

Abstract
Amantadine has been shown to reduce anesthesia and surgery-induced neuroinflammation and cognitive dysfunction. It is known that sepsis can impair brain function. We determined whether amantadine-attenuated sepsis-induced neuroinflammation and dysfunction of learning and memory and whether toll-like receptors (TLRs) play a role in the effects. Six- to eight-week-old mice were subjected to cecal ligation and puncture (CLP). Amantadine at 30 mg/kg/day was injected intraperitoneally for 3 days. CU-CPT22, a TLR1/TLR2 inhibitor, at 3 mg/kg/day was injected intraperitoneally for 2 days. Mice were subjected to Barnes maze and fear conditioning tests from 1 week after CLP. CLP induced neuroinflammation and cognitive dysfunction. CLP also increased the expression of toll-like receptor 2 (TLR2), TLR4, and TLR9, three major TLRs in the brain, in CD-1 male mice. Amantadine attenuated CLP-induced neuroinflammation and dysfunction of learning and memory but did not have significant effects on the expression of TLRs. CU-CPT22 also attenuated sepsis-induced neuroinflammation and cognitive dysfunction. Similarly, sepsis induced neuroinflammation and cognitive dysfunction in the C57BL/6J mice. Interestingly, sepsis also induced neuroinflammation and cognitive dysfunction in the TLR2 knockout mice. The effects of amantadine on the neuroinflammation and cognitive dysfunction were still apparent in these knockout mice. TLR2 contributes to sepsis-induced neuroinflammation and cognitive dysfunction. However, inhibiting TLR2 may not be a major mechanism for amantadine to inhibit sepsis-induced neuroinflammation and cognitive dysfunction.
Keywords: Amantadine, Cognitive function, Neuroinflammation, Sepsis, Toll-like receptors
Introduction
Sepsis-associated encephalopathy (SAE) can occur in up to 70% patients with sepsis and is a syndrome of diffuse brain dysfunction with various presentations [1]. Although delirium is a common acute presentation of SAE, up to 40% sepsis survivors can have cognitive dysfunction even 1 year after sepsis [1–4]. In addition to the significant morbidity from brain dysfunction, SAE is associated with increased mortality [1–4]. However, clinically practical methods to reduce SAE have not been developed yet.
Sepsis can cause mitochondrial dysfunction, inflammation, and oxidative stress, which have been proposed to be mechanisms for SAE [1, 2]. However, the exact mechanisms for SAE including the cognitive dysfunction at a delayed phase are not clear. We and others have shown that neuroinflammation is an underlying pathophysiological process for learning and memory dysfunction after surgery [5–8]. We have also shown that neuroinflammation lasted for a short period of time but is a critical process for the delayed phase of cognitive dysfunction after surgery [5, 9]. Amantadine, an anti-viral agent with anti-inflammatory property, reduces neuroinflammation and the delayed dysfunction of learning and memory after surgery [5].
Toll-like receptors (TLRs) play a central role in immune and inflammatory responses [6, 7, 10]. Various ligands act on TLRs that transmit extracellular signals into cells to activate transcription factors, such as nuclear factor κB (NFκB). These transcription factors then induce the production of inflammatory cytokines [7, 10, 11]. There are 13 types of TLRs. Among them, TLR2 and TLR4 are the major ones that can bind components of cells and tissues [6, 7, 10]. A recent study showed that TLR4 may be involved in cognitive dysfunction assessed at 24 h after tibial fracture and fixation [12].
Based on the above information, we hypothesize that TLR-mediated neuroinflammation contributes to sepsis-induced learning and memory impairment at a delayed phase and that amantadine attenuates this impairment by reducing neuroinflammation. To address these hypotheses, mice were subjected to cecal ligation and puncture (CLP), an animal model that best resembles human sepsis [13].
Materials and methods
The animal protocol was approved by the institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 80-23) revised in 2011. Efforts were made to minimize the number used and suffering of animals. Our manuscript was written up in accordance with the Animal Research: Reporting In Vivo Experiments.
Experimental protocols
Three experiments were performed. In experiment 1 (Fig. 1a), 6- to 8-week-old CD-1 male mice weighing 30–35 g from Charles River Laboratories International (Wilmington, MA) were randomly assigned to the following groups: (1) control (not being exposed to surgery or any drugs), (2) amantadine, (3) CLP, (4) CLP plus amantadine 1 (the first dose of amantadine was given intraperitoneally 15 min before the surgery to create CLP), and (5) CLP plus amantadine 2 (the first dose of amantadine was given intraperitoneally 6 h after the creation of CLP). One week after CLP, these mice were started to be tested in Barnes maze and then fear conditioning. We aimed to have 15 mice per group that would survive till the completion of these tests. Separate mice were assigned to the same groups as above but with an n = 6 per group per time point. These mice were sacrificed at 6 or 24 h after CLP to harvest brain tissues for Western blotting, immunohistochemistry, and ELISA.
Fig. 1.
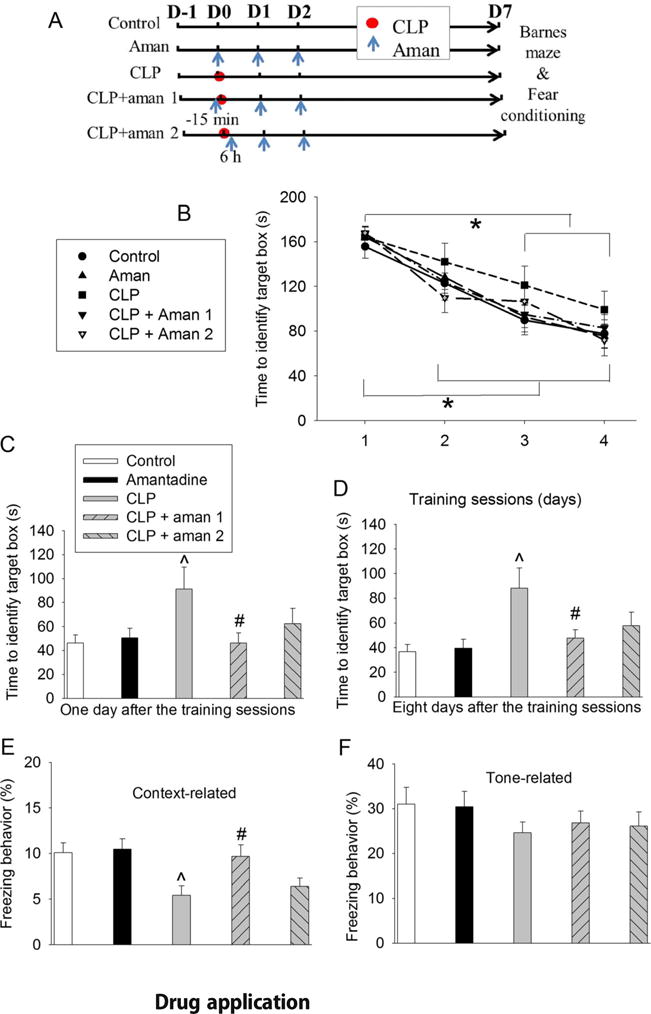
Amantadine attenuated sepsis-induced learning and memory dysfunction in CD-1 mice. Mice were subjected to Barnes maze and fear conditioning from 1 week after CLP. a Scheme of experimental protocol. b Training sessions of Barnes maze. c One day after the training sessions. d Eight days after training sessions. e Context-related fear conditioning. f Tone-related fear conditioning. Results are mean ± SD (n = 15). *P < 0.05 compared with the corresponding values on day 1. ^P < 0.05 compared with control. #P < 0.05 compared with CLP alone. Aman: amantadine; D: day
In experiment 2 (Fig. 5a), CU-CPT22 (#614305, Calbiochem, Billerica, MA), a TLR1/TLR2 antagonist [14], was used. Six- to eight-week-old CD-1 male mice were randomly divided into four groups: (1) control, (2) CU-CPT22, (3) CLP plus dimethyl sulfoxide (DMSO) (intraperitoneal injection of DMSO was administered 1 h before the surgery to create CLP), and (4) CLP plus CU-CPT22 (intraperitoneal injection of CU-CPT22 in DMSO was administered 1 h before the surgery to create CLP). Each group was aimed to have 15 mice. One week after CLP, these mice were started to be tested in Barnes maze and then fear conditioning. Separate mice were assigned to the same groups as above (n = 6 per group) and sacrificed at 24 h to harvest brain tissues after the surgery for ELISA.
Fig. 5.
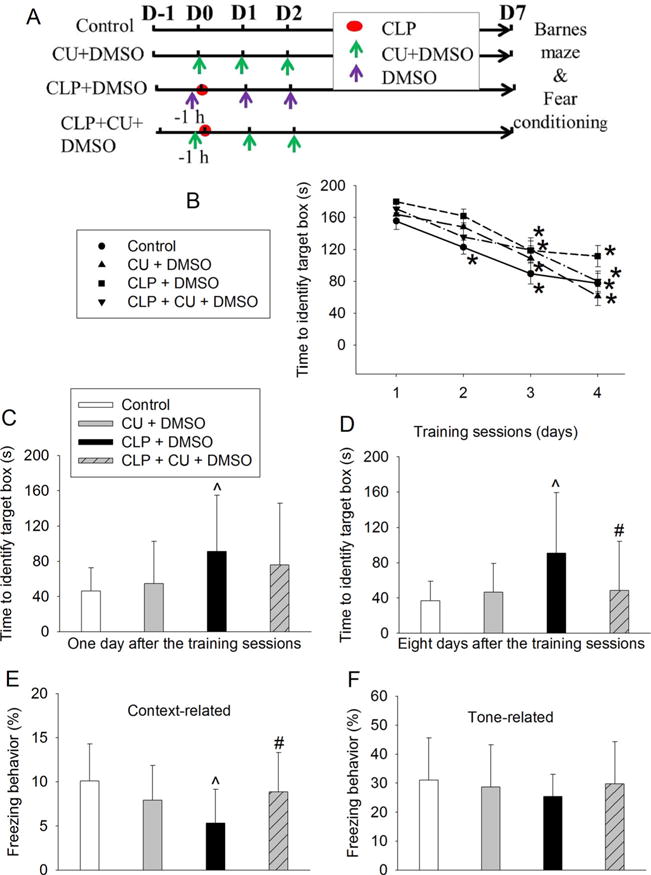
CU-CPT22 attenuated sepsis-induced learning and memory dysfunction in CD-1 mice. Mice were subjected to Barnes maze and fear conditioning from 1 week after CLP. a Scheme of experimental protocol. b Training sessions of Barnes maze. c One day after the training sessions. d Eight days after training sessions. e Context-related fear conditioning. f Tone-related fear conditioning. Results are mean ± SD (n = 15). *P < 0.05 compared with the corresponding values on day 1. ^P < 0.05 compared with control. #P < 0.05 compared with CLP alone. Aman: amantadine; CU: CU-CPT22; D: day
In experiment 3 (Fig. 6a), TLR2 knockout male mice (B6.129-Tlr2tm1Kir/J, stock number: 004650; Jackson Laboratory, Bar Harbor, ME) and C57BL/6J wild-type male mice (the mouse strain suggested by Jackson laboratory to be the control mice for the TLR2−/− mice) were used. The TLR2−/− mice were initially backcrossed with C57BL/6J wild-type mice. The C57BL/6J mice were from Charles River Laboratories (Wilmington, MA). Six- to eight-week-old male TLR2−/− mice were randomly divided into the following groups: (1) control, (2) CLP, and (3) CLP plus amantadine (intraperitoneal injection of amantadine was administered 15 min before the surgery). Six- to eight-week-old male C57BL/6J wild-type mice were randomly divided into control or CLP group. Each group was aimed to have at least 6 mice. One week after CLP, these mice were started to be tested in Barnes maze and then fear conditioning tests. Separate mice were assigned to the same groups as above (n = 6 per group) and sacrificed at 24 h to harvest brain tissues after the surgery for ELISA.
Fig. 6.
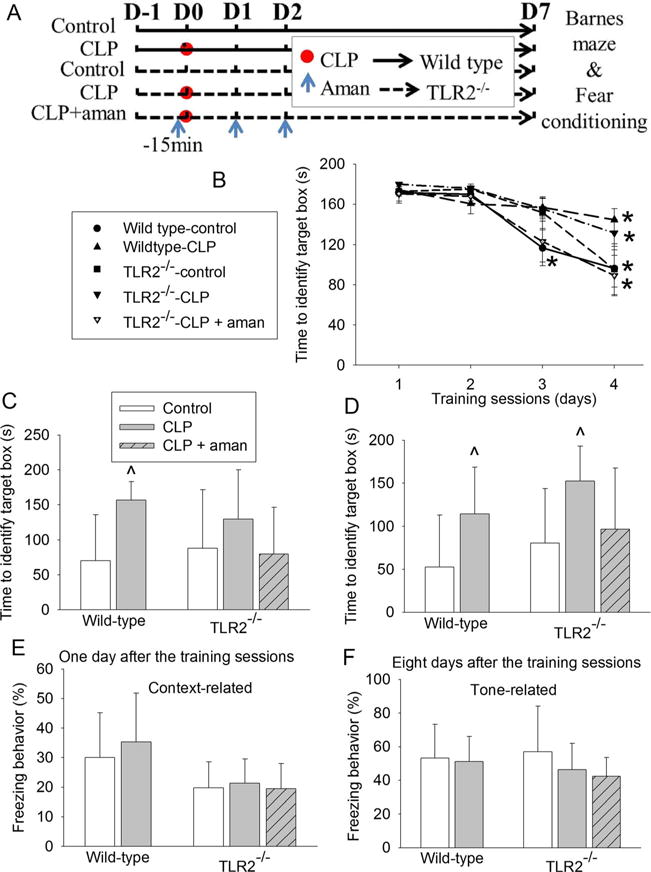
Effects of TLR2 knockout on sepsis-induced learning and memory dysfunction in mice. C57BL/6J mice (wild-type mice) and TLR2−/− mice were subjected to Barnes maze and fear conditioning from 1 week after CLP. a Scheme of experimental protocol. b Training sessions of Barnes maze. c One day after the training sessions. d Eight days after training sessions. e Context-related fear conditioning. f Tone-related fear conditioning. Results are mean ± SD (n = 7–9). *P < 0.05 compared with the corresponding values on day 1. ^P < 0.05 compared with control. Aman: amantadine; D: day
Anesthesia and surgery
Polymicrobial sepsis was induced by CLP according to the method described by Chaudry et al. [15]. Animals were anesthetized with 1.8% isoflurane; a 1-cm ventral midline incision was performed. The cecum was then carefully exposed, ligated just distal to the ileocecal valve with a 3–0 silk suture to avoid intestinal obstruction, and punctured twice using a 19-gauge needle. The punctured cecum was squeezed to expel a small amount of fecal material and returned to the abdominal cavity. The abdominal incision was closed in layers with a 7–0 silk suture and wound clips. The wound was infiltrated with 0.25% bupivacaine. All animals received subcutaneous administration of 1 ml normal saline (NS) immediately after the operation to provide fluid.
Drug application
Amantadine (A1260, Sigma-Aldrich, St. Louis, MO) was dissolved in NS and injected intraperitoneally at 30 mg/kg/day for 3 days with the first dose at 15 min before surgery or 6 h after surgery. Similar injections were performed in the amantadine only group. The amantadine dose was chosen based on previous studies [5, 16, 17].
CU-CPT22 solution (0.3 mg/ml) was prepared by dissolving CU-CPT22 powder in 2.5% dimethyl sulfoxide (DMSO; Fisher Scientific, Fair Lawn, NJ) and injected intraperitoneally at 3 mg/kg/day for 2 days with the first dose at 1 h before surgery. Similar injections were performed in the CU-CPT22 only group. The CU-CPT22 dose was chosen based on previous studies [14, 18]. In the CLP plus DMSO group, 2.5% DMSO was injected intraperitoneally at 10 ml/kg/day for 2 days with the first dose at 1 h before surgery.
Barnes maze
One week after surgery, the animals were subjected to Barnes maze as we previously described [5, 19] to test their spatial learning and memory. Animals were first placed in the middle of a circular platform with 20 equally spaced holes (SD Instruments, San Diego, CA). One of these holes was connected to a dark chamber called target box. Aversive noise (85 dB) and bright light (200 W) shed on the platform were used to encourage mice to find the target box. They had a spatial acquisition phase that lasted for 4 days with 3 min per trial, 2 trials per day and 15 min between each trial. Animals then went through the reference memory phase to test the short-term retention on day 5 and long-term retention on day 12. No test or handling was performed from day 5 to day 12. The latency to find the target box during each trial was recorded with the assistance of ANY-Maze video tracking system (SD Instruments).
Fear conditioning
One day after Barnes maze test, mice were subjected to fear conditioning test as we previously described [5, 19]. Each mouse was placed into a test chamber wiped with 70% alcohol and exposed to 3 tone-foot shock pairings (tone: 2000 Hz, 85 db, 30 s; foot shock: 0.7 mA, 2 s) with an intertrial interval 1 min in a relatively dark room (training sessions). The mouse was removed from this test chamber 30 s after the conditioning stimuli. The animal was placed back to the same chamber without the tone and shock 24 h later for 8 min. The animal was placed 2 h later into another test chamber that had different context and smell from the first test chamber in a relatively light room. This second chamber was wiped with 1% acetic acid. Freezing was recorded for 3 min without the tone stimulus. The tone was then turned on for 3 cycles, each cycle for 30 s followed by 1-min inter-cycle interval (4.5 min in total). Animal behavior in these two chambers was video recorded. The freezing behavior in the 8 min in the first chamber (context-related) and 4.5 min in the second chamber (tone-related) was scored by an observer who was blind to the group assignment.
Brain tissue harvest
Mice were killed by deep isoflurane anesthesia and transcardially perfused with NS at 6 or 24 h after anesthesia and surgery. Their left hippocampus was dissected out immediately for Western blotting, and the right cerebral hemisphere from Bregma −3 to −6 mm was used for immunohistochemistry. Another half of the mice in each group were euthanized under deep isoflurane anesthesia and transcardially perfused with NS at 24 h after anesthesia and surgery. Their whole hippocampus was dissected out immediately for ELISA tests.
Preparation of total cellular proteins and membrane proteins from hippocampus
Total cellular proteins and membrane proteins were prepared as we described before [20]. Briefly, to prepare total cellular protein extracts, brain tissues were homogenized in RIPA buffer (Cat. No. 89901; Thermo Scientific, Worcester, MA) containing protease inhibitor cocktail (Cat. No. P2714; Sigma, St. Louis, MO) and phosphatase inhibitor cocktail tablets (Cat. No. 04906845001; Roche Diagnostics Corporation, Mannheim, Germany). Homogenates were centrifuged at 13,000 rpm at 4 °C for 20 min. The supernatant was saved, and its protein concentration was determined by BCA Protein assay (Reagent A Cat. No.23228, Reagent B Cat. No. 23224; Thermo Scientific, Worcester, MA). To prepare the membrane protein fractions, brain tissues were homogenized in RIPA buffer (Cat. No. 89901; Thermo Scientific, Worcester, MA) containing protease inhibitor cocktail (Cat. No. P2714; Sigma, St. Louis, MO) and phosphatase inhibitor cocktail tablets (Cat. No. 04906845001; Roche Diagnostics Corporation, Mannheim, Germany) and homogenized with 20 full strokes in glass homogenizers. The lysates were centrifuged for 4200 rpm at 4 °C. The supernatant was centrifuged again at 33,300 rpm for 1 h at 4 °C. The pellet was re-suspended in the lysis buffer, and the protein concentrations of the samples were determined by BCA Protein assay (Reagent A Cat. No.23228, Reagent B Cat. No. 23224; Thermo Scientific, Worcester, MA).
Western blot analysis
Twenty micrograms of proteins per lane were separated on a polyacrylamide gel (Cat. No. 456-1025; Biorad, Hercules, CA) and then blotted onto a polyvinylidene difluoride membrane. The membranes were blocked with Protein-Free T20 Blocking Buffer (Cat. No. 37573, Thermo Scientific, Logan, UT) and incubated with the following primary antibodies overnight at 4 °C: rabbit monoclonal anti-TLR2 antibody (1:1000 dilution, Cat. No. ab108998, Abcam, Cambridge, MA), rabbit polyclonal anti-TLR4 antibody (1:1000 dilution, Cat. No. ab83444, Abcam, Cambridge, MA), mouse monoclonal anti-TLR9 antibody (1:500 dilution, Cat. No. ab12121, Abcam, Cambridge, MA), and mouse monoclonal anti-β-actin antibody (1:5000 dilution, Cat. No. ab6267, Abcam, Cambridge, MA). Appropriate secondary antibodies were used. Protein bands were visualized by Genesnap version 7.08 and quantified by Genetools version 4.01. The relative protein expression of TLR2, TLR4, and TLR9 was normalized to those of β-actin proteins from the same sample to control for errors in protein sample loading and transferring during Western analysis, respectively. The results from animals under various experimental conditions then were normalized by the mean values of the corresponding control animals.
Immunofluorescent staining
The staining was performed as we have described before [5, 21]. Briefly, mice were killed by deep isoflurane anesthesia and transcardially perfused with 4% paraformaldehyde at 24 h after the CLP. Brains were harvested, fixed in 4% paraformaldehyde at 4 °C for 18 h, and then embedded in paraffin. Coronal sections at 5 μm were mounted on slides. Antigen retrieval was performed in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) at 95 to 100 °C for 20 min. The sections were incubated with 5% normal donkey serum and 1% bovine serum albumin in Tris-buffered saline for 2 h at room temperature and then incubated at 4 °C overnight with the following primary antibodies: rabbit polyclonal anti-TLR2 antibody (1:200 dilution, Cat. No. PA1-41045; Thermo Scientific, Logan, UT, USA), goat polyclonal anti-ionized calcium binding adapter molecule 1 (Iba-1) antibody (1:1000 dilution, Cat. No. ab5076; Abcam, Cambridge, MA, USA), mouse monoclonal anti-neuronal nuclei (NeuN) antibody (1:100 dilution, Cat. No. MAB377; Calbiochem, Billerica, MA, USA), and mouse monoclonal anti-glial fibrillary acidic protein (GFAP) antibody (1:200 dilution, Cat. No. ab10062; Abcam, Cambridge, MA, USA). Sections were rinsed in Tris-buffered saline (TBS) with 0.025% triton-X 100. The donkey anti-rabbit IgG antibody conjugated with Alexa Fluor 594 (1:200 dilution, Cat. No. A21207; Invitrogen, Eugene, ON, USA), donkey anti-goat IgG antibody conjugated with Alexa Fluor 488 (1:200 dilution, Cat. No. A11055; Invitrogen, Eugene, ON, USA), donkey anti-mouse IgG antibody conjugated with Alexa Fluor 594 (1:200 dilution, Cat. No. A21203; Invitrogen, Eugene, ON, USA), or donkey anti-mouse IgG antibody conjugated with Alexa Fluor 594 (1:200 dilution, Cat. No. A21203; Invitrogen, Eugene, ON, USA) were incubated with the sections for 1 h at room temperature in the dark. After being washed in TBS, sections were mounted and cover-slipped with Vectashield mounting medium (H-1000; Vector Labs, Burlingame, CA, USA).
Images of immunostaining were acquired with a fluorescence microscope equipped with a charge-coupled device camera. A negative control without the incubation with the primary antibody was performed in all experiments.
ELISA assay of cytokines in the brain tissues
Interleukin (IL)-1β and IL-6 levels in the hippocampus at 24 h after the CLP were determined with Quantikine ELISA kits (Cat. No. MLB00C and Cat. No. M6000B; R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions and as we have described before [21, 22]. Briefly, brain tissues were homogenized on ice in 20 mM Tris-HCl buffer (pH 7.3) containing protease inhibitors (10 mg/ml aproteinin, 5 mg/ml peptastin, 5 mg/ml leupeptin, and 1 mM phenylmethanesulfonylfluoride). Homogenates were centrifuged at 10,000g for 10 min at 4 °C. The supernatant was then ultra-centrifuged at 150,000g for 2 h at 4 °C. Bradford protein assay of the supernatant was performed for each sample. The supernatant was used in ELISA. The optical density was measured at 450 nm (correction wavelength was set at 570 nm), and the amount of cytokines was calculated using the assay standard curves. The quantity of IL-1β and IL-6 in each brain sample was standardized to its protein contents.
Statistical analysis
Parametric results in normal distribution are presented as mean ± SD (n ≥ 5). The data from the training sessions of Barnes maze test within the same group were tested by one-way repeated measures analysis of variance followed by Tukey test. The data from the training sessions of Barnes maze test between groups were tested by two-way repeated measures analysis of variance followed by Tukey test. All other data were analyzed by one-way analysis of variance followed by the Tukey test if the data were normally distributed or by one-way analysis of variance on ranks followed by the Tukey test if the data were not normally distributed. Differences were considered significant at P < 0.05 based on two-tailed hypothesis testing. All statistical analyses were performed with SigmaStat (Systat Software, Point Richmond, CA).
Results
Sepsis induced impairment learning and memory and neuroinflammation that were attenuated by amantadine
No animals died in the control and amantadine groups (0%). The morality rates were 28.6, 16.7, and 31.8% for sepsis, sepsis plus amantadine 1, and sepsis plus amantadine 2 groups, respectively [X2(4, N = 90) = 12.073, P = 0.017]. These results suggest that sepsis increases the mortality.
Only the results of mice that survived the intended observation period were included and presented in the following sections.
The time for CD-1 mice to identify the target box was decreased with the increased training in the Barnes maze test. These times for the control and three amantadine groups on the training days 2, 3, and 4 were shorter than the times on day 1 while the times for mice with sepsis alone to identify the target box on the training day 3 and 4 were shorter than the time on day 1 (Fig. 1b). However, sepsis was not a factor to affect the time needed to identify the target box during the 4-day training [F(1,28) = 0.583, P = 0.451]. Of note, mice with sepsis alone took longer than control mice to identify the target box on day 1 and day 8 after the training sessions. This increase was attenuated by amantadine (Fig. 1c, d). Mice in the sepsis group had less freezing behavior than control mice in the context-related fear conditioning test, which was also attenuated by amantadine (Fig. 1e). There was no significant difference among the five groups of mice in the tone-related fear conditioning test (Fig. 1f).
Sepsis also increased the expression of IL-1β in the hippocampus. This increase was attenuated by amantadine. However, sepsis did not significantly increase IL-6 in the hippocampus (Fig. 2a, b).
Fig. 2.
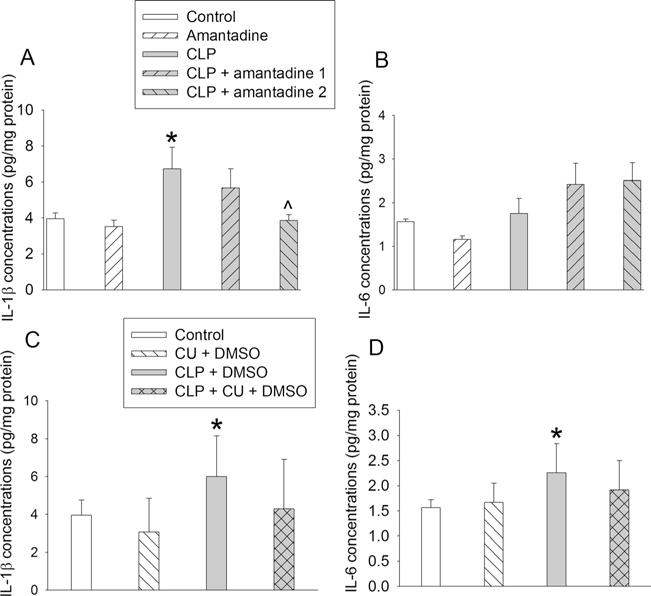
Sepsis increased IL-1β and IL-6 concentrations in the hippocampus of CD-1 mice. Hippocampus was harvested 24 h after CLP. a IL-1β levels in mice subjected to CLP and treated with amantadine. b IL-6 levels in mice subjected to CLP and treated with amantadine. c IL-1β levels in mice subjected to CLP and treated with CU-CPT22. d IL-6 levels in mice subjected to CLP and treated with CU-CPY22. Results are mean ± SD (n = 5–6). *P < 0.05 compared with control. ^P < 0.05 compared with CLP alone. CU: CU-CPT22
Sepsis increased the expression of TLRs and inhibition of TLR1/TLR2-attenuated sepsis-induced neuroinflammation and impairment of learning and memory
The expression of TLR2 and TLR9 in the plasma membrane was increased at 6 h after the onset of sepsis. The expression of TLR2, TLR4, and TLR9 in the plasma membrane was also increased 24 h after the onset of sepsis. Of note, the total amount of TLR4 and TLR9 at 6 h after the onset of sepsis and the total amount of TLR2 and TLR9 at 24 h after the onset of sepsis in the whole cell extracts were increased. Interestingly, amantadine did not appear to affect these increases of TLRs in the plasma membrane fraction or whole cell extracts (Fig. 3).
Fig. 3.
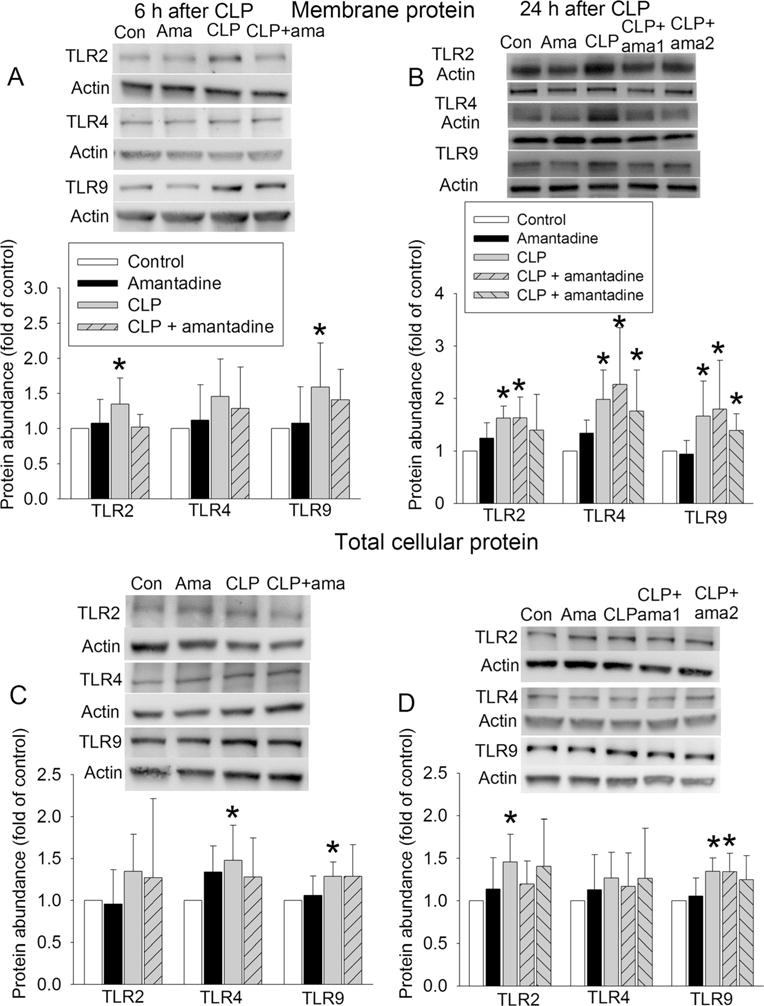
Sepsis increased the expression of TLRs in the hippocampus of CD-1 mice. Hippocampus was harvested 6 or 24 h after CLP. a TLR expression in the plasma membrane harvested 6 h after CLP. b TLR expression in the plasma membrane harvested 24 h after CLP. c TLR expression in the whole cell extract harvested 6 h after CLP. d TLR expression in the whole cell extract harvested 24 h after CLP. In each panel, representative images are presented in the top and quantification data are presented in the bottom. Results are mean ± SD (n = 5–6). *P < 0.05 compared with control. Ama: amantadine
Since TLR2 is a major TLR in the brain, we determined the cell types that expressed TLR2 by immunofluorescent staining. TLR2 staining was co-localized with NeuN and Iba-1, markers for neurons and microglia, respectively. However, TLR2 staining was not co-localized with GFAP, a marker for astrocytes (Fig. 4).
Fig. 4.
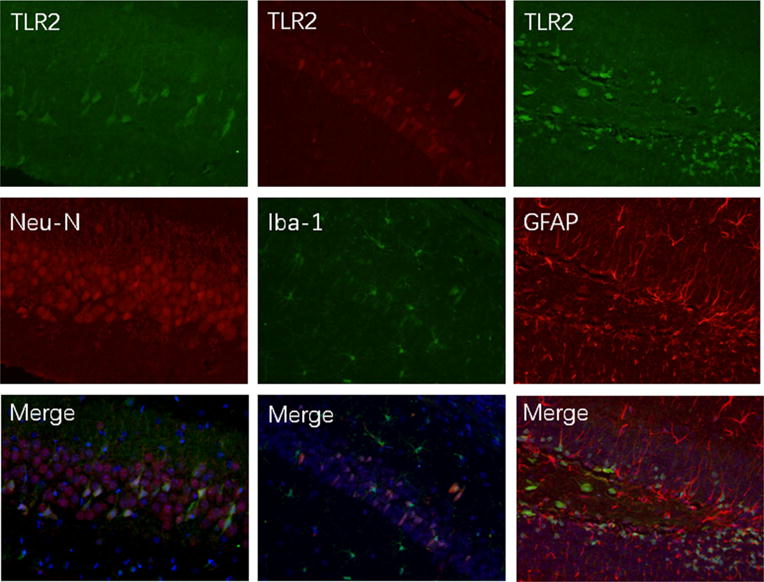
Co-expression of TLR2 with NeuN and Iba-1 in the brain sections of CD-1 mice
To determine whether TLR2 was involved in sepsis-induced neuroinflammation and cognitive dysfunction, we used CU-CPT22, a TLR1/TLR2 inhibitor [14], in the study. Consistent with the results presented above, mice in all four groups needed less time with increased training to identify the target box in the Barnes maze test (Fig. 5b). Sepsis increased the time to identify this box on day 1 and day 8 after the training sessions compared with control. This increase was attenuated by CU-CPT22 (Fig. 5c, d). The freezing behavior in the context-related fear conditioning was reduced by sepsis and this reduction was attenuated by CU-CPT22 (Fig. 5e). Tone-related fear conditioning behavior was not affected by any experimental conditions tested here (Fig. 5f). The IL-1β and IL-6 concentrations in the hippocampus were increased by sepsis and this increase was attenuated by CU-CPT22 (Fig. 2c, d).
Sepsis-induced neuroinflammation and learning and memory impairment in TLR2 knockout mice and amantadine appeared to inhibit these sepsis effects in these mice
C57BL/6J mice are suggested to be the control mice for TLR2−/− mice by Jackson Laboratory. Similar to the results of CD-1 mice, the time for CD57BL/6J mice to identify the target box was decreased with increased training in the Barnes maze test (Fig. 6b). Sepsis was not a significant factor to affect the performance of the C57BL/6J mice [F(1, 15) = 3.991, P = 0.064] or TLR2−/− mice [F(1,13) = 1.488, P = 0.244] during the training sessions. TLR2 knockout was also not a significant factor to affect the performance of mice [F(1, 14) = 0.611, P = 0.448] during the training sessions. Sepsis increased the time for C57BL/6J mice to identify the target box on day 1 and day 8 after the training sessions and for TLR2−/− mice to identify the box on day 8 after the training sessions. This increase was attenuated by amantadine (Fig. 6c, d). Sepsis did not appear to have an effect on context- and tone-related fear conditioning in C57BL/6J and TLR2−/− mice (Fig. 6e, f). Sepsis increased IL-1β and IL-6 in the hippocampus of C57BL/6J and TLR2−/− mice, and this increase was attenuated by amantadine (Fig. 7).
Fig. 7.
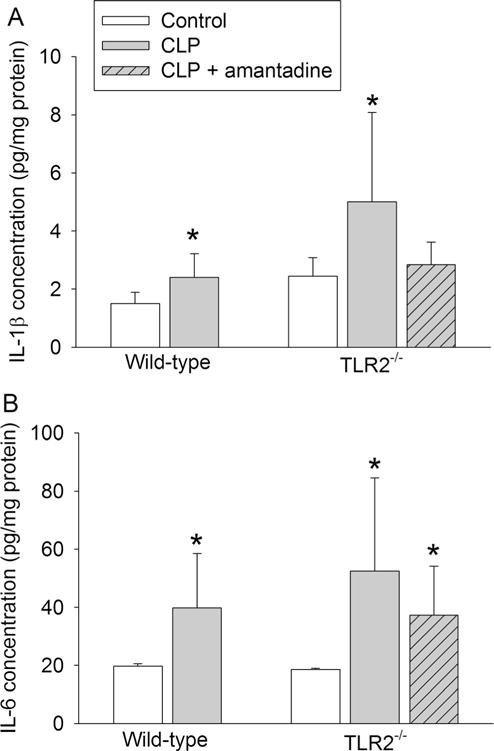
Effects of TLR2 knockout on sepsis-induced increase of IL-1β and IL-6 concentrations in mice. Hippocampus of C57BL/6J mice (wild-type mice) and TLR2−/− mice was harvested 24 h after CLP. a IL-1β levels in mice subjected to CLP and treated with amantadine. b IL-6 levels in mice subjected to CLP and treated with amantadine. Results are mean ± SD (n = 6–10). *P < 0.05 compared with control
Discussion
Our study clearly showed that mice after CLP developed learning and memory dysfunction because these mice took longer to identify the target box in the Barnes maze test and had less freezing behavior in the context-related fear conditioning than control mice. These findings are consistent with a previous study that used adult rats with CLP and tested their learning and memory by open field task and object recognition test [23]. Our study started Barnes maze test 1 week after CLP. It was more than 3 weeks after CLP when fear conditioning test was completed. Thus, our study has shown the delayed phase of cognitive dysfunction caused by sepsis.
Since inflammation is considered a mechanism for SAE [1, 2, 23] and our studies have shown a critical role of neuroinflammation in the delayed phase of postoperative cognitive dysfunction [5, 24], we propose that neuroinflammation is an underlying process for sepsis-induced cognitive dysfunction at a delayed phase. We focus on the role of TLRs in this dysfunction. TLRs play a critical role in innate immune response [6, 7, 10]. They are expressed in macrophages and dendritic cells [6, 7, 10]. In the brain, they are mainly expressed in the microglia. A small amount of TLRs may be expressed in neurons [25–27]. TLRs bind various components of pathogens or host cells. This binding leads to activation of NFκB, a transcription factor that plays a key role in inducing production of inflammatory cytokines [6, 7, 10, 11]. Since TLR2 and TLR4 are the major TLRs that can bind components of host’s cells and tissues [6, 7, 10], we determined the expression of these two TLRs. In addition, we selected TLR9, a receptor that can bind to pathogens including bacteria and viruses [28], to determine whether the effects of CLP on TLR2 and TLR4 were specific. Our results showed that sepsis increased the expression of all three TLRs in the plasma membrane, the functional site of TLRS. This increase was very fast, occurred at 6 h after CLP. Associated with this increase, there was also an increase of TLRs in the whole cell extracts, suggesting that this increase contributes to the increase of these TLRs in the plasma membrane.
Since the protein expression results did not give us an indication which TLRs might be involved in the CLP-induced neuroinflammation and cognitive impairment because all three measured TLRs were increased in expression after CLP, we arbitrarily decided to focus on TLR2. This decision was made because TLR2 is a major brain TLR, regulates neurogenesis, a process involved in cognition, and is a component of amyloid β peptide receptor complex [11, 29, 30]. Thus, it is likely that TLR2 may be involved in cognition. However, clear evidence for this involvement has not been reported and, thus, will be novel. In our study, in addition to the increased expression of TLR2, CU-CPT22 attenuated CLP-induced learning and memory dysfunction. Also, CLP induced neuroinflammation as reflected by increased IL-1β and IL-6 and CU-CPT22 attenuated this increase. These results provide initial evidence for the involvement of TLRs in SAE. Since CU-CPT22 is a cell permeable antagonist for TLR1/TLR2 [14] and TLR1 mostly binds to bacterial components [31], these results suggest a role of TLR2 in neuroinflammation and cognitive impairment caused by sepsis. Consistent with previous studies [25–27, 32], TLR2 was mostly expressed in the microglia and neurons of mice in our study. However, CLP still induces neuroinflammation and impairment of learning and memory in the TLR2 knockout mice, suggesting that other TLRs or mediators play a role in these sepsis effects. This complex situation is similar to that postoperative cognitive dysfunction. It has been shown that TLR4 is increased in the hippocampus of elderly rats at 1 to 3 days after splenectomy. These rats also had cognitive dysfunction [33, 34]. A recent study has shown that tibial fracture and fixation induced learning and memory impairment and increased TLR4 in the hippocampus of young adult mice. TLR4-deficient mice did not have significant learning and memory impairment 24 h after the surgery, although surgery-induced pattern of changes of the learning and memory parameters in these mice was similar to that in wild-type mice but with a smaller degree [12]. These results suggest that TLR4 plays a role in the cognitive dysfunction at the early phase after surgery. However, the expression of other TLRs after surgery is not determined in the previous studies [12, 33, 34]. Other TLRs may play a role because TLR4 knockout did not abolish surgery-induced neuroinflammation and dysfunction of learning and memory [12].
One of the major goals of this study was to determine whether amantadine attenuated neuroinflammation and dysfunction of learning and memory. Our results showed that amantadine with the first dose given 15 min before or 6 h after CLP attenuated CLP-induced learning and memory dysfunction, although giving the first dose at 15 min before CLP appeared to be more effective. Amantadine also appeared to inhibit neuroinflammation after CLP. However, amantadine did not affect TLR expression in the hippocampus, suggesting that inhibiting TLR expression may not be a mechanism for amantadine to inhibit neuroinflammation. In addition, amantadine may not work on TLR2 to reduce neuroinflammation and learning and memory dysfunction because amantadine remained to be effective to attenuate sepsis-induced neuroinflammation and dysfunction of learning and memory after CLP in the TLR2−/− mice. Amantadine can induce the expression of glial cell line-derived neurotrophic factor, which can inhibit microglial activation and neuroinflammation [5]. Our previous study has shown that this mechanism may play an important role in the attenuation of neuroinflammation and cognitive dysfunction after surgery [5].
We used CLP model, a widely used polymicrobial sepsis model [29]. Although this model is simple and simulates clinical sepsis, the amount of bacterial load is difficult to control [35]. In addition, the infection can be localized in some animals after CLP because adhesive small bowel can cover the ligated cecum [36]. These issues introduce variability among individuals. This feature may be a reason why the increase of IL-6 in Fig. 2d but not that in Fig. 2b in the mice with CLP reached statistical significance when compared to control mice. This small difference in the magnitude of IL-6 increase between two sets of experiments may not be due to the addition of DMSO, the solvent for CU-CPT22, in the experiments whose data are presented in Fig. 2d because DMSO used as a solvent has not been shown to increase inflammatory cytokine in the brain of rodents or affect cognition [37, 38].
Our study may have significant implications. Amantadine is already used clinically. Its clinical translation as a therapeutic agent for SAE can be quick if its effect on SAE is confirmed in human. Also, our study suggests a role of TLRs in sepsis-induced learning and memory dysfunction. TLRs may be molecular targets for reducing SAE.
Our study has limitations. Our results imply the involvement of TLRs in CLP-induced neuroinflammation and dysfunction of learning and memory but did not identify specific TLRs for these effects. Additional TLR-type specific inhibitors and molecular and genetic approaches to simultaneously knockdown or knockout the expression of a few TLRs may be needed to identify those TLRs that are involved in sepsis-induced neuroinflammation and dysfunction of learning and memory.
In conclusion, our results suggest that TLRs mediate sepsis-induced neuroinflammation and dysfunction of learning and memory. Amantadine effectively attenuates these sepsis effects possibly not through inhibition of TLR2.
Key messages.
Sepsis induces neuroinflammation and cognitive impairment, which were attenuated by amantadine. Toll-like receptors 2 mediates these sepsis effects but may not be the major target for amantadine to reduce these effects.
Acknowledgments
Grant support
This study was supported by grants (GM098308, HD089999 and AG056995 to Z Zuo) from the National Institutes of Health, Bethesda, MD, the Robert M. Epstein Professorship endowment, University of Virginia, Charlottesville, VA, and a grant (201704020222) from Guangzhou Science, Technology and Innovation Commission, Guangzhou, China.
The animal protocol was approved by the institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 80-23) revised in 2011.
Footnotes
Authors’ contributions ZZ conceived the project. WX, PH, WZ, and ZZ designed the study, WX, PH, and YL performed the experiments. WX and PH did the initial data analysis and drafted BMaterials and methods^ section. ZZ performed the final data analysis and wrote the manuscript.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557–566. doi: 10.1038/nrneurol.2012.183. [DOI] [PubMed] [Google Scholar]
- 2.Zampieri FG, Park M, Machado FS, Azevedo LC. Sepsis-associated encephalopathy: not just delirium. Clinics. 2011;66:1825–1831. doi: 10.1590/S1807-59322011001000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sprung CL, Peduzzi PN, Shatney CH, Schein RM, Wilson MF, Sheagren JN, Hinshaw LB. Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit Care Med. 1990;18:801–806. doi: 10.1097/00003246-199008000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Winters BD, Eberlein M, Leung J, Needham DM, Pronovost PJ, Sevransky JE. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med. 2010;38:1276–1283. doi: 10.1097/CCM.0b013e3181d8cc1d. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Tan H, Jiang W, Zuo Z. Amantadine alleviates postoperative cognitive dysfunction possibly by increasing glial cell line-derived neurotrophic factor in rats. Anesthesiology. 2014;121:773–785. doi: 10.1097/ALN.0000000000000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J, Sayed N, Hunter A, Au KF, Wong WH, Mocarski ES, Pera RR, Yakubov E, Cooke JP. Activation of innate immunity is required for efficient nuclear reprogramming. Cell. 2012;151:547–558. doi: 10.1016/j.cell.2012.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bi J, Shan W, Luo A, Zuo Z. Critical role of matrix metallopeptidase 9 in postoperative cognitive dysfunction and age-dependent cognitive decline. Oncotarget. 2017;8(31):51817–51829. doi: 10.18632/oncotarget.15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Jiang W, Zuo Z. Pyrrolidine dithiocarbamate attenuates surgery-induced neuroinflammation and cognitive dysfunction possibly via inhibition of nuclear factor kappaB. Neuroscience. 2014;261:1–10. doi: 10.1016/j.neuroscience.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann O, Braun JS, Becker D, Halle A, Freyer D, Dagand E, Lehnardt S, Weber JR. TLR2 mediates neuroinflammation and neuronal damage. J Immunol. 2007;178:6476–6481. doi: 10.4049/jimmunol.178.10.6476. [DOI] [PubMed] [Google Scholar]
- 12.Lu SM, Yu CJ, Liu YH, Dong HQ, Zhang X, Zhang SS, Hu LQ, Zhang F, Qian YN, Gui B. S100A8 contributes to postoperative cognitive dysfunction in mice undergoing tibial fracture surgery by activating the TLR4/MyD88 pathway. Brain Behav Immun. 2015;44:221–234. doi: 10.1016/j.bbi.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 13.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 14.Cheng K, Wang X, Zhang S, Yin H. Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew Chem. 2012;51:12246–12249. doi: 10.1002/anie.201204910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaudry IH, Wichterman KA, Baue AE. Effect of sepsis on tissue adenine nucleotide levels. Surgery. 1979;85:205–211. [PubMed] [Google Scholar]
- 16.Kim JH, Lee HW, Hwang J, Kim J, Lee MJ, Han HS, Lee WH, Suk K. Microglia-inhibiting activity of Parkinson’s disease drug amantadine. Neurobiol Aging. 2012;33:2145–2159. doi: 10.1016/j.neurobiolaging.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 17.Bido S, Marti M, Morari M. Amantadine attenuates levodopa-induced dyskinesia in mice and rats preventing the accompanying rise in nigral GABA levels. J Neurochem. 2011;118:1043–1055. doi: 10.1111/j.1471-4159.2011.07376.x. [DOI] [PubMed] [Google Scholar]
- 18.Ji YR, Kim HJ, Bae KB, Lee S, Kim MO, Ryoo ZY. Hepatic serum amyloid A1 aggravates Tcell-mediated hepatitis by inducing chemokines via toll-like receptor 2 in mice. J Biol Chem. 2015;290:12804–12811. doi: 10.1074/jbc.M114.635763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Wang Z, Zuo Z. Chronic intermittent fasting improves cognitive functions and brain structures in mice. PLoS One. 2013;8:e66069. doi: 10.1371/journal.pone.0066069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Park SH, Zhao H, Peng S, Zuo Z. A critical role of glutamate transporter type 3 in the learning and memory of mice. Neurobiol Learn Mem. 2014;114:70–80. doi: 10.1016/j.nlm.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Huang W, Zuo Z. Perioperative aspirin improves neurological outcome after focal brain ischemia possibly via inhibition of notch 1 in rat. J Neuroinflammation. 2014;11:56. doi: 10.1186/1742-2094-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Yin J, Li L, Deng J, Feng C, Zuo Z. Isoflurane postconditioning reduces ischemia-induced nuclear factor-κB activation and interleukin 1β production to provide neuroprotection in rats and mice. Neurobiol Dis. 2013;54:216–224. doi: 10.1016/j.nbd.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Della Giustina A, Goldim MP, Danielski LG, Florentino D, Mathias K, Garbossa L, Oliveira Junior AN, Fileti ME, Zarbato GF, da Rosa N, Martins Laurentino AO, Fortunato JJ, Mina F, Bellettini-Santos T, Budni J, Barichello T, Dal-Pizzol F, Petronilho F. Alphalipoic acid attenuates acute neuroinflammation and long-term cognitive impairment after polymicrobial sepsis. Neurochem Int. 2017;108:436–447. doi: 10.1016/j.neuint.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Cao L, Li L, Lin D, Zuo Z. Isoflurane induces learning impairment that is mediated by interleukin 1beta in rodents. PLoS One. 2012;7:e51431. doi: 10.1371/journal.pone.0051431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V, Konig J, Lehrach H, Nietfeld W, Trendelenburg G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 26.Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmun. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 27.Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Du X, Poltorak A, Wei Y, Beutler B. Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur Cytokine Netw. 2000;11:362–371. [PubMed] [Google Scholar]
- 29.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar a{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–281. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lien E, Ingalls RR. Toll-like receptors. Crit Care Med. 2002;30:S1–S11. [PubMed] [Google Scholar]
- 32.Mishra BB, Mishra PK, Teale JM. Expression and distribution of toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmun. 2006;181:46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, He H, Li D, Zhu W, Duan K, Le Y, Liao Y, Ou Y. The role of the TLR4 signaling pathway in cognitive deficits following surgery in aged rats. Mol Med Rep. 2013;7:1137–1142. doi: 10.3892/mmr.2013.1322. [DOI] [PubMed] [Google Scholar]
- 34.Yu L, Sun L, Chen S. Protective effect of senegenin on splenectomy-induced postoperative cognitive dysfunction in elderly rats. Exp Ther Med. 2014;7:821–826. doi: 10.3892/etm.2014.1501. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Singleton KD, Wischmeyer PE. Distance of cecum ligated influences mortality, tumor necrosis factor-alpha and interleukin-6 expression following cecal ligation and puncture in the rat. Euro Surg Res. 2003;35:486–491. doi: 10.1159/000073387. [DOI] [PubMed] [Google Scholar]
- 36.Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Huser N, Holzmann B, Stier A, Pfeffer K, Heidecke CD. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock. 2004;21:505–511. doi: 10.1097/01.shk.0000126906.52367.dd. [DOI] [PubMed] [Google Scholar]
- 37.Tian J, Dai H, Deng Y, Zhang J, Li Y, Zhou J, Zhao M, Zhao M, Zhang C, Zhang Y, Wang P, Bing G, Zhao L. The effect of HMGB1 on sub-toxic chlorpyrifos exposure-induced neuroinflammation in amygdala of neonatal rats. Toxicology. 2015;338:95–103. doi: 10.1016/j.tox.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Fan D, Li J, Zheng B, Hua L, Zuo Z. Enriched environment attenuates surgery-induced impairment of learning, memory, and neurogenesis possibly by preserving BDNF expression. Mol Neurobiol. 2016;53:344–354. doi: 10.1007/s12035-014-9013-1. [DOI] [PubMed] [Google Scholar]