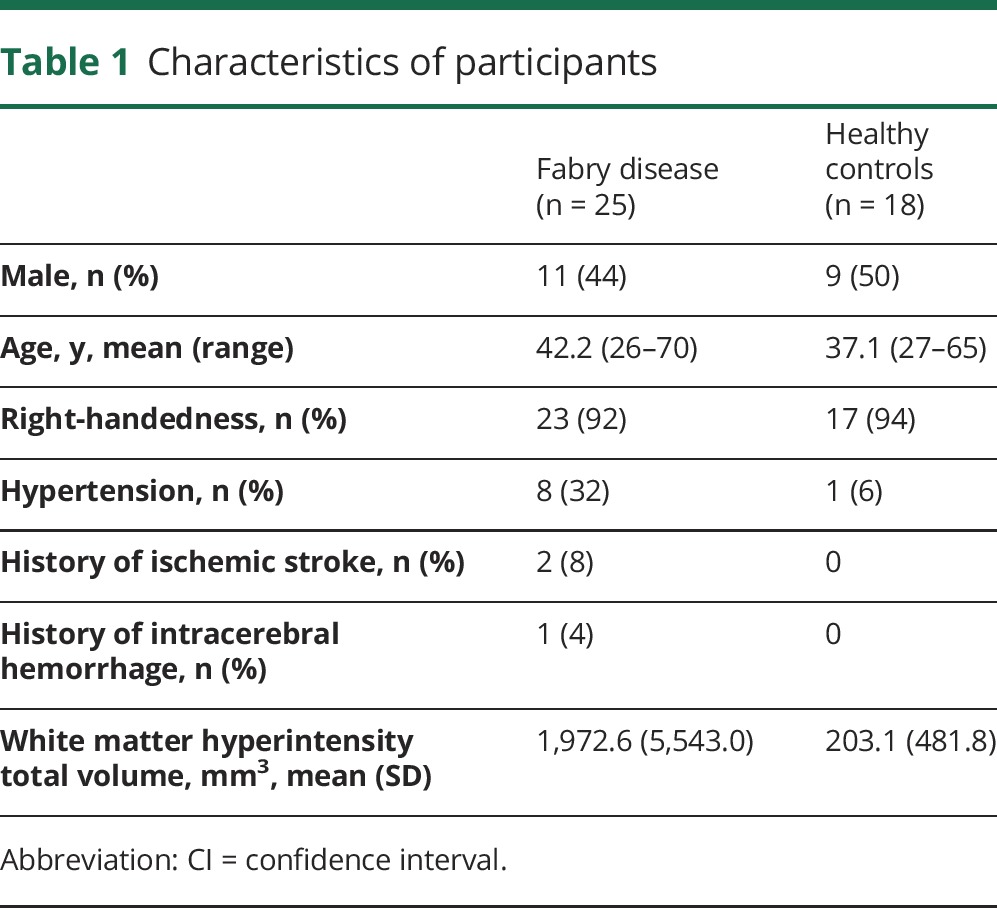
Twenty-five patients and 18 healthy controls were included in the study. The details of study participants are shown in table 1.
Table 1.
Characteristics of participants
Cerebral blood flow
The mean whole-brain CBF (derived from the mean of all available ROI from each patient) was 27.56 mL/100 mL/min (95% CI 23.78–31.34) for FD vs 22.39 mL/100 mL/min (95% CI 20.08–24.70) for healthy controls; p = 0.03 (table 2). In WM regions, CBF was higher in FD (22.42 mL/100 mL/min [95% CI 17.72–27.12] vs 16.25 mL/100 mL/min [95% CI 14.03–18.48]) (p = 0.05). By contrast, in deep GM there was no difference in CBF between groups (40.41 mL/100 mL/min [95% CI 36.85–43.97] for FD vs 37.46 mL/100 mL/min [95% CI 32.57–42.35] for healthy controls) (p = 0.38] (table 2). CBF was increased in all individual ROIs in the FD group compared to the healthy control group, except for the parietal lobe and thalamic pulvinar (table 2), though the difference was only statistically significant for the corpus callosum splenium. Representative CBF maps from a healthy control and a participant with Fabry disease are shown in figure 2. The clinical characteristics and profile of patients by sex are detailed in table 3 and table e-1 (links.lww.com/WNL/A361). No difference was seen between enzyme replacement therapy (ERT) treated and nontreated patients with FD for measure of whole-brain CBF, GM CBF, WM CBF, or lysoGb3 on univariate analysis (table e-2).
Table 2.
Comparison of mean cerebral blood flow (CBF) in whole-brain, white matter, and deep matter for participants with Fabry disease and healthy controls

Figure 2. Representative cerebral blood flow (CBF) maps from a healthy control (top row) and a participant with Fabry disease (bottom row).

CBF is shown on a hotwire scale between 0 and 100 mL/min/100 g. Note that CBF is clearly increased in the participant with Fabry disease (bottom row).
Table 3.
Clinical features of the patients with Fabry disease

Correlation of CBF with WM hyperintensities and biomarkers
The mean volume of WMH in patients with FD was 1,972.6 mm3, compared to 203.1 mm3 in healthy controls (p = 0.004). In the normal control group, 11 of 18 (61%) had no WMH, while in the FD group, only 5 of 25 (20%) had no WMH. In patients with FD with WMH (n = 20), whole-brain CBF correlated with WMH volume (r = 0.59, p = 0.006). In patients with FD, lyso-Gb3 levels did not correlate with CBF in the whole-brain (Spearman rho = −0.023, p = 0.9), in WM (Spearman rho = 0.02, p = 0.94), or in deep GM (Spearman rho = 0.005, p = 0.8). There was no correlation between lyso-Gb3 levels and total volume of WMH (Spearman rho = 0.1, p = 0.6).
Discussion
In our sample of 25 patients with FD (including both male and female patients), we found increased whole-brain CBF in comparison to 18 age-matched healthy controls. The mean of CBF measurements from WM regions was statistically significantly higher than in the mean value from deep GM regions, although the only individual WM region with increased CBF in patients with FD was the splenium of the corpus callosum. In patients with FD with WMH (n = 20), whole-brain CBF correlated with WMH volume (r = 0.59, p = 0.006), but none of the CBF measures (in whole-brain, WM, or deep GM) correlated with plasma lyso-Gb3.
Our CBF measures were broadly comparable to previously published data using a similar technique, suggesting that our data are reliable. A previous QUASAR study measured CBF in 13 healthy patients, with average CBF 38 mL/100 mL/min for GM and 23 mL/100 mL/min for WM.16 The GM value was almost equal to that observed in our healthy controls, but in our healthy control cohort WM CBF was slightly lower (16.46 mL/100 mL/min); however, our healthy controls (mean 37.1 years) are older than those in the previous study, and the method of quantifying CBF was different, based on a mean derived from ROIs rather than from calculated CBF measures through all acquired slices.
Our results are in keeping with previous reports using PET studies suggesting increased CBF in FD11,12,17,18; however, these earlier studies were limited to male patients, while we included a representative cohort of both male and female FD patients with FD. Moreover, the previous studies have not fully established the pattern of perfusion changes in WM and GM in FD.
Our findings not only confirm that FD is associated with increased CBF, but also suggest that the WM is particularly vulnerable to hemodynamic alterations. The correlation between CBF and WMH in FD suggests that increased CBF might be relevant to the pathogenesis of WM injury, which—from the high prevalence in MRI studies (typically around 40%)19—might be central to the cerebrovascular complications in both male and female patients, though we are not aware of evidence linking WMH to future stroke risk in FD. Although, intuitively, sphingolipid deposition in cerebral vessels in FD disease might be expected to reduce CBF, several mechanisms could contribute to elevated CBF in FD. Chronic nitric oxide pathway dysregulation in FD11 can generate peroxynitrite, which induces cerebral vasodilation and resists vasoconstriction by humoral mediators, increasing CBF.11 Alternatively, the sphingolipid pathway abnormality in FD could lead to abnormal excess compensatory neuronal activity, increased neuronal–blood flow coupling, and cerebral hyperperfusion, as is seen in other lysosomal storage disease such as Salla disease, a defect of sialic acid metabolism.20 One possibility is that metabolic insufficiency resulting from neuronal dysfunction in FD could cause a compensatory increase in CBF, which may be an adaptive or protective effect to reduce chronic cerebral hypoxic injury.4 Although the mechanisms of possible hyperperfusion in FD remain unclear, raised CBF may injure vascular walls of WM perforating arteries by shear stress,21 which can stimulate endothelial cells to translate the mechanical stimulus into biochemical signals, causing vascular remodeling by upregulation of growth factors or components of the vessel wall21; this could in turn lead to WMH or ischemic stroke through an occlusive vasculopathy.5 Increased CBF could also increase capillary leakage and macromolecular content of the extracellular space, causing parenchymal cerebral damage and potentially functional impairment of cognition.8
Our findings may also have relevance for FD pathogenesis since the WMH in age-related sporadic cerebral small vessel disease may be indistinguishable (on structural MRI) from those seen in FD, causing diagnostic challenges, especially in older individuals with vascular risk factors. Perfusion studies in sporadic SVD have shown reduced CBF in both normal-appearing and abnormal WM,22,23 suggesting that WMH result from reduced perfusion along penetrating arterioles causing ischaemic injury in deep WM regions with lowest perfusion pressure. Our data thus suggest a fundamental difference in mechanism underlying WMH in FD compared to sporadic SVD, which may make MRI perfusion a useful noninvasive method to distinguish between these 2 processes.
We found increased CBF in all brain regions except for the parietal lobe WM and thalamic pulvinar. By contrast with our findings, a previous small study of MRI perfusion using arterial spin tagging in 3 patients reported increased blood flow in the pulvinar in FD24 associated with the pulvinar sign of T1 hyperintensity. We systematically measured thalamic pulvinar CBF in a larger cohort and found no difference; moreover, none of our patients showed the T1 hyperintense pulvinar sign, which in the previous study was suggested to be pathognomonic of FD.24 More recent studies suggest that, although characteristic of FD, the pulvinar sign is only present in 25% of male patients, and is absent in female patients with FD.25 Our findings of apparently greater WM involvement is different from those of a previous study using PET, which found the abnormality in resting CBF in FD to be equally distributed between WM and GM,11 suggesting a global disease process rather than a selective vulnerability. However, the previous study included only male patients, and used a different method of measuring CBF. Furthermore, another study by the same group found hyperperfusion in WM of patients with FD, as well as reduced metabolism in the WM.26 The authors speculate that increased perfusion is a key mechanism underlying WM injury in FD, a hypothesis supported by our findings.
Our study has strengths. Patients were prospectively recruited and studied using a standardized MRI and clinical protocol. A single trained observer undertook all of the MRI analysis, and an expert neuroradiologist verified all ROI placements. Our sample size is large for a disease as rare as FD, though it is still possible that we were unable to show statistically significant changes in some brain regions (e.g., deep GM) because of limited participant numbers. We also included both male and female patients, unlike previous studies confined to male participants. Some limitations of our data should also be noted, including the exclusion of patients with MRI contraindications, for example those with severe cardiac involvement and implantable cardiac devices. Our findings may therefore not be generalizable to the full spectrum of patients with FD. Our study was cross-sectional and cannot establish the causal relationship between CBF and WMH. Moreover, it is possible that ERT had differential effects on CBF and on lyso-Gb3; ideally a cohort of untreated patients is required to study the relationship between CBF and plasma lyso-Gb3. We have not investigated how CBF is linked to cognitive function, which merits further study.
CBF is increased in FD, with a predilection for WM rather than GM regions. Furthermore, CBF is modestly correlated with WMH volume, suggesting a pathophysiologic link among blood flow changes, SVD, and WM injury. The lack of correlation with lyso-Gb3 suggests that CBF might provide useful information about FD progression, not directly related to circulating levels of the abnormally accumulated sphingolipid. However, our study was not able to investigate the relationship between CBF changes to longer term clinically meaningful outcome for patients (or the effects of ERT).
Since MRI ASL sequence is noninvasive, free from ionizing radiation, and reproducible at different centers, CBF may be an attractive biomarker in FD with relevance for diagnosis, and for detecting and monitoring cerebrovascular involvement. Other noninvasive methods might also have potential in FD; for example, cortical vascular dysfunction in the territory of the posterior circulation has also been described in a transcranial Doppler study in adult patients with FD.27 Furthermore, as CBF changes may potentially be reversible,11 CBF measured using an ASL technique may be an efficient surrogate marker for studies of new treatment strategies in FD.
Glossary
- ASL
arterial spin labeling
- CBF
cerebral blood flow
- CI
confidence interval
- ERT
enzyme replacement therapy
- FD
Fabry disease
- Gb3
globotriaosylceramide
- GM
gray matter
- PD
proton density
- QUASAR
Quantitative Signal Targeting With Alternating Radiofrequency Labeling of Arterial Regions
- ROI
region of interest
- SVD
small vessel disease
- TE
echo time
- TR
repetition time
- WM
white matter
- WMH
white matter hyperintensities
Footnotes
Editorial, page 721
Author contributions
P.P. was involved in data acquisition, statistical analysis, and drafting the manuscript. A.M. was involved in data acquisition, statistical analysis, and drafting the manuscript. I.D. was involved in data analysis and revising the manuscript. F.B. was involved in data acquisition and revising the manuscript. F.J. was involved in statistical analyses. C.W.-K. was involved in data acquisition and revising the manuscript. X.G. was involved in data acquisition and revising the manuscript. D.H. was involved in referring patients and revising the manuscript. L.C. was involved in project supervision and revising the manuscript. E.M. was involved in study concept and design, project supervision, and revising the manuscript. R.L. was involved in study concept and design, project supervision, obtaining funding, and revising the manuscript. D.J.W. was involved in study concept and design, data acquisition, data analysis, obtaining funding, project supervision, and drafting and revising the manuscript.
Study funding
Shire Pharmaceuticals provided a project grant to Dr. Lachmann and Prof. Werring. This work was undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Disclosure
P. Phyu, A. Merwick, I. Davagnanam, F. Bolsover, F. Jichi, C. Wheeler-Kingshott, X. Golay, D. Hughes, L. Cipolotti, and E. Murphy report no disclosures relevant to the manuscript. R. Lachmann reports funding from Shire, Ltd. D. Werring reports funding from Shire, Ltd. Go to Neurology.org/N for full disclosures.
References
- 1.Zarate YA, Hopkin RJ. Fabry's disease. Lancet 2008;372:1427–1435. [DOI] [PubMed] [Google Scholar]
- 2.Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest 2004;34:236–242. [DOI] [PubMed] [Google Scholar]
- 3.Grewal RP. Stroke in Fabry's disease. J Neurol 1994;241:153–156. [DOI] [PubMed] [Google Scholar]
- 4.Bolsover FE, Murphy E, Cipolotti L, Werring DJ, Lachmann RH. Cognitive dysfunction and depression in Fabry disease: a systematic review. J Inherit Metab Dis 2014;37:177–187. [DOI] [PubMed] [Google Scholar]
- 5.Mendez MF, Stanley TM, Medel NM, Li Z, Tedesco DT. The vascular dementia of Fabry's disease. Demen Geriatr Cogn Disord 1997;8:252–257. [DOI] [PubMed] [Google Scholar]
- 6.Fellgiebel A, Keller I, Marin D, et al. Diagnostic utility of different MRI and MR angiography measures in Fabry disease. Neurology 2009;72:63–68. [DOI] [PubMed] [Google Scholar]
- 7.Fazekas F, Enzinger C, Schmidt R, et al. MRI in acute cerebral ischemia of the young: the Stroke in Young Fabry Patients (sifap1) Study. Neurology 2013;81:1914–1921. [DOI] [PubMed] [Google Scholar]
- 8.Reisin RC, Romero C, Marchesoni C, et al. Brain MRI findings in patients with Fabry disease. J Neurol Sci 2011;305:41–44. [DOI] [PubMed] [Google Scholar]
- 9.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altarescu G, Moore DF, Pursley R, et al. Enhanced endothelium-dependent vasodilation in Fabry disease. Stroke 2001;32:1559–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore DF, Scott LT, Gladwin MT, et al. Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy. Circulation 2001;104:1506–1512. [DOI] [PubMed] [Google Scholar]
- 12.Moore DF, Altarescu G, Herscovitch P, Schiffmann R. Enzyme replacement reverses abnormal cerebrovascular responses in Fabry disease. BMC Neurol 2002;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segura T, Ayo-Martin O, Gomez-Fernandez I, Andres C, Barba MA, Vivancos J. Cerebral hemodynamics and endothelial function in patients with Fabry disease. BMC Neurol 2013;13:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petersen ET, Zimine I, Ho YC, Golay X. Non-invasive measurement of perfusion: a critical review of arterial spin labelling techniques. Br J Radiol 2006;79:688–701. [DOI] [PubMed] [Google Scholar]
- 15.Petersen ET, Mouridsen K, Golay X; Co-Authors of the QUASAR Test-Retest Study. The QUASAR reproducibility study, part II: results from a multi-center arterial spin labeling test-retest study. NeuroImage 2010;49:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersen ET, Lim T, Golay X. Model-free arterial spin labeling quantification approach for perfusion MRI. Magn Reson Med 2006;55:219–232. [DOI] [PubMed] [Google Scholar]
- 17.Moore DF, Altarescu G, Ling GS, et al. Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. Stroke 2002;33:525–531. [DOI] [PubMed] [Google Scholar]
- 18.Moore DF, Herscovitch P, Schiffmann R. Selective arterial distribution of cerebral hyperperfusion in Fabry disease. J Neuroimaging 2001;11:303–307. [DOI] [PubMed] [Google Scholar]
- 19.Fellgiebel A, Muller MJ, Mazanek M, Baron K, Beck M, Stoeter P. White matter lesion severity in male and female patients with Fabry disease. Neurology 2005;65:600–602. [DOI] [PubMed] [Google Scholar]
- 20.Suhonen-Polvi H, Varho T, Metsahonkala L, et al. Increased brain glucose utilization in Salla disease (free sialic acid storage disorder). J Nucl Med 1999;40:12–18. [PubMed] [Google Scholar]
- 21.Kolodny E, Fellgiebel A, Hilz MJ, et al. Cerebrovascular involvement in Fabry disease: current status of knowledge. Stroke 2015;46:302–313. [DOI] [PubMed] [Google Scholar]
- 22.Markus HS, Lythgoe DJ, Ostegaard L, O'Sullivan M, Williams SC. Reduced cerebral blood flow in white matter in ischaemic leukoaraiosis demonstrated using quantitative exogenous contrast based perfusion MRI. J Neurol Neurosurg Psychiatry 2000;69:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bastos-Leite AJ, Kuijer JP, Rombouts SA, et al. Cerebral blood flow by using pulsed arterial spin-labeling in elderly subjects with white matter hyperintensities. AJNR Am J Neuroradiol 2008;29:1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore DF, Ye F, Schiffmann R, Butman JA. Increased signal intensity in the pulvinar on T1-weighted images: a pathognomonic MR imaging sign of Fabry disease. AJNR Am J Neuroradiol 2003;24:1096–1101. [PMC free article] [PubMed] [Google Scholar]
- 25.Burlina AP, Manara R, Caillaud C, et al. The pulvinar sign: frequency and clinical correlations in Fabry disease. J Neurol 2008;255:738–744. [DOI] [PubMed] [Google Scholar]
- 26.Moore DF, Altarescu G, Barker WC, Patronas NJ, Herscovitch P, Schiffmann R. White matter lesions in Fabry disease occur in “prior” selectively hypometabolic and hyperperfused brain regions. Brain Res Bull 2003;62:231–240. [DOI] [PubMed] [Google Scholar]
- 27.Azevedo E, Mendes A, Seixas D, et al. Functional transcranial Doppler: presymptomatic changes in Fabry disease. Eur Neurol 2012;67:331–337. [DOI] [PubMed] [Google Scholar]
- 28.Hughes DA, Malmenäs M, Deegan PB, et al. ; FOS Investigators. Fabry International Prognostic Index: a predictive severity score for Anderson-Fabry disease. J Med Genet 2012;49:212–220. [DOI] [PubMed] [Google Scholar]