

SUMMARY
ZEB1 transcription factor is important in both development and disease, including many TGFβ-induced responses, and the epithelial-to-mesenchymal transition (EMT) by which many tumors undergo metastasis. ZEB1 is differentially phosphorylated in different cell types; however the role of phosphorylation in ZEB1 activity is unknown. Luciferase reporter studies and electrophoresis mobility shift assays (EMSA) show that a decrease in phosphorylation of ZEB1 increases both DNA-binding and transcriptional repression of ZEB1 target genes. Functional analysis of ZEB1 phosphorylation site mutants near the second zinc finger domain (termed ZD2) show that increased phosphorylation (due to either PMA plus ionomycin, or IGF-1) can inhibit transcriptional repression by either a ZEB1-ZD2 domain clone, or full-length ZEB1. This approach identifies phosphosites that have a substantial effect regulating the transcriptional and DNA-binding activity of ZEB1. Immunoprecipitation with anti-ZEB1 antibodies followed by western analysis with a phospho-Threonine-Proline-specific antibody indicates that the ERK consensus site at Thr-867 is phosphorylated in ZEB1. In addition to disrupting in vitro DNA-binding measured by EMSA, IGF-1-induced MEK/ERK phosphorylation is sufficient to disrupt nuclear localization of GFP-ZEB1 fusion clones. These data suggest that phosphorylation of ZEB1 integrates TGFβ signaling with other signaling pathways such as IGF-1. This article is protected by copyright. All rights reserved.
Keywords: transcription regulation, IGF1, ERK1, nuclear localization
INTRODUCTION
Zinc finger E-box-binding homeobox, ZEB1 (δEF1, Zfhx1a, Zfhep, TCF8) mediates a diverse array of processes including mesoderm-derived cell differentiation, eye development, neural development and lymphopoiesis, as well as cell proliferation and senescence (Broege et al., 2011; Gheldof et al., 2012). ZEB1 can act in the TGFβ pathway as a Smad-binding factor, and is strongly involved in the Epithelial-to-Mesenchymal Transition (EMT) during both normal development and disease (Sanchez-Tillo et al., 2012). EMT can be activated by diverse cytokine signals, including TGFβ, Notch, Wnt, and TKR (tyrosine kinase receptor) signaling, which regulate transcription factors such as ZEB1, ZEB2, Snail, Slug and Twist (Moustakas and Heldin, 2007; Polyak and Weinberg, 2009). At a molecular level, ZEB1 regulates transcription of target genes, repressing many genes implicated in immune cell, myoblast and skeletal differentiation (α4 integrin, CD4 and interleukin-2 (IL-2), p73, αMHC, α-collagen I) and EMT (E-cadherin, PATJ, CRB3), and activating mesenchymal marker genes (Broege et al., 2011). ZEB1 binds a consensus DNA sequence that corresponds to the E2-box sequence CACCTG which is a subset of E-box sequences (CANNTG) (Ikeda and Kawakami, 1995). Two isoforms of ZEB1 are able to autoregulate the ZEB1 promoter by binding to the E2-box located in its own gene, and repressing transcription (Manavella et al., 2007). Repression by ZEB1 is mediated through recruitment of the corepressor C-terminal-binding protein (CtBP1/2) and histone deacetylase (HDAC1) (Wang et al., 2009), and through the NC2 (DR1/DRAP1) negative cofactor of TATA-binding protein (Gheldof et al., 2012). However, it is likely that multiple upstream pathways are involved in ZEB1 regulation of differentiation and developmental events.
ZEB1 is regulated post-transcriptionally by the miRNA-200 family and other microRNAs (Gregory et al., 2008; Park et al., 2008), and post-translationally by SUMOylation (Long et al., 2005). However, other posttranslational modifications have been little studied. We used a sensitive immunoblot to show that cells express hyperphosphorylated and hypophosphorylated forms of ZEB1 in a cell-specific manner (Costantino et al., 2002). In addition, LC-MS/MS (Liquid Chromatography - Tandem Mass Spectrometry) studies show differential phosphorylation of ZEB1 across the cell cycle (Dephoure et al., 2008). Therefore, it was of interest to characterize the significance of phosphorylation in the biological function of this transcription factor. The present study finds that interaction of native ZEB1 with target gene DNA-binding sites in vitro is inhibited by phosphorylation. Further, repression of transcription by ZEB1 is regulated through phosphorylation of key Ser/Thr sites by kinases of signaling pathways such as IGF-1. Changes in transcriptional regulation by ZEB1 may also be mediated in part by changes in nuclear localization. Thus, the activation of ZEB1 is modulated by kinases allowing ZEB1 to serve as an integrating factor of external signals.
EXPERIMENTAL
Cell lines
CHO-K1 (Chinese Hamster Ovary), COS-7 (African green monkey kidney), and Jurkat (human lymphoblastic T-leukemia) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and propagated as previously described (Costantino et al., 2002; Manavella et al., 2007).
Reporter and expression plasmids
ZEB1 is a large transcription factor (1124 amino acids) having an N-terminal cluster of zinc fingers (ZD1), a central homeodomain (HD), and a C-terminal zinc finger domain (ZD2) followed by an acidic domain (EE) (Fig. 4A). Full-length rat ZEB1 cDNA and ZEB-1b cDNA (similar to rat Ensembl ZEB1-202 transcript having an alternate exon #1) (Cabanillas and Darling, 1996) were subcloned into pcDNA4/HisMaxB (Invitrogen, Carlsbad, CA, USA). The human ZEB1 gene promoter was isolated as indicated (Manavella et al., 2007). All the sequences and orientation of the clones were verified by sequencing. The CD4 promoter (CD4EPLuc) (Brabletz et al., 1999), and E-cadherin Luc promoter (Comijn et al., 2001) are described elsewhere. Another ZEB1 target, the p73 gene, was also used in these studies through the construct named 370 prom p73 intr which includes an intronic fragment of p73 gene inserted downstream of 370-bp of the p73 promoter (Fontemaggi et al., 2001). The ZEB1 ZD2E clone expresses the C-terminal half of the ZEB1 protein encompassing the ZD2 zinc finger domain and the acidic domain, as shown in Figure 4 and described previously (Cabanillas et al., 2001). The pcDNA1/ZD2 clone was generated after SmaI digestion of pcDNA1/ZD2E. GFP-ZEB1 clones (GFP-ZD2, GFP-ZD2-NLS, and GFP-ZD2-ZF) were made by subcloning portions of rat ZEB1 cDNA by standard methods into the vector eGFP-C3 (Clontech, Mountain View, CA, USA).
FIGURE 4. Phosphorylation within ZD2 inhibits binding to DNA and transcriptional activity.
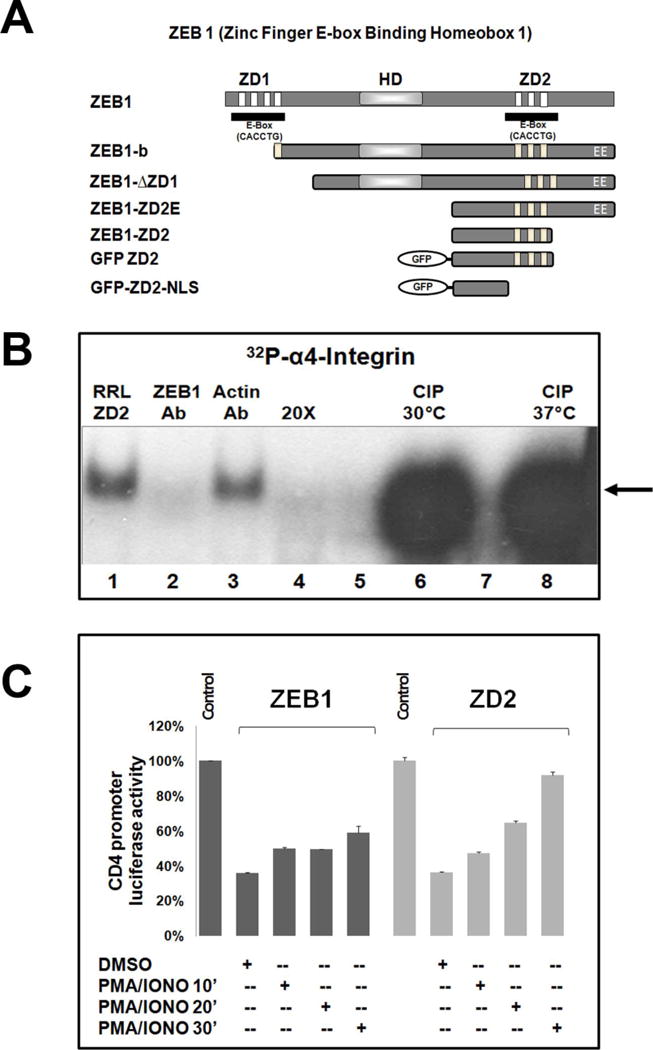
(A) Cartoons showing the ZEB1 structure representing its zinc finger domains (ZD1 and ZD2), the central homeodomain (HD) and an acidic domain (EE). The ZEB1 subclones used in different experiments are also illustrated. (B) EMSAs were performed with RRL programmed with ZD2 mRNA. ZD2-RRLs were incubated with [32P]α4-integrin (lane 1), and ZEB1 R17 antibody (lane 2), or an anti-actin antibody (lane 3), or were competed by 20X excess of unlabelled α4-integrin oligonucleotide (lane 4). ZD2-programmed RRL was treated with buffer alone (lanes 5, 7) or CIP at 30°C (lane 6) or 37°C (lane 8). (C) Luciferase reporter assay in CHO-K1 cells showing the CD4 promoter-luciferase activity co-transfected with the expression vectors for full-length ZEB1 or wtZD2 after PMA/Iono incubation for 10 min. to 30 min. The results indicate mean ± S.E.M. (n=2).
Dephosphorylation of protein in vitro
Jurkat, P19 and CHO-K1 nuclear extract proteins and rabbit reticulocyte lysates were dephosphorylated with highly purified Calf Intestinal Phosphatase (CIP, New England Biolabs) in 100 mM NaCl, 10 mM MgCl2, 50 mM Tris-HCl pH 7.9, 1 mM dithiothreitol, as described (Costantino et al., 2002), and used for EMSAs. Parallel control incubations with CIP plus 100 mM sodium phosphate (termed CIP+P) were used as a negative control since sodium phosphate buffer is a strong inhibitor of the phosphatase enzyme, but would be unlikely to inhibit any contaminating proteases or other activity in the CIP preparation.
Treatment of cells with kinase activators and inhibitors
Cells in culture were starved for 16 h in D-MEM/0.1% FBS and treated as follows: 10 or 40 ng/ml PMA plus 1 μg/ml ionomycin (Iono) for 15-30 minutes; 50 nM of Calphostin C for 30-60 min followed or not by PMA/Iono 30 min; 5 or 10 μM LY294002 for 1 h; 10, 25 or 50 μM PD98059 for 1 h; 1 or 10 μM SB20358 for 30 min; 50 ng/ml KT5720 for 30 min; or DMSO (solvent); and 10 nM IGF-1 (Millipore Co. Billerica, MA, USA) (Luo et al., 2005) or water (IGF-1 solvent) for 1 h. The amount of DMSO used was never higher than 0.1%. After treatment, cells were washed and used for isolation of nuclear extracts.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed as previously described (Manavella et al., 2007). For supershift analysis, polyclonal anti-ZEB1 antibody (anti-ZEB1-HD) against the homeodomain of ZEB1 (Smith and Darling, 2003), or two different commercial ZEB1 antibodies were used; ZEB1 R17, raised against a C-terminal region of ZEB1 and ZEB1 E20, raised against the N-terminus of the full-length protein (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Immunoblots and immunoprecipitation
Nuclear extracts from COS-7 and CHO-K1 were used for immunoblots with polyclonal anti-ZEB1-HD antibody. For immunoprecipitation, CHO-K1 cells were transfected with ΔZD1 or ZD2E DNAs, or the empty vector pcDNA1. Cell lysates were immunoprecipitated with ZEB1 R17 or anti-ZEB1-HD antibodies. Eluted immune-complexes were run in 10% SDS-PAGE gels. The blots were blocked and incubated with mouse monoclonal antibody to phospho-ThrPro MAPK/CDK substrate. Subsequently, blots were stripped, re-blocked, and incubated with ZEB1 R17 antibody.
Site-directed mutagenesis
Mutant constructs (e.g., mutant ZD2, termed mZD2; and mutant ZEB1, termed mZEB1) were created using the GeneTailor Site-Directed Mutagenesis System (Invitrogen) to modify the parent pcDNA1/ZD2 or full-length pcDNA1/ZEB1 clones following the manufacturer’s methods. Serine (Ser) or threonine (Thr) residues were changed to alanine or glutamic acid as indicated. The mutagenic primers (Integrated DNA Technologies, Coralville, IA, USA) are indicated in Supplementary Materials, Table 1. The identities of the mutated plasmids were confirmed by sequence analysis.
Luciferase reporter assays
CHO-K1 and COS-7 cells were transfected by lipofection with PEI (PolyEthylenImine, Polysciences Inc. Warrington, PA, USA) (Erbacher et al., 2004). CHO-K1 cells (5 × 104) were transfected with 0.8 μg of each DNA and 0.5 μg of CMVβ clone (β-galactosidase reporter vector, Clontech, Mountain View, CA, USA). For treatment with either activators or inhibitors of signaling pathways, thirty-two hours after adding the DNA, cells were cultured in starvation medium (D-MEM/0.1% FBS) and then treated as indicated above. Luciferase and β-galactosidase activities were evaluated as described (Cabanillas et al., 2001). Results were expressed as luciferase/β-galactosidase activity.
Confocal microscopy
5 × 104 CHO-K1 cells were grown on glass slides and transfected with 2.1 μg of each GFP-ZEB1 ZD2 fusion clone, or full-length ZEB1, mutant, or control cDNAs. Cells were serum-starved and then treated as follows: 10 μM PD98059 or 0.1% DMSO (solvent) for 1 h; and/or 10 nM IGF-1 or water (IGF-1 solvent) for 1 h, 40 ng/ml PMA plus 1 μg/ml Iono for 15 min. Cells were handled as indicated (Lorenzatti et al., 2011). For cells transfected with full-length ZEB1 constructs, the glass slides were incubated with polyclonal anti-ZEB1 antibody (anti-ZEB1-HD). Alexa 488-labeled goat anti-rabbit IgG was used as a secondary antibody. Cells were counterstained with DAPI. Confocal images were taken with an Olympus FluoView FV1000 Laser Scanning confocal microscope.
Statistical analysis
Means were compared by one-way ANOVA and Student-Newman-Keuls (SNK) tests of the differences among means. Data represent means ± S.E.M. of 3 or more experiments unless specified. P<0.05 was considered significant.
In silico Analysis
Prediction of ZEB1 potential phosphorylation sites were performed using NetPhos 2.0 (Blom et al., 2004), and kinase predictions were made by Kinase Phos (Huang et al., 2005) and PredPhospho (Kim et al., 2004). Putative nuclear localization signals (NLS) were identified through use of the program PSORT II (http://psort.nibb.ac.jp).
RESULTS
Dephosphorylation Increases DNA-binding by ZEB1
We previously found that ZEB1 is differentially phosphorylated in a cell-specific manner (Costantino et al., 2002). This is supported by several proteomic studies which identify ZEB1 phosphopeptides (Mayya et al., 2009) (Dephoure et al., 2008). To determine whether the phosphorylation state of ZEB1 influences its biological role as a transcription factor, we first tested potential changes in DNA-binding after dephosphorylation using previously characterized native response elements from the p73 gene (Fontemaggi et al., 2001), the α-4 integrin gene (Sanchez-Tillo et al., 2012), or its own promoter, ZEB1 (Manavella et al., 2007). Nuclear extracts (NE) from Jurkat and CHO-K1 cells were used for EMSA. The NE were treated with calf intestinal phosphatase (CIP) alone, or in the presence of phosphate buffer (as a negative control that blocks the activity of CIP enzyme). ZEB1 bound to each of the response elements, and in each case dephosphorylation with CIP greatly enhanced binding (Fig. 1A, lanes 2 and 4; Fig. 1B lane 2 and 8; Fig. 1C lane 2). This complex was sequence-specific as shown by competition with unlabeled oligonucleotide, and a lack of competition with either mutated p73E5 oligonucleotide or an excess of an unrelated oligonucleotide (actin) (Fig. 1C). This CIP-treated complex included ZEB1 as indicated by the ability of the anti-ZEB1 antibody to block complex formation (Fig. 1A and B). In addition, EMSAs with COS-7 cell NE also show increased specific binding of ZEB1 to both CD4 and α-4 integrin promoter sites after phosphatase treatment (Supplementary material S1). These data indicate that at least some ZEB1 protein is phosphorylated in vivo, and removing those phosphate groups increases binding to DNA.
FIGURE 1. The phosphorylation state of ZEB1 changes its DNA-binding activity.
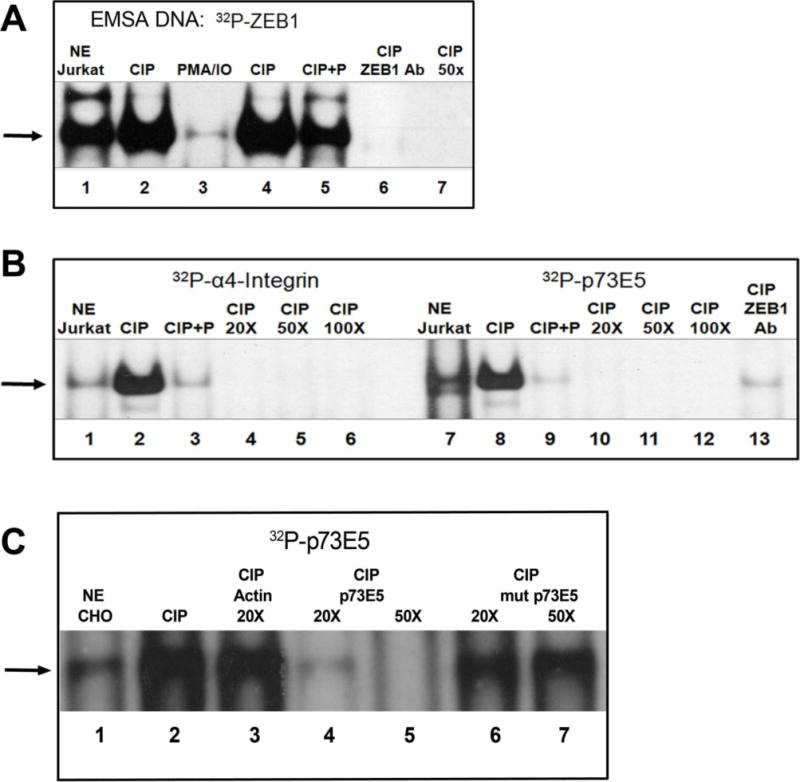
EMSAs were performed with Jurkat and CHO-K1 nuclear extracts (NE) and DNA oligonucleotides [32P]ZEB1, [32P]α4-integrin or [32P]p73E5. NE were treated with calf intestinal phosphatase (CIP) or CIP plus phosphate (CIP + P) except in “NE Jurkat”, “NE CHO” and “PMA/IO” lanes. (A) Lane 1, untreated NE; Lane 2, CIP treated; Lane 3, PMA/IO: cells were preincubated for 30 minutes with PMA+Iono as in M&M; Lanes 4 + 5, CIP-treated without or with phosphate buffer; Lane 6, ZEB1 antibody (R17) was incubated with CIP-treated NE; Lane 7, [32P]ZEB1 probe was competed with a 50-fold (50X) molar excess of unlabeled ZEB1 oligonucleotide. (B) Untreated (lanes 1, 7) or CIP-treated NE was incubated with [32P]α4-integrin probe or [32P]p73E5 probe. Binding was competed by 20X, 50X or 100X excess of cold oligonucleotides. N-terminus ZEB1 antibody (E20) was used to block ZEB1-binding. (C) [32P]p73E5 was incubated with untreated (Lane 1) or CIP-treated CHO NE. Binding was completed by 20X excess of unrelated Actin oligonucleotide, 20-50X excess of p73E5 oligonucleotide, or 20-50X excess of mutated p73E5 oligonucleotide.
Signaling pathways involved in ZEB1 phosphorylation
We investigated the signaling kinases involved in ZEB1 phosphorylation. We examined the effects of short term activation or inhibition of kinase in cells on the ability of endogenous ZEB1 to bind target genes by EMSAs. Both COS-7 and CHO-K1 cells were incubated with kinase inhibitors prior to isolating NE for these experiments. Pharmacological inhibition of protein kinase C (PKC) (by Calphostin C), PI3K (by LY294002) or MEK/ERK (by PD98059) increased binding of ZEB1 to its target genes (Fig. 2). For both PD98059 and LY294002 there was a dose-dependent increase of ZEB1 binding, starting with the lowest doses. Importantly, inhibition of p38 MAPK (by SB20358) and PKA (KT5720) did not alter binding (Fig. 2B, lanes 3 and 5), demonstrating the specificity of the effect. Conversely, we find that activation of PKC decreases the ability of ZEB1 to bind DNA. Jurkat, CHO-K1 and COS-7 cells were treated with the PKC-activators PMA plus Ionomycin (PMA/Iono) for 30 minutes. Nuclear extracts from these cells were used for either EMSA or immunoblots. Binding of ZEB1 to DNA was reduced using NE from PMA/Iono-treated Jurkat or COS-7 cells (Fig. 1A; Fig. 2C; and Supplementary material S1C and S1D). Moreover, the sequential incubation of CHO-K1 or COS-7 cells with Calphostin C (Cal C) followed by PMA/Iono demonstrated the reversible nature of phosphorylation’s effect on binding to DNA (Fig. 2C lanes 2-4; and Supplementary material S1D). Immunoblots were used to evaluate the abundance of ZEB1 protein after the treatment of the cell lines. Fig. 2D shows that neither activators nor inhibitors changed levels of ZEB1 expression. Since the short pre-treatment with PMA/Iono did not disrupt expression of ZEB1 protein, the dramatic decrease of ZEB1 binding to DNA is likely due to protein modification, not degradation. Separately, immunoblots were used to confirm the appropriate activities of the kinase activator or inhibitor treatments (Supplementary material S2). Taken together, these results support the conclusion that specific kinase activities regulate the ability of ZEB1 to bind DNA.
FIGURE 2. Signaling pathways regulate of ZEB1 binding to target genes.
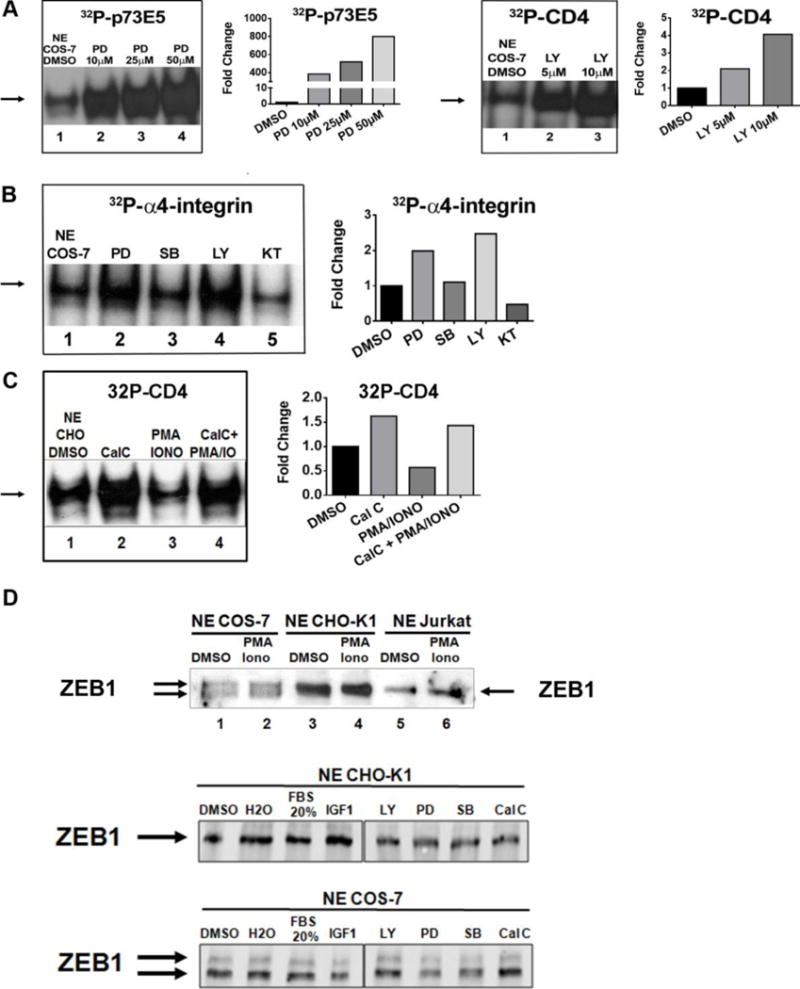
(A) EMSAs were performed with COS-7 NE incubated with [32P]p73 E5 or [32P]CD4. Cells were pre-incubated with DMSO (solvent), 10-50 μM PD98059 (PD) or 5–10 μM LY294002 (LY) prior to making NE as indicated in M&M. (B) EMSAs were performed with COS-7 NE incubated with [32P]α4-integrin. Cells were incubated with DMSO, PD98059 (PD), SB203580 (SB), LY294002 (LY) or KT5720 (KT) prior to making NE. (C) EMSAs were performed with CHO-K1 NE incubated with [32P]CD4. Cells were incubated with DMSO (solvent) (lane 1), and consecutively with Calphostin C (CalC) (lane 2), followed by PMA+Iono (lane 3), followed by CalC-PMA+Iono (lane 4). (A–C) Graphs represent quantification of the results shown in the adjacent EMSA, which are representative of three independent experiments. (D) Expression of endogenous ZEB1 protein is not altered by 20% Fetal Bovine Serum (FBS), IGF-1, PMA/Iono, LY, PD, SB or CalC. Immunoblots of ZEB1 in NE from cells incubated with indicated reagents as in M&M. Arrows indicate ZEB1 positions.
Phosphorylation changes transcriptional activity of ZEB1
In order to test whether the changes in kinase activity alters the transcriptional activity of ZEB1, we used luciferase reporter assays. We cotransfected ZEB1-responsive promoters (from the ZEB1, p73 or CD4 genes) with or without a ZEB1 expression vector into COS-7 and CHO-K1 cells that were subsequently treated with PMA/Iono, and the resulting luciferase activity was assessed. Relative to a promoter-less vector, each of these promoter-luciferase constructs was active, and showed no significant change of luciferase activity after a 15-minute PMA/Iono treatment (Fig. 3A). In the absence of PMA/Iono, cotransfection of ZEB1 repressed each of the promoters to 50-60% of the untreated control (Fig. 3B–D). Importantly, PMA/Iono treatment significantly inhibited the ability of cotransfected ZEB1 to repress these promoters (Fig. 3B–D). A similar pattern of results was obtained with a minimal CD4 promoter construct having only two copies of the ZEB1 binding site (not shown).
FIGURE 3. PMA/Iono treatment of cells inhibits ZEB1-mediated repression.
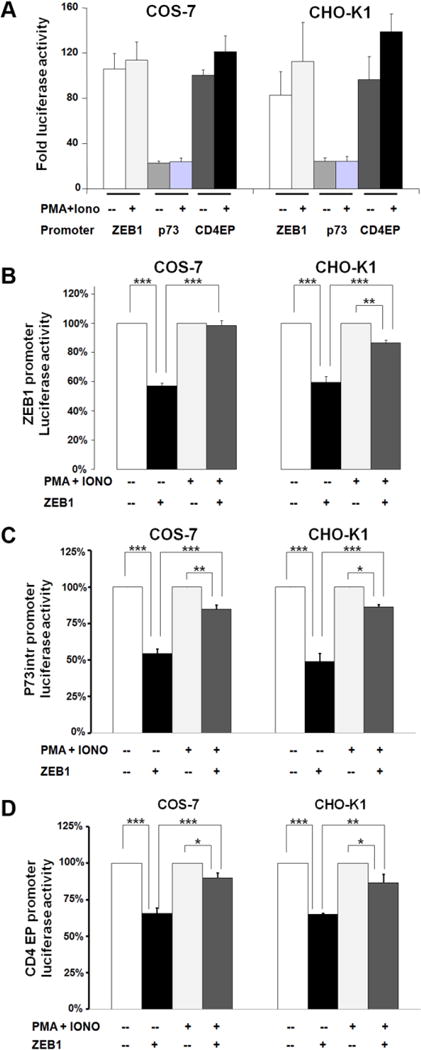
COS-7 and CHO-K1 cells were co-transfected and treated with PMA/Iono for 30 minutes as indicated in M&M. (A) In the absence of cotransfected ZEB1, PMA/Iono has no effect. Results are expressed as mean ± S.E.M. of the fold luciferase activation normalized to the vector pGL3-basic. (B-D) The effect of kinase activation on ZEB1-mediated repression is tested with (B) the ZEB1 promoter, (C) the p73intr promoter, or (D) the CD4 promoter. Results are expressed as mean ± S.E.M. (n=4-6) of promoter activation. DMSO or PMA+Iono treatment without transfected ZEB1 is set as 100%. *P<0.05; ** P<0.01; ***P<0.001.
ZD2 domain of ZEB1 is sufficient for regulation by phosphorylation
ZEB1 contains two separate zinc finger domains (ZD1 and ZD2) widely flanking a central homeodomain-like sequence (HD), and an acidic domain (EE). We were interested in localizing the minimal region of ZEB1 that is responsive to phosphorylation. A set of ZEB1 cDNAs deleting either the amino or the carboxyl terminus were constructed (Fig. 4A). cDNAs were transcribed and translated in vitro in rabbit reticulocyte lysates (RRL) (Cabanillas et al., 2001; Darling et al., 1998), and translated proteins treated with or without CIP, and used for EMSA as above. There was increased DNA binding when the ZEB1-b or ZD2E proteins were dephosphorylated by CIP (Supplementary material S3). Dephosphorylation of even the smallest domain of ZEB1 tested, ZD2, showed a dramatic increase of binding to the α4-integrin-response element (Fig. 4B lanes 6 and 8). Addition of the anti-ZEB1 R17 antibody, but not the anti-actin antibody, disrupted formation of the complex, and the complex was competed by an excess of the same unlabelled oligonucleotide, demonstrating specificity (Fig. 4B). This demonstrates that the protein region around the second zinc finger domain is sufficient for responding to phosphorylation, at least with respect to DNA-binding activity. Accordingly, luciferase reporter assays were performed with vectors expressing full-length ZEB1 or the ZD2 region, each of which repressed expression of the CD4-luciferase reporter. The ZD2 clone was designed to include a nuclear localization signal and the binding site for the corepressor CtBP1 (Broege et al., 2011), with the intent to retain the ability to regulate transcription. When transfected cells were incubated with PMA/Iono for 10, 20, or 30 minutes, repression by either ZEB1 or wtZD2 was partially or fully reversed (Fig. 4C). The effect of PMA/Iono was dependent on treatment time and in the case of ZD2 almost completely blocked repression. These results indicate that the ZD2 region of ZEB1 is sufficient for responding to PMA/Iono treatment, and emphasizes the effects of phosphorylation on both DNA-binding and transcriptional regulation. This prompted us to focus on ZD2 as a discrete region of ZEB1 that is functionally responsive to phosphorylation.
Identification of a functional phosphorylation site in ZD2
CLUSTALW alignment of human, rat and mouse sequences (Fig. 5A) shows that the ZD2 region is highly conserved among species, including conservation of putative phosphorylation sites. Bioinformatic prediction of phosphorylation sites in the wtZD2 sequence (rat ZEB1 amino acids 728 to 994) with NetPhos 2 (Blom et al., 2004), KinasePhos (Huang et al., 2005), and PredPhospho (Kim et al., 2004) gave similar sets of high-scoring phosphorylation sites, most of which were systematically mutated to alanine residues, which cannot be phosphorylated (Fig. 5B). Two sets of alanine mutations were made; set 1 (mutant ZD2-1; mZD2-1) has mutations in the flanking region (aa 851–873) and set 2 (mZD2-2) has mutations within the zinc fingers (aa 895 through 961). Each set has overlapping sub-groups of mutations, as shown in Fig. 5B. Transfection of these mutant ZD2 clones, followed by immunoblots, showed similar levels of expression as compared to wtZD2 (Supplementary material S4). Luciferase reporter assays were performed to test the ability of the alanine mutants to repress ZEB1 target genes. Wild type ZD2 expression reduced luciferase activity to 55-60% of the control (Fig. 5C). None of the mutations in set 2 significantly enhanced repression as compared to wtZD2, indicating that phosphorylation of these sites within the zinc fingers is not critical. However, both mZD2-1A and mZD2-1B repressed significantly more strongly than wtZD2, further decreasing the luciferase activity to about 35% of the control (Fig. 5C). This may be due to an inability of endogenous kinases to regulate these mutants by phosphorylation.
FIGURE 5. Mutagenesis of phosphorylation sites for transcriptional regulation of ZD2.
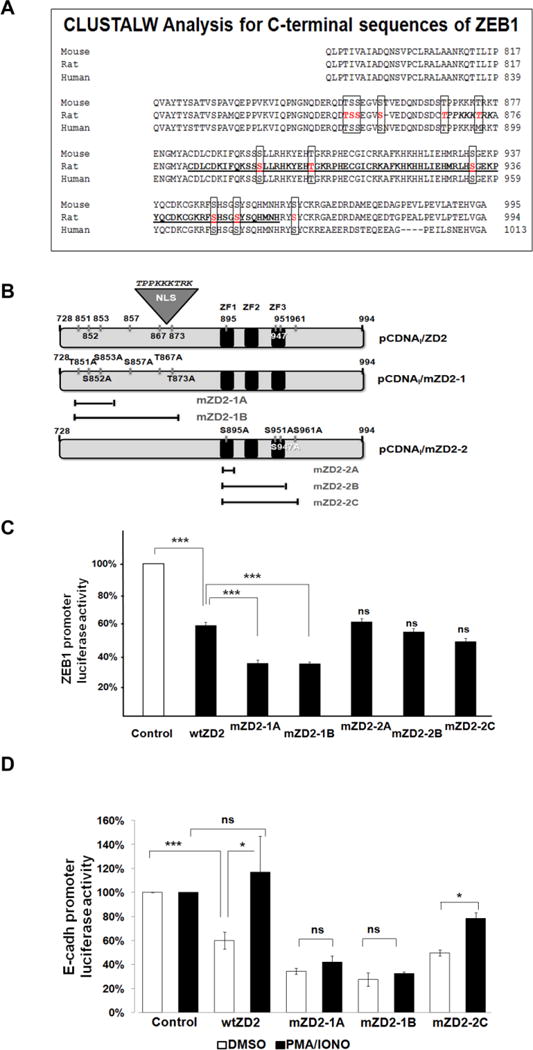
(A) Alignment by CLUSTALW of three mammalian sequences (human, rat and mouse) for a carboxy-terminal fragment of ZEB1 (788-994 in rat) showing the conserved putative phosphorylation sites in text boxes. Amino acids were numbered according to UniProtKB/Swiss-Prot Q62947 for rat ZEB1. (B) Schematic representation of the rat wtZD2 and mutant constructs. The wtZD2 clone is identical to ZEB1-ZD2 in Figure 4A. CtBP binding site is indicated. (C) Luciferase reporter assays in CHO-K1 cells testing the ZEB1 promoter-luciferase activity co-transfected with mutant ZD2-1 and mZD2-2 constructs (mZD2-1A/B, mZD2-2A/B/C). The activity of the promoter in the absence of ZD2 was set as 100%. The results represent mean ± S.E.M. of 3-4 experiments. (D) Effect of PMA/Iono treatment on the transcriptional role of mZD2-1 and mZD2-2 on the E-cadherin promoter. The results represent mean ± S.E.M. of 2 experiments. NS = not significant compared to DMSO or control. *P<0.05; ***P<0.001.
Importantly, activation of PKC also gave different responses with the different ZD2 mutations. The E-cadherin-luciferase reporter plasmid showed the expected repression when cotransfected with the wild type ZD2. As above, both the mZD2-1A and -1B constructs caused greater repression than wtZD2 (Fig. 5D, white bars). Treatment of these cells with PMA/Iono relieved repression by either wtZD2 or mZD2-2C (Fig. 5D), as well as mZD2-2A and mZD2-2B clones (not shown). Conversely, PMA/Iono had no effect on repression by mZD2-1A or mZD2-1B mutant clones, identifying critical phosphorylation sites. The inability of mZD2-1A to respond to PMA/Iono treatment suggests that PKC acts (directly or indirectly) through the mutated triplet, residues Thr-851, Ser-852, and/or Ser-853.
Signaling pathways involved in ZD2 phosphorylation
Recent reports (Chua et al., 2007; Lorenzatti et al., 2011) establish an association between IGF-1 activation and ZEB1 expression. We explored this relationship in the context of ZEB1 protein phosphorylation. CHO-K1 cells were transfected with the E-cadherin-luciferase reporter, and either the wtZD2 region of ZEB1, or its mutants. The IGF-1 pathway was activated by incubation of the transfected cells with human recombinant IGF-1 at physiological doses (Luo et al., 2005). As described above, wtZD2 or its mutants repressed the E-cadherin promoter, with the mZD2-1A and -1B mutants showing greater repression. Repression by either wtZD2 or mZD2-2C was largely relieved by IGF-1, which significantly increased the luciferase activity from 40% to 70% of the control (Fig. 6A). Conversely, repression by the mutants mZD2-1A or -1B was not relieved by IGF-1. These results suggest that IGF-1 acts through Thr-851, Ser-852, or Ser-853 (which are mutated in both mZD2-1A and -1B), and thus inhibits ZD2 from repressing the promoter.
FIGURE 6. The phosphorylation sites in mZD2-1 are relevant for ZEB1 transcriptional regulation by MAPKs.
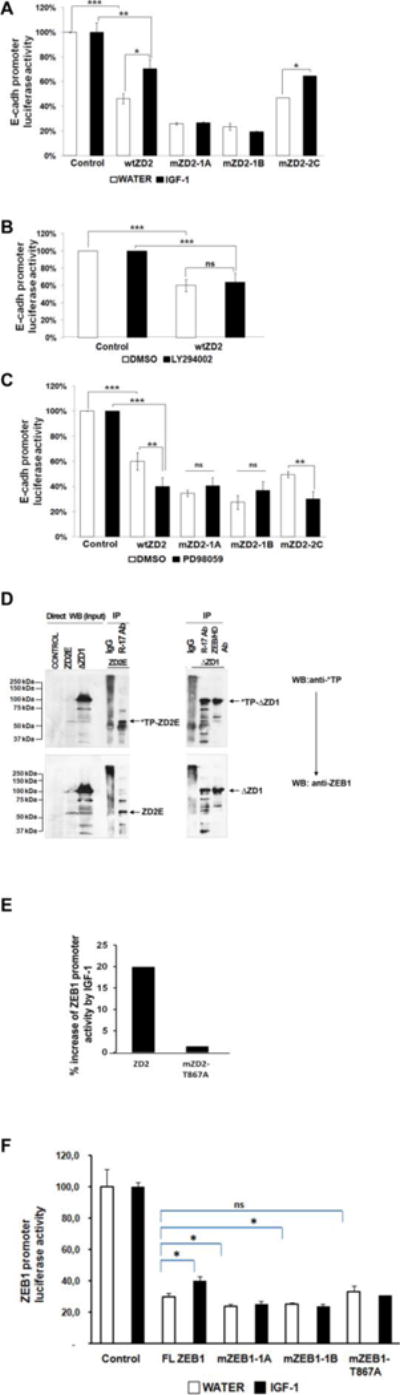
Luciferase reporter assays in CHO-K1 cells showing the effect of transfected wtZD2 or the mutants, mZD2-1A, -B and mZD2-C, on the ZEB1 target promoter E-cadherin (A, B and, C) were performed (n=2). Treatments: (A) IGF-1, (B) LY294002 or, (C) PD98059. (D) Whole-cell lysates of CHO-K1 cells transfected with ΔZD1 or ZD2E expression vectors were immunoprecipitated with two anti-ZEB1 antibodies: anti-ZEB1-HD and ZEB1 R-17 or an IgG control antibody. The blot was revealed with phospho-TP MAPK antibody and R-17 as control. (E) Luciferase reporter assays in CHO-K1 cells showing the effect of IGF-1 on the transcriptional activity induced by wtZD2, mZD2-T867A and mZD2-T867E clones. The percentage of increase of ZEB1 promoter activity by IGF-1 effect is shown. (F) Luciferase reporter assays in CHO-K1 cells incubated with or without IGF-1 showing the effect of FLZEB1 and the mutants, mZEB1-1A, mZEB1-1B and mZEB1-T867A on the ZEB1 target promoter. The activities of the promoter in the absence of ZEB1 (Control) were set as 100% *P<0.05; ** P<0.01.
IGF-1 induces activation of two different pathways downstream of IRS-1; the PI3K and the MAP-kinase (MAPK) MEK/ERK pathways (Samani et al., 2007; Witsch et al., 2010). The participation of PI3K in regulation of the ZEB1 ZD2 domain was tested by assessing the effect of LY294002 on E-cadherin promoter activity. Incubation with this PI3K inhibitor had no significant effect on the repression of E-cadherin promoter by wtZD2 (Fig. 6B), indicating that the PI3K pathway is not involved. This is in agreement with the lack of a consensus phosphorylation site in ZD2 for Akt (RXXS/T). However, we note that full-length ZEB1 has several Akt consensus sites in the first zinc finger domain, which may mediate the observed effect of LY294002 on DNA-binding of full-length ZEB1 (Fig. 2A).
The involvement of MEK/ERK signaling in ZEB1 activity was also tested. Incubation of CHO-K1 cells with the specific MEK1 inhibitor PD98059 at different concentrations enhanced repression by wtZD2 (Fig. 6C). PD98059 significantly increased repression of the E-cadherin promoter by either wtZD2 or the mZD2-2C mutant. However, no effect of PD98059 was found with the mZD2-1A or -1B mutants, experimentally implicating ZEB1 Thr-851, Ser-852, or Ser-853 sites as downstream of MEK/ERK. The activity of the ZD2 region of ZEB1 was inversely regulated by IGF-1 compared to the MEK1 inhibitor, both being dependent on the same phosphorylation sites. This suggests that the IGF-1/MEK/ERK pathway is one mechanism for phosphorylation, and regulation of ZEB1. However, the cluster of Thr-851/Ser-852/Ser-853 does not have the typical recognizable motif of MAPK substrates (Thr-Pro) suggesting that phosphorylation of these sites is secondary to a different MAPK site. A perfect MAP kinase substrate recognition motif is present nearby at the Thr-867 site which is the only Thr-Pro site in the ZD2 sequence, and which is mutated in mZD2-1B (Fig. 5). To test whether this MAPK site is phosphorylated, an immunochemical approach was taken with a phosphorylation-specific monoclonal antibody that recognizes the phospho-Thr-Pro motif. CHO-K1 cells were transfected with vectors expressing regions of ZEB1 encompassing ZD2 (either ΔZD1 or ZD2E; Fig. 4A) and the cell lysates were immunoprecipitated with one of two anti-ZEB1 antibodies. Immunoblots were probed with an anti-phospho-Thr-Pro MAPK antibody, or anti-ZEB1 as a control (Fig. 6D). ZD2E contains only one Thr-Pro sequence (at Thr-867), whereas ΔZD1 contains one additional Thr-Pro at position 348-349. Fig. 6D (upper panel) shows an anti-phospho-ThrPro reactive band of about 100 kDa in the lysates of cells transfected with ΔZD1, but not in control lysates. This phospho-Thr-Pro reactive protein is present after immunoprecipitation with either ZEB1 antibody. Similarly, the lysate from cells transfected with ZD2E reveals a positive band of 60 kDa with the phospho-Thr-Pro antibody (upper panel) that was consistent with the size of this C-terminal fragment of ZEB1. The 60 kDa phospho-Thr-Pro reactive protein is detected after immunoprecipitation with anti-ZEB1 R-17 antibody. No specific bands were immunoprecipitated with the IgG control. This indicates that transfected ZEB1 clones produce proteins which are immunoprecipitated with anti-ZEB1 antibodies, and also bound by the anti-phospho-Thr-Pro antibody, directly probing for phosphorylation of ZEB1.
To confirm the dependence of IGF-1 signaling on Thr-867, we mutated this site in the ZD2 clone and tested it in luciferase reporter assays (Fig. 6E). As expected, the mutant mZD2-T867A (ZD2 sequence mutated at Thr-867 to alanine) was not responsive to IGF-1, demonstrating that phosphorylation at this consensus site is required for regulation by IGF-1. Therefore, three lines of evidence; immunoreactivity with a specific phospho-Thr-Pro antibody, the loss of IGF-1 responsiveness by the T867A mutation, and the increased repression due to the MEK inhibitor PD98059, all support the idea that IGF-1 regulates the ZD2 domain of ZEB1 through ERK1/2.
In addition, we note that the ability of IGF-1 to reverse repression by ZD2 also depends on phosphorylation within Thr-851/Ser-852/Ser-853 (mutated in mZD2-1A; Fig. 6A). These are PKC consensus sequences, and mutation (in mZD2-1A) disrupts the effect of the PKC activators PMA/Iono (Fig. 5D). However, these mutations also disrupt the responses to IGF1 or PD98059 despite the fact that the ERK1/2 site is not mutated (Figs. 6A and 6C). Apparently, phosphorylation of the ERK1/2 site at Thr-867 must be followed by PKC-mediated phosphorylation at Thr-851/Ser-852/Ser-853, the latter of which is necessary to inhibit transcriptional repression by ZD2.
Thr-851/Ser-852/Ser-853/Thr-867 regulate the transcriptional role of full-length ZEB1
In order to confirm our observations in the context of full-length ZEB1 (FL ZEB1) we made new alanine mutant clones that target these sites. Thus, the clones used in transient transfection assays were: mZEB1-1A (FL ZEB1 mutated at Thr-851, Ser-852, and Ser-853), or mZEB1-1B (FL ZEB1 mutated at Thr-851/Ser-852/Ser-853 and Thr-867). As shown in Fig. 6F, full-length ZEB1 repressed the ZEB1 promoter and IGF-1 significantly relieved that repression. However, mutated FL ZEB1 (ZEB1-1A and mZEB1-1B) repressed luciferase activity significantly more, yet were not responsive to IGF-1. Importantly, a single mutation in FL ZEB1 (mZEB1-T867A) was repeatedly not responsive to IGF-1 treatment. These results are similar to our previous studies on the ZD2 region (Fig. 5). These data demonstrate that these phosphorylation sites influence the transcriptional activity of full length ZEB1 transcription factor, emphasizing the role of T867 in the response to IGF-1, and of Thr-851/Ser-852/Ser-853 in the basal inhibition by ZEB1.
Phosphorylation of ZEB1 by ERK1/2 changes the nuclear localization of the protein
Phosphorylation can regulate nuclear localization of proteins. As the initial step to investigate the regulation of ZEB1 subcellular localization, we identified the nuclear localization signal (NLS) of ZEB1. Bioinformatic analysis of ZEB1 (using PSORT II) suggests the presence of one NLS prior to the first zinc finger domain (ZD1), and a second NLS (PKKKTRK) at amino acids 869-875 immediately after the key phosphorylation site at Thr-867 adjacent to the second zinc finger domain (Fig. 5B). To test for NLS activity throughout ZEB1, ten GFP-fusion clones were created containing fragments scanning the entire ZEB1 cDNA, especially focusing on the basic regions identified by PSORT II (Supplementary material S5). Surprisingly, the predicted NLS prior to ZD1 was not active; however, a cryptic NLS was identified within ZD1 (Supplementary material S5). This cryptic NLS was localized between amino acids 111-241, which contains a sequence similar to the bipartite NLS in the EGR1 protein and other proteins (Mingot et al., 2009). Importantly, fluorescence microscopy of transfected cells also confirmed the presence of an active NLS between amino acids 819 - 881 in the ZD2 region (Supplementary material S5), which encompasses the NLS consensus sequence at amino acids 869 - 875.
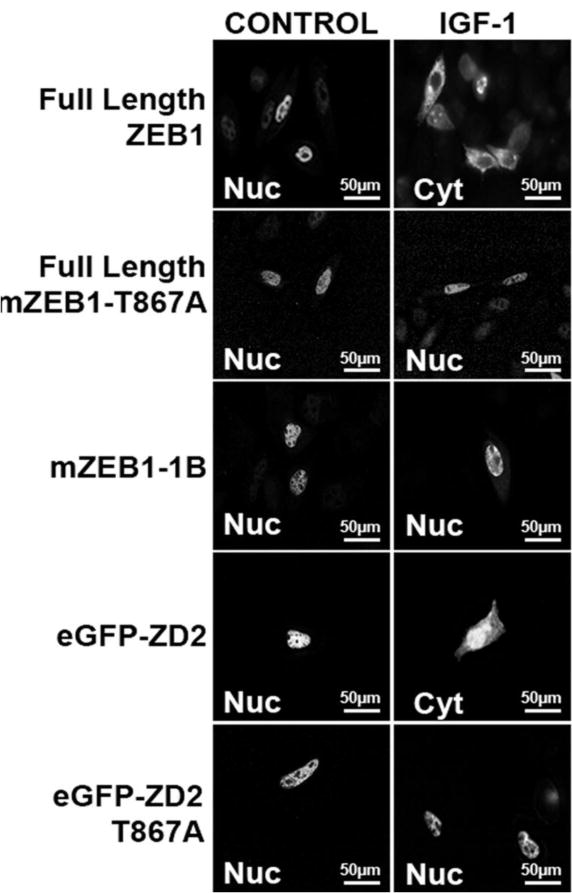
Since the Thr-867 phosphorylation site is located at the second NLS, we hypothesized that MEK/ERK may also regulate ZEB1 localization. Transfected cells were treated to activate or inhibit MAPK activity and assess the location of the GFP-ZEB1 fusion proteins. eGFP alone was used as the negative control, and showed cytosolic expression (Supplementary material S6A). In addition, a fusion protein with the NLS from GKLF (gut-enriched Kruppel-like Factor) was used as the positive control for nuclear localization (Shields and Yang, 1997). GFP-GKLF nuclear fluorescence was not modified by incubation with PD98059 or IGF-1 (Fig. 7A). Two GFP-ZEB1 fusion clones were used (Fig. 4A); GFP-ZD2 contains the same sequence as the wtZD2 cDNA clone, and GFP-ZD2-NLS contains only the portion of ZD2 prior to the zinc fingers, including Thr-867 and the NLS consensus sequence. Importantly, both GFP-ZD2 and GFP-ZD2-NLS are nuclear (Supplementary material S5), demonstrating that the basic residues within the zinc fingers are not necessary for nuclear localization as reported for Snail protein (Mingot et al., 2009). Also, the zinc fingers of ZD2 are not sufficient for nuclear targeting (Supplementary material S5, GFP-ZD2-ZF clone). The localization of GFP-ZD2 or GFP-ZD2-NLS was not altered when transfected cells were treated with PD98059, Cal C or LY294002 demonstrating that inhibition of certain kinases does not prevent nuclear localization (Fig. 7A). However, incubation with IGF-1 increased relative expression in the cytoplasm of both GFP-ZD2 and GFP-ZD2-NLS, indicating that the IGF-1 pathway can inhibit nuclear localization of ZD2. Preincubation with PD98059 to block MEK/ERK activity did block the IGF-1 response since both GFP-ZD2 and GFP-NLS remained nuclear (Fig. 7A). This indicates that IGF-1 acts through the signaling intermediates MEK/ERK to influence nuclear localization. The only MAPK substrate within ZD2 is Thr-867/Pro-868, so MEK/ERK phosphorylation of this site seems to shift the subcellular localization of ZEB1, which would reinforce the loss of its transcriptional activity. Indeed, when the mutant clone GFP-ZD2 T867A was transfected to CHO-K1 cells, there was no response to incubation with IGF-1 and the clone remained in the nuclear compartment (Fig. 7B). Importantly, the full-length mZEB1-T867A mutant remained nuclear after IGF-1 treatment (Fig. 7B). A color reproduction with the counterstaining of DAPI is shown in Supplementary material S6B. These results indicate that the phosphorylation of Thr-867 by MEK/ERK is a necessary step in regulating the subcellular localization of ZEB1.
FIGURE 7. Phosphorylation of ZD2 alters nuclear localization.
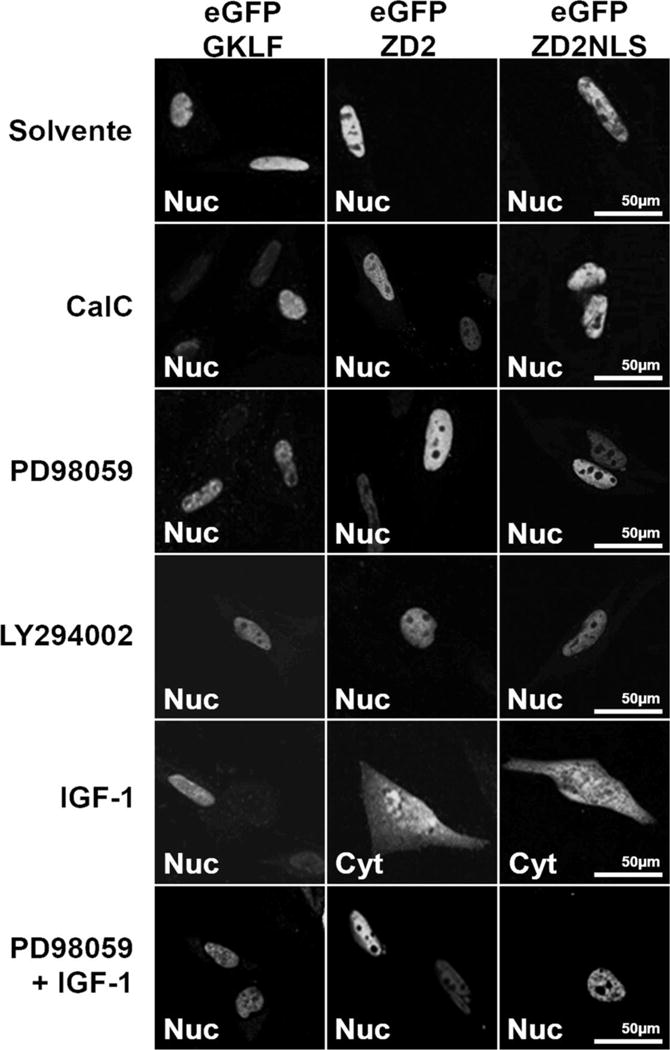
(A) Confocal microscopy of CHO-K1 cells transfected with the pEGFP-GKLF, GFP-ZD2 or GFP-ZD2-NLS expression vectors treated as indicated.
(B) Confocal microscopy of CHO-K1 cells transfected with Full-Length ZEB1, mZEB1-T867A, mZEB1-1B, eGFP-ZD2 or eGFP-ZD2 T867A clones and treated with IGF-1 as indicated. The location of ZEB1 signal after IGF-1 incubation is indicated by Cyt (cytoplasmic) or Nuc (nuclear) inside the panels. A color reproduction with the counterstaining of DAPI is shown in Supplementary material S6B.
Overall, these data suggest a model whereby kinase signaling impacts several functions of ZEB1. Several kinases contribute to rapid disruption of native ZEB1 function by repressing in vitro DNA-binding. IGF-1/ERK1/2 signaling can inhibit nuclear localization of full-length ZEB1. In addition, signaling through PKC or ERK1/2 inhibits transcriptional repression, apparently by sequential ERK1/2 phosphorylation of Thr-867 enabling subsequent PKC phosphorylation at Thr-851/Ser-852/Ser-853 (Fig. 8).
FIGURE 8. Kinase activity inhibits ZEB1 function through the ZD2 region.
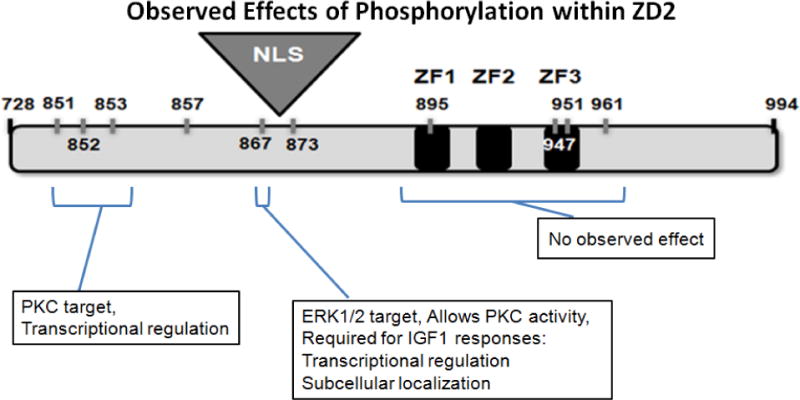
Specific phosphosites are critical for regulation of ZEB1 activities. ERK1/2 phosphorylation of T867 allows PKC activity at adjacent sites, blocking ZEB1-mediated transcriptional repression.
DISCUSSION
Post-translational regulation of ZEB1 by kinases has not been described. Nonetheless, rat ZEB1 has numerous predicted phosphorylation sites, and may be phosphorylated by various kinases at different sites within the protein. We took advantage of the observation that native ZEB1 is partially phosphorylated in vivo (Mayya et al., 2009) (Dephoure et al., 2008) (Costantino et al., 2002) to test the effect of removal of phosphates on in vitro DNA-binding. Surprisingly, this showed an increase of DNA-binding activity when native ZEB1 was de-phosphorylated. This occurred using several previously characterized response elements, and hence is not DNA sequence-specific. To identify kinase pathways that may regulate ZEB1, cell cultures were treated briefly with various inhibitors prior to isolating nuclear extracts for EMSA. These experiments show that inhibitors of PKA or p38 MAPK had no effect, whereas inhibitors of PKC, PI3K, or MEK/ERK kinases each increased native ZEB1 binding to DNA. This is consistent with other transcription factors which are phosphorylated by multiple kinases (Holmberg et al., 2002). For example, Snail participates in TGFβ signaling and EMT analogous to ZEB1, and is phosphorylated by GSK-3b, PDK1 and Pak1 (Du et al., 2010; Yang et al., 2005; Zhou et al., 2004). The significance of ZEB1 phosphorylation is suggested by our observation that treatment of cells with PMA/Iono for 15 minutes to activate PKC, inhibited transcriptional repression by transfected full-length ZEB1. Taken together, these studies suggest that the activity of endogenous full-length ZEB1 is regulated through kinase pathways.
We focused on defining the molecular regulation of the region around the second zinc finger domain, ZD2, in part because this region has previously been reported to replicate the transcriptional repression caused by the full-length ZEB1 protein (Gheldof et al., 2012; Williams et al., 1991). We find that in both DNA-binding and transfection experiments the ZD2 region matches the response of the full-length ZEB1 to phosphorylation (Fig. 4). Therefore, we sought to identify specific signaling intermediates regulating activity through the ZD2 region. Surprisingly, alanine mutation of four kinase target sequences within or immediately after the zinc fingers (in mZD2-2C) did not disrupt the response to phosphorylation. In contrast, mutation of kinase target sites before the zinc fingers did block kinase signaling; neither mZD2-1A nor mZD2-1B responded to kinase activation by PMA/Iono or IGF-1 (Fig. 5D and 6A). Inhibition of MEK/ERK (which is part of the IGF-1 pathway) enhanced repression by transfected wtZD2, but had no effect on the mZD2-1A or mZD2-1B mutants. Consistent with this, mZD2-1B includes a mutated Thr-867 that is within an ERK1/2 target sequence. Phosphorylation of Thr-867 was detected by immunoprecipitation of ZEB1 followed by western analysis with a phospho-Thr-Pro-specific antibody (Fig. 6D). These data define a rapid pathway by which IGF-1 acts through MEK/ERK to phosphorylate Thr-867 for repression of ZEB1 activity. This suggests a mechanism by which IGF-1 can modulate TGFβ signaling. Similarly, FGF signaling through ERK has been suggested to influence the TGFβ pathway through phosphorylation of CtBP1 (Shirakihara et al., 2011).
In addition to IGF-1/MEK/ERK regulation, brief PMA/Iono treatment also inhibits ZEB1 (and wtZD2) activity. This indicates that the PKC pathway can repress ZEB1, apparently through phosphorylation of the Ser/Thr amino acids mutated in mZD2-1A (Thr-851, Ser-852, and Ser-853), which is unresponsive to PMA/Iono (Fig. 4D). PKC signaling is key for many cellular functions, including embryogenesis, proliferation, differentiation, and programmed cell death (Buchner, 2000). Interestingly, mutation of the Thr-851, Ser-852, and Ser-853 triplet (which are not ERK consensus sequences) also blocks the IGF1/MEK/ERK signal, suggesting that the PKC pathway and MEK/ERK may act sequentially within this small region of ZEB1 or the full-length ZEB1 itself. Mutation of the triplet is sufficient to block the response of ZD2 to either increased or decreased MEK/ERK signaling (Fig. 6A and 6C, respectively). Also, IGF1/MEK/ERK signaling is blocked by the single T867 mutation of either ZD2 (Fig. 6E). That is, phosphorylation of Thr-867 by MEK/ERK appears to be necessary to allow phosphorylation of Thr-851/Ser-852/Ser-853 by the PKC pathway, which in turn inhibits ZEB1 activity. This sequential phosphorylation model is not an uncommon feature for integrating signaling pathways (Murbartian et al., 2005; Reuben et al., 2004; Zhao et al., 2005).
Separately, we note that the inhibitor LY294002 had no effect on the wtZD2 clone, but did activate DNA-binding and transcriptional repression by the native ZEB1. This suggests that PI3K may regulate ZEB1 through sites in the N-terminal half of the protein, outside of the wtZD2 construct.
There are several instances where repression of ZEB1 by phosphorylation could potentially contribute to known physiological responses. ZEB1 is an important repressor of the IL2 gene in T lymphocytes, and activation of T cells by PMA/Iono rapidly increases IL2 gene expression (Dreikhausen et al., 2003; Wang et al., 2009), possibly in part by phosphorylation of ZEB1. Similarly, ZEB1 contributes to regulation of cell differentiation by repression of the p73 gene; hence, the rapid stimulation of p73 mRNA expression during PMA-induced differentiation of HL60 monocytes (Fontemaggi et al., 2001) may be due in part to phosphorylation of ZEB1.
Phosphorylation can regulate the nuclear localization of a protein (Dominguez et al., 2003; Gurel et al., 2008). Cytoplasmic localization of ZEB2/SIP1 has been reported in several cancers (Oztas et al., 2010), and similarly, we observe strong cytoplasmic localization of both ZEB1 and ZEB2 in poorly differentiated oral squamous cell carcinoma (manuscript in preparation). In the current study, we have identified an active NLS immediately N-terminal to the zinc fingers in the wtZD2 construct (aa 869-875; Fig. 5). Treatment of transfected cells with IGF-1 caused cytoplasmic localization of both the GFP-ZD2 and GFP-ZD2-NLS fusion proteins as well as full-length ZEB1, in a MEK/ERK dependent fashion. The mutated ZEB1 and ZD2 at Thr-867 are accordingly not responsive to IGF-1 indicating the dependence on this site for nuclear localization. Hence, the IGF-1/MEK/ERK pathway not only inhibits DNA-binding of ZEB1 (and ZD2), but reinforces that by also inhibiting nuclear localization.
ZEB1 is important for development and differentiation of multiple different tissues, as well as EMT in normal development and during metastasis in cancer. Many of these roles are due to ZEB1 acting in the TGFβ signaling pathway (Moustakas and Heldin, 2007; Polyak and Weinberg, 2009). Repression of ZEB1 by MEK/ERK, PKC or PI3K pathways would balance activation by TGFβ signaling (Gheldof et al., 2012), allowing ZEB1 to integrate these pathways. We provide the first demonstration that ZEB1 activity is directly regulated by kinase signaling, and identify two relevant kinase pathways. We suggest that phosphorylation of ZEB1 can act to integrate TGFβ signals with other cytokines and growth factors in the local environment of the cell, such as IGF-1. The role of ZEB1 phosphorylation is only beginning to be understood. More work is needed to understand the function of each phosphorylation site and their implications in the many cellular processes in which ZEB1 takes part.
Supplementary Material
Acknowledgments
The authors thank Dr. Giulia Fontemaggi, Dr. Thomas Brabletz, and Dr. Geert Berx for the generous gifts of plasmids. We greatly thank to Dr. Pilar Crespo from Laboratorio de Microscopía Óptica, CIBICI-CIQUIBIC, Universidad Nacional de Córdoba for her invaluable help with confocal microscopy and image processing, and Dr. Marianne Mills for initial nuclear localization experiments.
Contract grant sponsor: Consejo Nacional de Investigaciones Científicas y Técnicas de Argentina (CONICET)
Contract grant number: PIP 11220100100480
Contract grant sponsor: Agencia Nacional de Promoción Cientifica y Tecnológica (ANPCYT)
Contract grant number: PICT 2010-2082
Contract grant sponsor: Secretaría de Ciencia y Técnica de la Universidad Nacional de Córdoba (SECyT)
Contract grant number: 05/C566.
Contract grant sponsor: National Institutes of Health
Contract grant number: P30ES014443, and DE019243
ABBREVIATIONS
- CIP
calf intestine phosphatase
- EMSA
electrophoresis mobility shift assays
- EMT
epithelial-mesenchymal transition
- ERK
extracellular-signal-regulated kinase
- IGF-1
insulin-like growth factor 1
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- NLS
nuclear localization signal
- PI3K
phosphoinositide 3-kinase
- PKA
protein kinase A
- PKC
protein kinase C
- PMA
Phorbol-12-Myristate-13-Acetate
- RTK
receptor tyrosine kinase
- TGFβ
transforming growth factor beta
- wt
wild type
- ZD1
first zinc finger domain
- ZD2
second zinc finger domain
- ZEB1
Zinc finger E-box-binding homeobox
Footnotes
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jcp.25338]
Additional Supporting Information may be found in the online version of this article.
The authors have no conflicts of interest.
References
- Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4(6):1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Hlubek F, Lohberg C, Meiler J, Suchy U, Kirchner T. Negative regulation of CD4 expression in T cells by the transcriptional repressor ZEB. Int Immunol. 1999;11(10):1701–1708. doi: 10.1093/intimm/11.10.1701. [DOI] [PubMed] [Google Scholar]
- Broege AM, Anose BM, Sanders MM. Regulating the expression of the ZEB1 transcription factor in health and disease. Current Trends in Endocrinology. 2011;5:75–91. [Google Scholar]
- Buchner K. The role of protein kinase C in the regulation of cell growth and in signalling to the cell nucleus. J Cancer Res Clin Oncol. 2000;126(1):1–11. doi: 10.1007/pl00008458. [DOI] [PubMed] [Google Scholar]
- Cabanillas A, Smith G, Darling D. T3-activation of the rat growth hormone gene is inhibited by a zinc finger/homeodomain protein. Mol Cell Endocrinol. 2001;181(1–2):131–137. doi: 10.1016/s0303-7207(01)00531-7. [DOI] [PubMed] [Google Scholar]
- Cabanillas AM, Darling DS. Alternative splicing gives rise to two isoforms of Zfhep, a zinc finger/homeodomain protein that binds T3-response elements. DNA Cell Biol. 1996;15(8):643–651. doi: 10.1089/dna.1996.15.643. [DOI] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- Costantino ME, Stearman RP, Smith GE, Darling DS. Cell-specific phosphorylation of Zfhep transcription factor. Biochem Biophys Res Commun. 2002;296(2):368–373. doi: 10.1016/s0006-291x(02)00880-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua H, Bhat-Nakshatri P, Clare S, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26(5):711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- Darling D, Gaur N, Zhu B. A zinc finger homeodomain transcription factor binds specific thyroid hormone response elements. Mol Cell Endocrinol. 1998;139(1–2):25–35. doi: 10.1016/s0303-7207(98)00076-8. [DOI] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105(31):10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Montserrat-Sentis B, Virgos-Soler A, Guaita S, Grueso J, Porta M, Puig I, Baulida J, Franci C, Garcia de Herreros A. Phosphorylation Regulates the Subcellular Location and Activity of the Snail Transcriptional Repressor. Mol Cell Biol. 2003;23(14):5078–5089. doi: 10.1128/MCB.23.14.5078-5089.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreikhausen UE, Gorf K, Resch K, Szamel M. Protein kinase Cbeta1, a major regulator of TCR-CD28-activated signal transduction leading to IL-2 gene transcription and secretion. Int Immunol. 2003;15(9):1089–1098. doi: 10.1093/intimm/dxg112. [DOI] [PubMed] [Google Scholar]
- Du C, Zhang C, Hassan S, Biswas MH, Balaji KC. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010;70(20):7810–7819. doi: 10.1158/0008-5472.CAN-09-4481. [DOI] [PubMed] [Google Scholar]
- Erbacher P, Bettinger T, Brion E, Coll JL, Plank C, Behr JP, Remy JS. Genuine DNA/polyethylenimine (PEI) complexes improve transfection properties and cell survival. J Drug Target. 2004;12(4):223–236. doi: 10.1080/10611860410001723487. [DOI] [PubMed] [Google Scholar]
- Fontemaggi G, Gurtner A, Strano S, Higashi Y, Sacchi A, Piaggio G, Blandino G. The Transcriptional Repressor ZEB Regulates p73 Expression at the Crossroad between Proliferation and Differentiation. Mol Cell Biol. 2001;21(24):8461–8470. doi: 10.1128/MCB.21.24.8461-8470.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheldof A, Hulpiau P, Roy F, Craene B, Berx G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cellular and Molecular Life Sciences. 2012;69(15):2527–2541. doi: 10.1007/s00018-012-0935-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory P, Bert A, Paterson E, Barry S, Tsykin A, Farshid G, Vadas M, Khew-Goodall Y, Goodall G. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- Gurel Z, Ronni T, Ho S, Kuchar J, Payne KJ, Turk CW, Dovat S. Recruitment of ikaros to pericentromeric heterochromatin is regulated by phosphorylation. J Biol Chem. 2008;283(13):8291–8300. doi: 10.1074/jbc.M707906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg CI, Tran SE, Eriksson JE, Sistonen L. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem Sci. 2002;27(12):619–627. doi: 10.1016/s0968-0004(02)02207-7. [DOI] [PubMed] [Google Scholar]
- Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005;33:W226–229. doi: 10.1093/nar/gki471. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Kawakami K. DNA binding through distinct domains of zinc-finger-homeodomain protein AREB6 has different effects on gene transcription. Eur J Biochem. 1995;233(1):73–82. doi: 10.1111/j.1432-1033.1995.073_1.x. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee J, Oh B, Kimm K, Koh I. Prediction of phosphorylation sites using SVMs. Bioinformatics. 2004;20:3179–3184. doi: 10.1093/bioinformatics/bth382. [DOI] [PubMed] [Google Scholar]
- Long J, Zuo D, Park M. Pc2-mediated Sumoylation of Smad-interacting Protein 1 Attenuates Transcriptional Repression of E-cadherin. J Biol Chem. 2005;280(42):35477–35489. doi: 10.1074/jbc.M504477200. [DOI] [PubMed] [Google Scholar]
- Lorenzatti G, Huang W, Pal A, Cabanillas AM, Kleer CG. CCN6 (WISP3) decreases ZEB1-mediated EMT and invasion by attenuation of IGF-1 receptor signaling in breast cancer. J Cell Sci. 2011;124(Pt 10):1752–1758. doi: 10.1242/jcs.084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol. 2005;170(3):455–464. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manavella PA, Roqueiro G, Darling DS, Cabanillas AM. The ZFHX1A gene is differentially autoregulated by its isoforms. Biochem Biophys Res Commun. 2007;360(3):621–626. doi: 10.1016/j.bbrc.2007.06.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, Rodionov V, Han DK. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009;2(84):ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- Mingot JM, Vega S, Maestro B, Sanz JM, Nieto MA. Characterization of Snail nuclear import pathways as representatives of C2H2 zinc finger transcription factors. J Cell Sci. 2009;122(Pt 9):1452–1460. doi: 10.1242/jcs.041749. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98(10):1512–1520. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murbartian J, Lei Q, Sando JJ, Bayliss DA. Sequential Phosphorylation Mediates Receptor- and Kinase-induced Inhibition of TREK-1 Background Potassium Channels. J Biol Chem. 2005;280(34):30175–30184. doi: 10.1074/jbc.M503862200. [DOI] [PubMed] [Google Scholar]
- Oztas E, Avci ME, Ozcan A, Sayan AE, Tulchinsky E, Yagci T. Novel monoclonal antibodies detect Smad-interacting protein 1 (SIP1) in the cytoplasm of human cells from multiple tumor tissue arrays. Experimental and molecular pathology. 2010;89(2):182–189. doi: 10.1016/j.yexmp.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Reuben PM, Sun Y, Cheung HS. Basic calcium phosphate crystals activate p44/42 MAPK signal transduction pathway via protein kinase Cmicro in human fibroblasts. J Biol Chem. 2004;279(34):35719–35725. doi: 10.1074/jbc.M403406200. [DOI] [PubMed] [Google Scholar]
- Samani AA, Yakar S, LeRoith D, Brodt P. The Role of the IGF System in Cancer Growth and Metastasis: Overview and Recent Insights. Endocr Rev. 2007;28(1):20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- Sanchez-Tillo E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69(20):3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields JM, Yang VW. Two potent nuclear localization signals in the gut-enriched Kruppel-like factor define a subfamily of closely related Kruppel proteins. J Biol Chem. 1997;272(29):18504–18507. doi: 10.1074/jbc.272.29.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakihara T, Horiguchi K, Miyazawa K, Ehata S, Shibata T, Morita I, Miyazono K, Saitoh M. TGF-beta regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011;30(4):783–795. doi: 10.1038/emboj.2010.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GE, Darling DS. Combination of a zinc finger and homeodomain required for protein-interaction. Mol Biol Rep. 2003;30(4):199–206. doi: 10.1023/a:1026330907065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lee S, Teh CE, Bunting K, Ma L, Shannon MF. The transcription repressor, ZEB1, cooperates with CtBP2 and HDAC1 to suppress IL-2 gene activation in T cells. Int Immunol. 2009;21(3):227–235. doi: 10.1093/intimm/dxn143. [DOI] [PubMed] [Google Scholar]
- Williams T, Moolten D, Burlein J, Romano J, Bhaerman R, Godillot A, Mellon M, Rauscher F, 3rd, Kant J. Identification of a zinc finger protein that inhibits IL-2 gene expression. Science. 1991;254(5039):1791–1794. doi: 10.1126/science.1840704. [DOI] [PubMed] [Google Scholar]
- Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010;25(2):85–101. doi: 10.1152/physiol.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 2005;65(8):3179–3184. doi: 10.1158/0008-5472.CAN-04-3480. [DOI] [PubMed] [Google Scholar]
- Zhao KW, Li D, Zhao Q, Huang Y, Silverman RH, Sims PJ, Chen GQ. Interferon-alpha-induced expression of phospholipid scramblase 1 through STAT1 requires the sequential activation of protein kinase Cdelta and JNK. J Biol Chem. 2005;280(52):42707–42714. doi: 10.1074/jbc.M506178200. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.