

Abstract
The stereoselective synthesis of syn-β-fluoroaziridine building blocks via chiral aryl iodide-catalyzed fluorination of allylic amines is reported. The method employs HF-pyridine as a nucleophilic fluoride source together with mCPBA as a stoichiometric oxidant, and affords access to arylethylamine derivatives featuring fluorine-containing stereocenters in high diastereo- and en-antioselectivity. Catalyst-controlled diastereoselectivity in the fluorination of chiral allylic amines enabled the preparation of highly enantioenriched 1,3-difluoro-2-amines bearing three contiguous stereocenters. The enantioselective catalytic method was applied successfully to other classes of multi-functional alkene substrates to afford anti-β-fluoropyrrolidines, as well as a variety of 1,2-oxyfluorinated products.
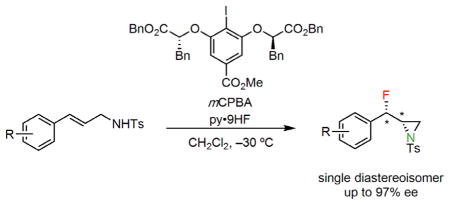
Graphical Abstract
The unique ability of fluorine substituents to modulate the physical and biological properties of organic molecules1 has inspired widespread efforts to develop methods for the selective construction of C–F bonds.2 Given the crucial importance of basic nitrogen groups in bioactive compounds, the stereoselective construction of fluoroamines has emerged as a particularly promising direction in organofluorine chemistry. For example, introduction of a stereodefined β-fluorine in the piperidine ring of MK-0731 was found to attenuate the basicity of the amine while reducing cellular efflux by P-glycoprotein and improving metabolic stability (Figure 1a).3 Strategic incorporation of fluorine-bearing stereocenters into β-peptide structures has been demonstrated to induce powerful and predictable conformational effects (Figure 1b).4
Figure 1.

Fluoroamination of alkenes. a, Examples of compounds where incorporation of a stereodefined C–F bond in a β-relationship to a nitrogen has been shown to impart beneficial properties to the molecule. b, β-Fluoroaziridination via sulfonamide trapping of a C(sp3)–I(III) intermediate.
As a result of the strong interest in accessing compounds possessing the vicinal fluoroamine substructure, a variety of approaches have been devised for their synthesis. Thanks to the availability of effective organocatalytic methods for α-fluorination of carbonyl compounds,5 one-pot α-fluorination/reductive amination protocols are possible for accessing enantioenriched vicinal fluoroamines. 6 An alternative metal-free approach involves the fluoroamination of alkenes promoted by hypervalent iodine compounds.7 For example, a method to generate fluorinated piperidines enantioselectively using stoichiometric iodine(III) reagents has been reported by Nevado and coworkers.7a Other notable approaches include the use of anionic phase-transfer8 and Lewis basic9 catalysts together with Selectfluor in the stereoselective fluorination of enamides and indoles, respectively. Transition-metal catalysis has also been applied to fluoroamination reactions.10 In a particularly significant advance, an enantio- and regioselective iron(II)-catalyzed intermolecular fluoroamination of alkenes was described recently by Xu et al. 10d These examples highlight the significant progress made toward accessing β-fluoroamines, as well as the continuing need for general catalytic methods for their stereocontrolled synthesis.
We and others have developed a variety of oxidative fluorofunctionalization reactions of alkenes based on aryl iodine(I–III) catalysis.11 In the presence of even weakly nucleophilic groups proximal to the alkene, the C(sp3)–I(III) intermediate that is proposed to be generated after initial fluoride addition (e.g., A, Figure 1b) can be intercepted either temporarily or permanently to give rise to a variety of different pathways and products. This principle has been exploited with C2-symmetric aryl iodide catalysts in the highly enantioselective syntheses of anti-1,2-difluorinated cinnamamides,11g chiral β, β-difluoroarylethanes11m and 4-fluoroisochromanones.11j We hypothesized that enantioen-riched syn-β-fluoroaziridines might be generated in an analogous manner by trapping the key ArI(III) intermediate A with appropriately positioned nitrogen nucleophiles (Figure 1b). This methodology would provide access to versatile precursors to β-fluoroamines, as the strained aziridine may be engaged in regioselective ring opening with a wide variety of nucleophiles.12 We describe here the successful development and application of this principle to the highly stereoselective fluoroaziridination of cinnamyl amine derivatives, and elaboration of the products to a variety of novel fluorinated phenethylamine derivatives.13
Cinnamyl tosylamide derivative 2a was evaluated as a model substrate in the proposed fluoroaziridination reaction. Evaluation of a variety of reagent combinations and experimental protocols led to the identification of optimal conditions similar to those reported in our previously described alkene 1,1-difluorination and fluorolactonization reactions (see Supplementary Information).11j,m Thus, in the presence of chiral aryl iodide catalysts, mCPBA as a stoichiometric oxidant and HF-pyridine as a nucleophilic fluoride source and acid promoter, 2a was observed to undergo selective oxidation to generate the corresponding β-fluoroaziridine product (3a) as a single detectable diastereoisomer. Catalyst 1a proved superior with respect to both yield and enantioselectivity (94% ee) among all chiral aryl iodides that were evaluated (see Supplementary Information). Judicious optimization of HF loading (15 equiv. HF) and temperature (–30 °C) proved crucial for obtaining the desired aziridine product in high yield; at higher HF loadings and/or temperatures, the 1,3-difluoride product resulting from opening of the putative aziridinium ion intermediate was obtained. Sulfonamides proved to be uniquely effective in the fluoroaziridination reaction, as carbonyl-based protecting groups (amides, carbamates) led to the formation of 1,2-oxy-fluorinated products instead. Variation of the sulfonyl protecting group had no effect on the enantioselectivity of the reaction, but the toluenesulfonamides generally afforded the highest yields (see Supplementary Information) and were therefore selected for investigation of substrate scope.
A variety of electron-deficient cinnamyl tosylamides were found to undergo clean conversion to the corresponding β-fluoroaziridine products (3a–m) as single diastereoisomers and with high enantioselectivity (Figure 2a). The syn relative stereochemistry of the products was established by X-ray crystallographic analysis of 3b, and is consistent with the intermediacy of a fluoroiodinated adduct (A, Figure 1b). Substrates bearing electron-donating substituents were poor substrates for the fluoroaziridination reaction (see below), but it proved possible to generate products bearing O-aryl substituents by employing triflate-protected derivatives (3e and 3h). This result bears significance given the prevalence of 3- and 4-hydroxy-substitution patterns in biologically important arylethylamines. Trisubstituted cinnamyl tosylamides proved less effective as substrates, as reflected by the tertiary fluoride 3n being isolated in moderate yield and en-antioselectivity.
Figure 2.

Evaluation of substrate scope for syn-β-fluoroaziridination. a, Substrate scope evaluation for electron-deficient cinnamyl tosylamides. Isolated yields are indicated below each product (3); conditions: substrate (0.52 mmol), catalyst (10 mol%), mCPBA (0.6 mmol), pyr•9HF (7.5 mmol HF) in DCM (3.0 mL) cooled to –30 °C, 18 h. b, Product selectivity as a function of substituent. The ratio of fluoroaziridine product yield to the sum of fluoroaziridine and 1,1-difluoromethylated product was determined by 19F NMR analysis of the crude mixture.
Substrates lacking electron-withdrawing substituents were found to undergo a phenonium ion rearrangement pathway to afford 1,1-difluoromethylated products in competition with the desired fluoroaziridination reaction (Figure 2b).11m For example, nearly exclusive formation of the 1,1-difluoromethylated product was observed in the case of the unsubstituted phenyl substrate (R = H). Indeed, a good correlation between the electronic properties of the substituents (σ+) and the selectivity for the desired product was observed.14
The β-fluoroaziridine product 3c was selected as a model for synthetic elaboration studies. We found that ring opening with different classes of nucleophiles could be effected smoothly to yield a variety of α-fluoroarylethyl amine products in excellent yields and without compromise of enantiomeric integrity (Figure 3a). Fluoride opening could be accomplished by simply subjecting 3c to HF-pyridine at room temperature to yield the corresponding 1,3-difluoride (4a). Treatment of 3c with other strong Brønsted acids such as hydrobromic and trifluoroacetic acid also resulted in clean ring opening, affording 4b and 4c respectively. In contrast, strongly basic nucleophiles were found to promote elimination pathways predominantly. In accordance with literature precedent, regioselective ring opening of 3c with trimethylsilyl azide and trimethylsilylcyanide in the presence of catalytic base was observed to yield 4d and 4e respectively,15 while 4f was obtained in quantitative yield by heating 3c with thiophenol in dimethyl sulfoxide.16 While removal of tosyl protecting groups is generally challenging, preparation, elaboration and deprotection of the analogous nosyl derivatives (e.g. 3o) proved straightforward (Figure 3b).17 While the fluoroaziridination reaction was found to be restricted to cinnamyl amine derivatives, the aromatic group in 3h could be degraded oxidatively to a functional carboxylic acid handle in a simple two-step sequence (Figure 3c).
Figure 3.

Synthetic elaboration studies. a, Reactions of fluoroaziri-dine 3c (upgraded from 97 to 99% ee by recrystallization). b, Reactions of the nosyl-protected fluoroaziridine 3o. c, Oxidative degradation of aromatic group in 3h to a carboxylic acid. Isolated yields are indicated below each product (4); experimental details are provided in the Supplementary Information. TBD, triazabicyclodecene; TBAF, tetrabutylammonium fluoride.
1,3-Difluorides (6a–e) bearing three contiguous stereocenters were generated when the reaction protocol was applied to chiral substrates bearing an aliphatic allylic substituent, as in 5a (Figure 4a). Higher HF loadings (25–50 equiv. HF) were required to achieve full conversion of the starting material, and under these conditions the fluoride addition products were obtained exclusively. Completely regioselective ring opening of the putative 1,2-disubstituted aziridinium ion intermediate was observed, presumably due to the β-fluorine inductively deactivating ring opening at the homobenzylic position. The degree and sense of diastereoselectivity in the reaction was found to be subject to high levels of catalyst control. In the presence of an achiral catalyst such as iodobenzene, subjection of enantiopure 5a to the fluoroaziridination protocol resulted in generation of the 1,3-difluoride 6a in low, 1.7:1 d.r. (anti/syn). In contrast, chiral catalyst 1a promoted formation of either diastereomer of 6a with high selectivity depending on the enantiomer of catalyst employed. Consistent results were obtained in the catalyst-controlled difluorination of other substituted cinnamyl sulfonamides (5b–5e). X-Ray crystal structure analyses of both diastereomers of 6a revealed that the alkyl chains adopt dramatically different conformations in the solid state to minimize unfavorable dipolar interactions between the 1,3-difluoro substituents. These observations add to the increasing body of evidence that 1,3-difluorides can serve as potentially valuable conformational control elements.
Figure 4.

Extension of the fluoroamination reaction protocol. a, Formation of 1,3-difluoro-2-amines bearing three contiguous ste-reocenters from enantioenriched allylic sulfonamides. b, Fluoroam-ination of bis-homoallylic sulfonamide 7 results in formation of the corresponding anti-β-fluoropyrrolidine. Isolated yields are indicated below all products; the yield in parentheses corresponds to re-crystallized product. Full experimental details are provided in the Supplementary Information.
Variation of the spacing between the alkene and the sulfonamide was examined with an interest in generating fluorinated N-heterocycles of varying size. Substrates bearing either two or four methylene groups between the alkene and sulfonamide did not form the desired 4- or 6-membered N-heterocycles. Instead, low reactivity and conversion of the starting material to multiple uncharacterized fluorinated products were observed. In contrast, the fluoroamination protocol was applied successfully to the formation of 5-membered N-heterocycles. For example, β-fluoropyrrolidine 8 was obtained in good yield and in high levels of diastereo- and enantioselectivity (Figure 4b). The enantiomeric purity of 8 was readily upgraded to >99% by recrystallization. While a syn relationship is observed in the case of the fluoroaziri-dine products (2a–n), anti-fluoroamination of the alkene in 7 was observed.7a The fluoroaziridination and fluoropyrrolidination reactions therefore must proceed by fundamentally different mechanisms, yet both pathways can be promoted with high enantioselectivity using the same aryl iodide catalyst 1a.
For several classes of cinnamyl derivatives, neighboring group participation by O-centered functional groups was found to result in stereoselective generation of oxyfluorinat-ed products. Benzyl ether 9 underwent a fluorinative rearrangement reaction to generate the 1,3-difluoride 10, consistent with formation and fluorinative ring opening of the corresponding oxiranium ion intermediate. As noted above, allylic amine substrates bearing carbonyl-containing nitrogen protecting groups such as carbamates and acetamides were found to undergo cyclization via reaction at the carbonyl oxygen rather than the nitrogen, resulting in the formation of a variety of novel 1,2-oxyfluorinated products (Figure 5). For example, when the reaction protocol was applied to Cbz-protected cinnamyl amine 11, formal 1,2-oxyfluorination of the alkene was observed to generate the cyclic carbamate 12 as a single observable diastereoisomer in good yield and in 93% ee. Allylic acetates such as 13 were observed to undergo reaction via an analogous pathway, resulting in the formation of mixtures of hydroxy and acetoxy products that presumably arise from hydrolytic ring opening of an acetoxonium ion intermediate. Treatment of the product mixture with acetic anhydride yielded the corresponding 1-fluoro-2,3-diacetoxylated product 14 in good yield and as a single observable diastereoisomer in 94% ee.
Figure 5.

Catalytic diastereo- and enantioselective 1,2-oxyfluorination reactions. Isolated yields are indicated below each product. Experimental details are provided in the Supplementary Information.
The catalytic enantioselective fluorofunctionalization reactions described herein provide access to a variety of new fluorine-containing chiral compounds with potentially unique conformational and functional properties. Efforts to evaluate and exploit the utility of these polyfunctional fluorinated compounds are currently underway.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM043214), and by an NSF pre-doctoral fellowship to S.M.B. We thank Dr. Shao-Liang Zheng (Harvard University) for determination of all of the X-ray crystal structures.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Experimental procedures; characterization data (PDF)
Crystallographic data for 3b (CIF)
Crystallographic data for 4a (CIF)
Crystallographic data for syn-6a (CIF)
Crystallographic data for anti-6a (CIF)
Crystallographic data for 8 (CIF)
Crystallographic data for 12 (CIF)
Crystallographic data for 16 (CIF)
References
- 1.For some leading reviews, see: Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. J Med Chem. 2015;58:8315. doi: 10.1021/acs.jmedchem.5b00258.Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c.Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f.
- 2.For some leading reviews, see: Yang X, Wu T, Phipps RJ, Toste FD. Chem Rev. 2015;115:826. doi: 10.1021/cr500277b.Lectard S, Hamashima Y, Sodeoka M. Adv Synth Catal. 2011;352:2708.
- 3.(a) Morgenthaler M, Schweizer E, Hoffman-Roder F, Benini F, Martin RE, Jaeschke G, Wagner B, Fischer H, Bendels S, Zimmerili D, Schneider J, Hiedrich F, Kansy M, Muller K. ChemMedChem. 2007;2:1100. doi: 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]; (b) Cox CD, Coleman PJ, Breslin MJ, Whitman DB, Garbaccio RM, Fraley ME, Buser CA, Walsh ES, Hamilton K, Schaber MD, Lobell RB, Tao W, Davide JP, Diehl RE, Abrams MT, South VJ, Huber HE, Torrent M, Prueksaritanont T, Li C, Slaughter DE, Mahan E, Fernandez-Metzler C, Yan Y, Kuo LC, Kohl NE, Hartman GD. J Med Chem. 2008;51:4239. doi: 10.1021/jm800386y. [DOI] [PubMed] [Google Scholar]
- 4.(a) Seebach D, Dubost E, Mathad RI, Jaun B, Limbach M, Löweneck M, Flögel O, Gardiner J, Capone S, Beck AK, Widmer H, Langenegger D, Monna D, Hoyer D. Helv Chim Acta. 2008;91:1736. [Google Scholar]; (b) Mathad RI, Gessier F, Seebach D, Jaun B. Helv Chim Acta. 2005;88:266. [Google Scholar]
- 5.For seminal examples utilizing organocatalysts, see: Shibata N, Suzuki E, Takeuchi Y. J Am Chem Soc. 2000;122:10728.Cahard D, Audouard C, Plaquevent JC, Roques N. Org Lett. 2000;2:3699. doi: 10.1021/ol006610l.Kim DY, Park EJ. Org Lett. 2002;4:545. doi: 10.1021/ol010281v.Enders D, Hüttl MR. Synlett. 2005;6:991.Marigo M, Fielenbach D, Braunton A, Kjærsgaard A, Jørgensen KA. Angew Chem, Int Ed. 2005;44:3703. doi: 10.1002/anie.200500395.Steiner DD, Mase N, Barbas CF., III Angew Chem, Int Ed. 2005;44:3706. doi: 10.1002/anie.200500571.Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f.
- 6.(a) Fadeyi OO, Lindsley CW. Org Lett. 2009;11:943. doi: 10.1021/ol802930q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schulte ML. Org Lett. 2011;13:5684. doi: 10.1021/ol202415j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Appayee C, Brenner-Moyer SE. Org Lett. 2010;12:3356. doi: 10.1021/ol101167z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Kong W, Feige P, de Haro T, Nevado C. Angew Chem Int Ed. 2013;52:2469. doi: 10.1002/anie.201208471. [DOI] [PubMed] [Google Scholar]; (b) Chen H, Kaga A, Chiba S. Org Biomol Chem. 2016;14:5481. doi: 10.1039/c5ob01854d. [DOI] [PubMed] [Google Scholar]
- 8.(a) Honjo T, Phipps RJ, Raunivar V, Toste FD. Angew Chem Int Ed. 2012;51:9684. doi: 10.1002/anie.201205383. [DOI] [PubMed] [Google Scholar]; (b) Phipps RJ, Hiramatsu K, Toste FD. J Am Chem Soc. 2012;134:8376. doi: 10.1021/ja303959p. [DOI] [PubMed] [Google Scholar]
- 9.Lozano O, Blessley G, Matrinez del Campo T, Thompson AL, Giuffredi GT, Bettati M, Walker M, Borman R, Governeur V. Angew Chem Int Ed. 2011;50:8105. doi: 10.1002/anie.201103151. [DOI] [PubMed] [Google Scholar]
- 10.(a) Wu T, Yin G, Liu G. J Am Chem Soc. 2009;131:16354. doi: 10.1021/ja9076588. [DOI] [PubMed] [Google Scholar]; (b) Qiu S, Xu T, Zhou J, Guo Y, Liu G. J Am Chem Soc. 2010;132:2856. doi: 10.1021/ja909716k. [DOI] [PubMed] [Google Scholar]; (c) Zhang H, Song Y, Zhao J, Zhang J, Zhang Q. Angew Chem Int Ed. 2014;53:11079. doi: 10.1002/anie.201406797. [DOI] [PubMed] [Google Scholar]; (d) Lu D-F, Zhu C-L, Sears JD, Xu H. J Am Chem Soc. 2016;138:11360. doi: 10.1021/jacs.6b07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Dohi T, Kita Y. Chem Commun. 2009:2073. doi: 10.1039/b821747e. [DOI] [PubMed] [Google Scholar]; (b) Martín Romero R, Wöste TH, Muñiz K. Chem Asian J. 2014:9. doi: 10.1002/asia.201301637. [DOI] [PubMed] [Google Scholar]; (c) Yoshimura A, Zhdankin VV. Chem Rev. 2016;116:3328. doi: 10.1021/acs.chemrev.5b00547. [DOI] [PubMed] [Google Scholar]; (d) Suzuki S, Kamo T, Fukushi K, Hiramatsu T, Tokunaga E, Dohi T, Kita Y, Shibata N. Chem Sci. 2014;5:2754. [Google Scholar]; (e) Haubenreisser S, Wöste T, Martínez C, Ishihara K, Muñiz K. Angew Chem Int Ed. 2016;55:413–417. doi: 10.1002/anie.201507180. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mizar P, Laverny A, El-Sherbini M, Farid U, Brown M, Malmedy F, Wirth T. Chem Eur J. 2014;20:9910. doi: 10.1002/chem.201403891. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Banik SM, Medley JW, Jacobsen EN. J Am Chem Soc. 2016;138:5000. doi: 10.1021/jacs.6b02391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Molnár IG, Gilmour R. J Am Chem Soc. 2016;138:5004. doi: 10.1021/jacs.6b01183. [DOI] [PubMed] [Google Scholar]; (i) Wöste TH, Muñiz K. Synthesis. 2016;48:816. [Google Scholar]; (j) Woerly EM, Banik SM, Jacobsen EN. J Am Chem Soc. 2016;138:13858. doi: 10.1021/jacs.6b09499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Muñiz K, Barreiro L, Romero RM, Martínez C. J Am Chem Soc. 2017;139:4354. doi: 10.1021/jacs.7b01443. [DOI] [PubMed] [Google Scholar]; (l) Gelis C, Dumoulin A, Neuville L, Masson G. Org Lett. 2017;19:278. doi: 10.1021/acs.orglett.6b03631. [DOI] [PubMed] [Google Scholar]; (m) Banik SM, Medley JW, Jacobsen EN. Science. 2016;353:51. doi: 10.1126/science.aaf8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yudin AK. Aziridines and Epoxides in Organic Synthesis. Wiley-VCH; Feb, 2006. [Google Scholar]
- 13.(a) Trachsel D. Drug Testing and Analysis. 2012;4:577. doi: 10.1002/dta.413. [DOI] [PubMed] [Google Scholar]; (b) Nichols DE. Journal of Pharmaceutical Sciences. 1981;70:839. doi: 10.1002/jps.2600700802. [DOI] [PubMed] [Google Scholar]
- 14.A traditional Hammett plot depicting the log(product ratio) vs σ+ has a similar appearance to Figure 2b and is provided in the Supporting Information.
- 15.Wu J, Hou X-L, Dai L-X. J Org Chem. 2000;65:1344. doi: 10.1021/jo9913816. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Sun X, Sun W. Org Biomol Chem. 2006;4:4231. doi: 10.1039/b611707d. [DOI] [PubMed] [Google Scholar]
- 17.Fukuyama T, Jow C-K, Cheung M. Tetrahedron Lett. 1995;36:6373. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.