

Abstract
Background
Deleterious genetic variation can increase in frequency as a result of mutations, genetic drift, and genetic hitchhiking. Although individual effects are often small, the cumulative effect of deleterious genetic variation can impact population fitness substantially. In this study, we examined the genome of commercial purebred chicken lines for deleterious and functional variations, combining genotype and whole-genome sequence data.
Results
We analysed over 22,000 animals that were genotyped on a 60 K SNP chip from four purebred lines (two white egg and two brown egg layer lines) and two crossbred lines. We identified 79 haplotypes that showed a significant deficit in homozygous carriers. This deficit was assumed to stem from haplotypes that potentially harbour lethal recessive variations. To identify potentially deleterious mutations, a catalogue of over 10 million variants was derived from 250 whole-genome sequenced animals from three purebred white-egg layer lines. Out of 4219 putative deleterious variants, 152 mutations were identified that likely induce embryonic lethality in the homozygous state. Inferred deleterious variation showed evidence of purifying selection and deleterious alleles were generally overrepresented in regions of low recombination. Finally, we found evidence that mutations, which were inferred to be evolutionally intolerant, likely have positive effects in commercial chicken populations.
Conclusions
We present a comprehensive genomic perspective on deleterious and functional genetic variation in egg layer breeding lines, which are under intensive selection and characterized by a small effective population size. We show that deleterious variation is subject to purifying selection and that there is a positive relationship between recombination rate and purging efficiency. In addition, multiple putative functional coding variants were discovered in selective sweep regions, which are likely under positive selection. Together, this study provides a unique molecular perspective on functional and deleterious variation in commercial egg-laying chickens, which can enhance current genomic breeding practices to lower the frequency of undesirable variants in the population.
Electronic supplementary material
The online version of this article (10.1186/s12711-018-0390-1) contains supplementary material, which is available to authorized users.
Background
In animal breeding, the number of deleterious genetic variants that are segregating in a population is affected by several factors, e.g. genetic drift, mutation rate, and selection. As a result, small effective population size and artificial selection can impact population fitness in domesticated populations substantially [1] and can lead to a high risk of inbreeding depression, which is the result of the accumulation of deleterious alleles that increase in frequency, mainly due to genetic drift [1]. Deleterious alleles are expected to be purged from the population by purifying selection, and thus, generally remain at low frequencies in a population [2]. However, many evolutionary forces shape the landscape of deleterious alleles in a population, including recombination and genetic hitchhiking, which is a change in allele frequency due to the allele being passed along together with a variant that is under selection [3]. Recent examples have shown a large impact of such deleterious alleles in several livestock populations [4, 5]. Therefore, effective purging of these deleterious variants is desired. However, most of these variants are rare, and selection on rare variants is usually inefficient, especially if the relationship between genotype and phenotype is poorly characterised [6, 7].
In this study, we examined chicken layer lines that have been primarily selected for production traits, including mortality, egg production, egg composition, shell quality [8], and traits related to animal welfare [9]. In spite of the many positive consequences of this artificial selection, several health issues are associated with intense selection for production traits in laying hens, including excessive comb growth, brittle bones, feather pecking, and ovarian cancer [10–12]. To date, the underlying genetic architecture of these deleterious effects has not been characterised. Therefore, it is essential to better understand the relationship between genotype and phenotype, which is, to a large extent, still a black box [13].
Purebred chickens are routinely genotyped by breeding companies using SNP genotyping panels to accelerate genetic progress by applying genomic selection [14]. Although genomic selection itself may not be very efficient in eliminating low-frequency deleterious variants, the large number of routinely genotyped and pedigreed individuals does allow for the identification of deleterious variation. A powerful method is to systematically assess missing homozygosity in the genome by identifying haplotypes that cause early lethality by statistical depletion, or even absence, of the homozygous state, suggesting that they carry a lethal recessive mutation [4]. This approach can detect even very rare (frequency < 2%) deleterious haplotypes if a large number, at least several thousands, of animals are genotyped in a population. One disadvantage of this method is that low-frequency deleterious variants that reside on common haplotypes will be missed [5]. An alternative method that does allow such rare deleterious alleles to be identified is to sequence the entire genome of tens to hundreds of animals from a population. Whole-genome sequencing (WGS) can be used to identify potential phenotype-altering variants, which can range from embryonic lethal (EL) to only mildly deleterious mutations in coding regions, and to predict their effects using various tools [15]. The use of WGS data from a population can lead to the discovery of variants that are beneficial for breeding programs [16, 17], e.g. by looking for regions in the genome that are under (recent) positive selection. A challenge for this approach is to differentiate true selected variants and variants that increased in frequency as a result of genetic drift. In addition, the incompleteness of current genome annotations in most livestock species hampers the identification of such variants.
In this study, we combined two complementary approaches to identify deleterious and functional variation (positively selected variants in relation to traits under selection) in purebred commercial layer lines. First, we showed that missing homozygosity can result from early embryonic lethality. Second, we mined the genomes of 250 whole-genome-sequenced individuals for deleterious (including embryonic lethal) and functional variants. The result is a comprehensive catalogue of putative deleterious and functional variants, which will be an important resource for future functional studies in chicken and should facilitate the purging of deleterious variants in breeding populations.
Methods
Animals, genotypes and pre-processing
We genotyped six different commercial chicken breeds using the 60 K Illumina SNP BeadChip: one purebred white layer dam line (WA), one purebred white layer sire line (W1), two crossbred lines (CB: W1-WA, W1-WD) and two brown layer lines (B1, B2) (see Additional file 1: Table S1). All animals from multiple generations were genotyped as part of a routine data collection from Hendrix-Genetics breeding programs. Chromosomal positions were determined based on the Gallus gallus GalGal5 reference assembly [18]. SNPs with an unknown position on the Galgal5 reference assembly and SNPs on sex chromosomes were discarded. Pre-processing was performed using PLINK v1.90b3.30 [19, 20] based on the following criteria: each SNP had to have a minor allele frequency higher than 0.01 (1%) and a call rate higher than 0.85 and animals with a call rate lower than 0.7 were discarded from the analysis. We did not filter for deviations from Hardy–Weinberg equilibrium (HWE) because haplotypes that exhibit a deficit in homozygosity were expected to deviate from HWE.
Phasing and identification of missing homozygous haplotypes
We used the BEAGLE version 4.0 genetic analysis software for phasing of the SNP genotypes [21]. We used a sliding-window approach using window sizes ranging from 0.25 to 1 Mb in steps of 0.5 times the window size. Haplotypes with a frequency higher than 0.5% were retained for identification of missing homozygotes. The expected number of homozygotes was estimated using the parental haplotype information with the formula described by Fritz et al. [22]. The number of heterozygous offspring from carrier matings was also calculated to verify whether there was a deviation from HWE. An exact binomial test was applied to compare the number of observed versus expected homozygotes. Haplotypes were considered significantly depleted of homozygotes if the p value for this test was less than 0.005.
Population sequencing and mapping
We used WGS data from three commercial white layer lines, two dam lines (WA: 71, WD: 78) and one sire line (W1: 101), and sequenced a total of 3.502 Tbp (tera base pairs) from 35.94 billion paired-end 100 bp reads sequenced on an Illumina HiSeq machine. We used Sickle software to trim the sequences [23], BWA-MEM (version 0.7.15, [24]) to map the WGS data to the chicken reference genome (Galgal5) [18], the Samtools dedup function to discard duplicate reads [25], and GATK IndelRealigner to perform local realignments of reads around indels [26].
Variant detection and post-processing
We performed population-based variant calling using Freebayes software taking the aligned BAM files as input with the following settings: —min-base-quality 10—min-alternate-fraction 0.2—haplotype-length 0—pooled-continuous—ploidy 2—min-alternate-count 2 [27]. Post-processing was performed using bcftools [25] and variants that were located within 3 bp of an indel, or with a phred quality score and call rate below 20 and 0.7, respectively, were discarded. Moreover, genotype calls were filtered for sample depth (min: 4, max: AvgDepth * 2.5).
Candidate gene identification
We imputed the 250 WGS animals to 60 K genotypes, to match 60 K-based haplotypes to the available sequence data. The software Confirm-gt [21] was used to match chromosome, strand, and allele to the phased 60 K reference population. BEAGLE version 4.0 was used for imputation and phasing. Carriers of haplotypes that were significantly depleted of homozygotes were examined for causal variants by selecting protein-altering variants carried uniquely by the haplotype carriers. We used the variant effect predictor (VEP, Ensembl-release 86) to predict the impact of the candidate variants identified [28]. The impact of the missense variants were assessed using the SIFT and PROVEAN software tools [29, 30].
Population statistics
Principle component analysis was performed using PLINK on the filtered vcf files and plotted using the R package ggplot2. PLINK was used with the—het option to calculate the inbreeding coefficient of each individual to assess the level of genetic diversity within each line.
Functional annotation of variants
Annotation of the freebayes-called variants was performed using Variant Effect Predictor [28]. Variant effect prediction for protein-altering variants was performed using SIFT [29] and PROVEAN [30]. The following variant classes were considered as potentially causing loss of function: splice acceptor, splice donor, inframe indels, frameshift, stop loss, stop gain, and start lost variants. Moreover, only variants that were annotated in genes and which were (mostly) 1:1 orthologous in Ensembl (release 86) were retained to minimize the effect of off-site mapping of sequence reads, as this leads to miscalls, which can be particularly problematic for large gene families (e.g. olfactory receptors). In addition, compensation of function by (recent) paralogous genes will likely ameliorate the effects of damaging mutations in these genes. Also, since gene models might be incorrect, variants that did not have a combined RNA-seq expression coverage of at least 200 in the Ensembl (release 86) merged RNA-seq dataset were discarded. The number and load of deleterious variants for each line were inferred from the final set of deleterious variants.
Spectrum of allele frequencies for different classes of variants
We determined the distribution of allele frequencies for different classes of variants (synonymous, missense tolerated, missense deleterious, stop-gained) to test whether predicted deleterious mutations have generally lower allele frequencies. We generated a histogram with 20 bins (with steps of 0.05 allele frequency) starting from a very low (0–0.05) to very high allele frequency (0.95–1) for the different classes of variants using the PyVCF and SciPy software packages.
Candidate embryonic lethal variants in protein coding genes
To identify putative embryonic lethal (EL) variants, we selected all LoF and deleterious missense variants, for which no individuals that were homozygous for the alternate allele were observed. For every EL candidate we examined whether the gene is known to cause early lethality in mice obtained from the MGI database release 6.10 (i.e. phenotypes from null-mutant mice) [31]. We manually examined all predicted EL variants in JBrowse [32] to exclude false positives that derived from sequencing and mapping errors. Significant differences in hatchability between carrier by carrier versus carrier by non-carrier phenotypes were assessed using a two-sample t-test, assuming equal variances.
Relative position of indels and stop-gained variants in the protein
We divided proteins from Ensembl release 86 in 10 bins (from N- to C-terminal end) and we determined the relative position of the indel and stop-gained variants by dividing the position of the affected amino acid by the total protein length.
Fixed and line-specific “evolutionary-intolerant” variants
We considered all alleles with a frequency higher than 0.9 (within each line) as fixed or nearly fixed variation. To identify regions under selection, we used an approach similar to that described by Elferink et al. [33], but we applied a window size of 20 kb with a minimum number of 20 variants in each window. We selected a threshold of zHp ≤ − 2.7 representing the extreme lower end of the zHp distribution (see Additional file 2: Figure S1). Windows below this threshold were assumed to be enriched for regions of selective sweeps. We selected line-specific high-frequency variants (i.e. absent in the other two populations) with an allele frequency higher than 0.7.
Gene-set enrichment analysis
We tested whether certain gene families are enriched for deleterious mutations. Therefore, gene-set enrichment analysis was performed using the DAVID functional annotation and classification tools [34]. Enrichment clusters (as produced by DAVID) with a score higher or equal to 1.3 were considered to be enriched [34].
Deleterious alleles in regions of low recombination
The recombination rate is the genetic length in centimorgans divided by the physical genomic distance in mega base pairs and was calculated for bins of approximately 750 kb on macrochromosomes 1 to 5 using the linkage map of Elferink et al. [35]. Microchromosomes were excluded because of their extreme high recombination rates [36]. The ratio of predicted deleterious to predicted tolerated mutations (prediction by SIFT) was calculated within each bin by dividing the number of deleterious missense mutations by the sum of the synonymous and tolerated missense mutations over all three breeding lines. Pearson correlation was used to infer the relationship between the ratio of predicted deleterious to predicted tolerated mutations and the recombination rate.
Results
Screening for haplotypes that exhibit missing or deficient homozygosity
In layer breeding programs, genetic improvement is primarily achieved on elite purebred lines. These purebred lines are then crossed to produce parent stock production animals that are again crossed to produce the final laying hen production animals, which benefit from the full exploitation of heterosis [37]. To successfully screen these purebred lines for missing homozygosity, we assumed that not all deleterious variation has been purged, and that some low-frequency deleterious variation remains in the population. Since we examined carrier by carrier (C × C) matings, 25% of the offspring were expected to be homozygous for the carrier haplotype. In total, we examined six lines for missing homozygosity, one purebred white layer dam line (WA), one purebred white layer sire line (W1), two crossbred lines (CB: W1-WA, W1-WD) and two brown layer lines (B1, B2). In total, information was available for 22,323 (post-filtering) animals genotyped on the Illumina 60 K chicken SNP BeadChip (52,232 SNPs), which provided the statistical power required to detect even very rare haplotypes (see Additional file 1: Table S1). We performed phasing of all data to determine the haplotypes and used an overlapping sliding-window approach to identify haplotypes with a significant deficit in homozygotes.
We identified 9, 13, 7, and 50 haplotypes that exhibited a statistical deficit in homozygosity (DH) in the WA, W1, CB, and B1-B2 lines, respectively (Table 1) and (see Additional file 3: Table S1, S2, S3, and S4). The length of these haplotypes ranged from 0.25 to 1 Mb and the frequency of putative deleterious haplotypes ranged from 0.5 to 18.3%. The percentage of heterozygous progeny from C × C matings for these haplotypes was generally higher than 50%, which supports the deviation from HWE due to missing homozygous offspring (Table 1). The frequency of these haplotypes was generally low (< 5%) but two haplotypes that showed a deficit in homozygosity had relatively high frequencies (> 10%) in the crossbred line (on Gallus gallus chromosome (GGA)1: 180.25–180.75 Mb and GGA5: 5.5–6.0 Mb).
Table 1.
Statistics for missing and depleted homozygous SNP haplotypes in four lines of layer chickens
| Lines | WA | W1 | CB | B1-B2 |
|---|---|---|---|---|
| Samples | 4409 | 7197 | 3983 | 6737 |
| Trios | 2291 | 3619 | 3539 | 3118 |
| Number of haplotypes | 9 | 13 | 7 | 50 |
| Number of loci | 9 | 13 | 7 | 45 |
| Average haplotype length | 24.22 | 33.3 | 22.29 | 23.20 |
| Average number of haplotypes per window | 17.11 | 15.08 | 12.43 | 15.40 |
| Average haplotype frequency | 2.6% | 3.1% | 8.3% | 1.5% |
| Average homozygous expected | 6.06 | 8.13 | 30.71 | 8.08 |
| Average carrier matings with genotyped offspring | 3.11 | 4.23 | 53.71 | 3.12 |
| Average carrier matings in pedigree | 9.00 | 12.38 | 54.71 | 6.62 |
| Average carrier progeny | 24.22 | 32.54 | 119.71 | 32.32 |
| Percentage heterozygote carrier progeny | 60.1% | 51.3% | 70.5% | 46.0% |
| Average number of genes in window | 20.9 | 20.0 | 9.14 | 6.30 |
Averages for all parameters are provided for each line. The number of loci represents the unique number of genomic windows containing significant haplotypes
We examined the sequence of the carriers for haplotypes showing a deficit in homozygosity (from the WA and W1 lines) for protein altering variants that were shared by the carriers for each putative deleterious haplotype but for which no homozygous individuals were observed. We identified two candidate mutations (see Additional file 1: Table S2) that segregated in the purebred (WA and W1) and crossbred lines. These two haplotypes, which were initially identified in the crossbreds (GGA2: 56.0–56.5, GGA3: 94.125–94.875 Mb), contain protein altering mutations in the ADNP2 (C198S) and SOX11 (A261G) genes. Both these genes are considered to be essential for normal development and associated with early lethality in mice (inferred from null-mutants, [38, 39]). Only the alanine to glycine mutation in the SOX11 genes was predicted to be mildly deleterious by SIFT and PROVEAN (see Additional file 1: Table S2).
A catalogue of genomic variation in three white-layer lines
We also explored the use of WGS data for direct inference of deleterious variation using sequence data from three commercial white layer lines, one sire line (W1), and two dam lines (WA and WD). We sequenced 250 animals from these lines (WA: 71, WD: 78, and W1: 101), for a total volume of 3.502 Tbp (tera base pairs) from 35.94 billion paired-end 100 bp reads. Mapping was performed with BWA-MEM (version 0.7.15, [24]) to the Gallus gallus build 5 reference genome [18] with an average mappability and coverage of 99.76%, and 11.4 (range: 8.3X to 22.9X), respectively (Pipeline overview [see Additional file 2: Figure S2]). We performed population-based variant calling using Freebayes [27] to identify 10,260,277 (post-filtering) variants in the three lines (see Additional file 1: Table S3). From the total 10,260,277 (post-filtering) identified variants, 9,469,408 (98.5% biallelic) were SNPs and 790,869 were indels. The average SNP density was 11.0 per kb (see Additional file 1: Table S3). We identified 2,143,367 novel variants (20.89%) that were not annotated in dbSNP (build 147), of which the majority was breeding line specific (WA, WD, or W1) (see Additional file 1: Table S4). An average call rate of 0.95 and an average transition/transversion (TS/TV) ratio of 2.53 were found for the entire variant set (see Additional file 2: Figure S3 and Additional file 1: Table S5), which are congruent with previous findings in other avian species [40, 41]. Sample origin was validated using principal component analysis (PCA) (see Additional file 2: Figure S4).
We assessed the level of genetic diversity by calculating the F statistic within the three lines (WA, WD, and W1) and observed that it was lower in the WA line than in the other two lines (see Additional file 2: Figure S5). Accordingly, we found a smaller number of line-specific SNPs in the WA line compared to the other two lines (see Additional file 1: Table S4). Moreover, we observed that WA animals carried on average fewer deleterious variants than the other two lines. However, the mutation load, calculated as the ratio of deleterious (SIFT < 0.01) to synonymous variants, was higher in the WA line than in the WD and W1 lines, which was in line with the lower genetic diversity within this line (Fig. 1).
Fig. 1.
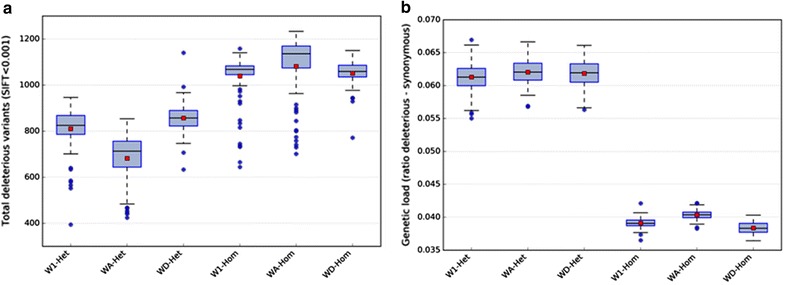
a Distribution of the number of heterozygous (-Het) and homozygous (-Hom) individuals for putative deleterious variants. b Mutation load, calculated as the ratio of deleterious to synonymous variants for heterozygous and homozygous individuals for putative deleterious variants
Variant effect prediction assigned a range of functional classes to the identified variants (see Additional file 1: Table S6). Of the 120,149 coding (35,963 protein-altering) variants that we identified, the large majority were synonymous and non-synonymous mutations. Furthermore, 2.04% (2437) of the variants were classified as potentially introducing a loss-of-function (frameshift, inframe deletion, inframe insertion, splice acceptor, splice donor, start lost, stop gained, and stop lost variants). Of the 33,492 missense mutations, 5546 and 3053 were predicted to be deleterious by the SIFT and PROVEAN software, respectively, of which 1847 were predicted by both methods (see Additional file 2: Figure S6). A final set of 4219 putative deleterious variants, distributed across nine classes of deleterious variants, was obtained after filtering (see “Methods”) and (see Additional file 1: Table S7).
Evidence for purifying selection on deleterious mutations
We found that the spectrum of allele frequencies of deleterious variants differed from that of neutral variants, and was skewed towards a higher proportion of low-frequency alleles (Fig. 2) and (see Additional file 2: Figure S7). Their relative low frequency supports the hypothesis that the predicted deleterious variants are subject to purifying selection.
Fig. 2.
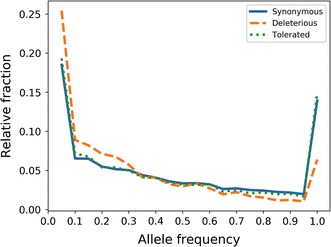
Allele frequency distribution for different functional classes of putative deleterious variants. Deleterious variants (deleterious missense and stop-gained) show distinct allele frequency spectra compared to variants considered to be neutral (synonymous, missense tolerated). Missense variants are classified by SIFT (deleterious: SIFT score ≤ 0.05, tolerated: SIFT score > 0.05)
Relative position of indels and stop-gained variants in the protein
The impact of LoF variants on the protein is potentially determined by the position of the variant in the amino acid sequence. We found that frameshift and stop-gained variants were enriched at the N- and C-terminal ends of the protein, a pattern that was not present for inframe indels, which rather showed a more or less uniform distribution of location across the protein (Fig. 3a). Frameshift or stop-gained variants at the N-terminus could be “rescued” by alternate start-codons, while variants at the C terminus are less likely to be disruptive because they may still result in a more-or-less functional protein. Moreover, deleterious missense mutations occurred more often at the N- and C-terminal ends of the protein, while synonymous mutations occurred less frequently at those positions (see Additional file 2: Figures S8 and S9). Overall, coding indels were enriched for in-frame indels (e.g. 3, 6, 9 bp), because these are more likely to be evolutionary-tolerated (and therefore not purged from the population), which usually does not apply to frameshift indels (Fig. 3b).
Fig. 3.
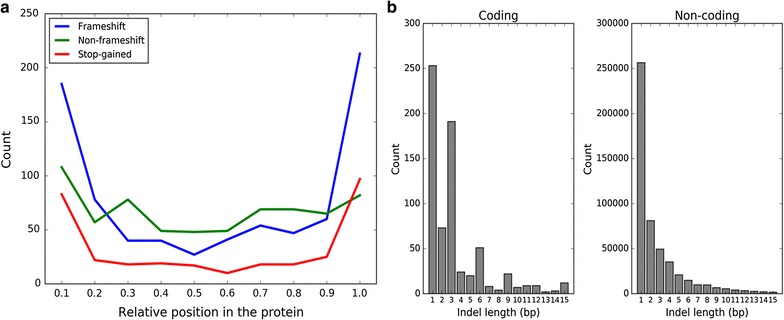
a Relative position of frameshift, non-frameshift indels, and stop-gained variants. Frameshift variants are enriched in N- and C-terminal parts of the protein. Frameshift variants at the N-terminal sites are potentially “rescued” by alternate start-codons. Frameshift variants at the C terminal end are likely not disruptive since a functional protein might still be translated. b Distribution of lengths of coding and non-coding indels. In-frame indels (i.e. indels with lengths of 3, 6, and 9 nucleotides) are enriched in coding regions
Less effective purging in regions of low recombination
Next, we examined whether the ratio of deleterious to tolerated mutations was affected by the recombination rate. A significant negative correlation (r = − 0.26, p = 2.89×10e−9) was found between the recombination rate and the ratio of deleterious to tolerated alleles, providing evidence of more effective purging in regions with high recombination rates (Fig. 4). Enrichment of deleterious over tolerated variants was especially evident in regions of very low recombination (recombination rate less than 2%, [see Additional file 2: Figure S10]).
Fig. 4.
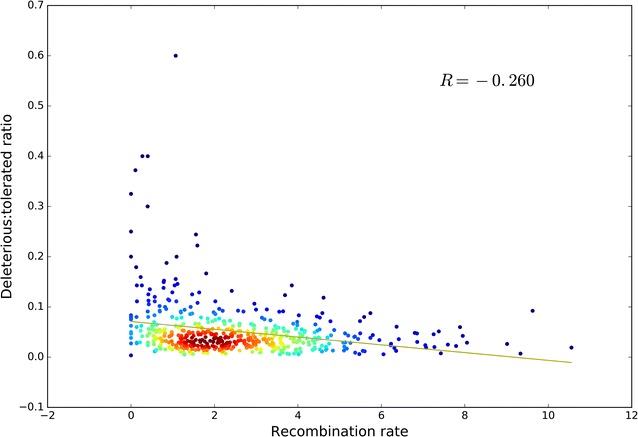
Pearson correlation between recombination rate and the ratio of putative deleterious to tolerated alleles for regions that harbour such alleles. Results indicate that regions of low recombination are generally enriched for deleterious variants (R = − 0.26, P = 2.89e−09)
Candidate EL variants in protein coding genes
To identify variants that likely result in early lethality during development (EL), we selected all putative LoF and deleterious missense variants that met the following two criteria: (1) no homozygous individuals for the allele were observed; and (2) the affected gene caused early lethality in null-mutant mice [31]. Based on these criteria, we identified 11 frameshift, five inframe indels (predicted as deleterious by PROVEAN), six stop-gained, five splice acceptor, eight splice donor, and 121 deleterious missense variants (see Additional file 4: Table S1). The majority of these 152 candidate EL variants (86.6%) were specific to one line and contained frameshift mutations in the APAF1 and NHLRC2 genes, which are both associated with embryonic lethality and malformations in cattle [42, 43]. Of the five in-frame indels, two exhibited relatively high carrier frequencies (> 5%) in the WD line and affected the genes CHTF18 and FLT4. We also identified 13 candidate splice donor and acceptor variants that could potentially lead to mis-splicing, resulting in an incomplete or incorrect protein. Two splice variants exhibited relatively high allele frequencies (> 5%) and affected the POLR1B and HP1BP3 genes. Moreover, one high-frequency (22.3%) stop-gained variant affected the C-terminal end of the SCRIB protein and, thus, might not be disruptive as an almost complete functional protein should be translated (see Additional file 4: Table S1).
Missense variants
The large majority (~ 84%) of the 122 candidate EL missense variants were specific to a line (WA: 19, WD: 46, and W1: 37). Twenty-five variants were predicted to be highly deleterious (PROVEAN score < − 5, Table 2, and [see Additional file 2: Figure S11]). One specific missense variant in the OFD1 gene, which causes a tyrosine to cysteine substitution (Y19G), is a strong candidate for embryonic lethality in homozygous carriers, in spite of its relative high frequency (8.9%). The tyrosine at position 19 of OFD1 is highly conserved among vertebrates and, thus, this missense mutation is predicted to be highly deleterious (PROVEAN: − 7.42, SIFT: 0.0). From the 18 carrier animals (15 sires and 3 dams), we identified three C × C matings in the breeding data that showed a significant (p = 0.0165) increase in the percentage of embryos that died during development (see Additional file 1: Table S8).
Table 2.
Missense variants predicted to be highly deleterious (PROVEAN score < − 5.0) and their phenotypic consequences in null mutant mice based on the MGI database
| Chr. | Position | Ref. | Alt. | Heterozygotes | Line | Symbol | AA position | AA change | SIFT score | Provean score | MGI phenotype |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 49464407 | C | T | 7 | W1 | NAGA | 271 | R/C | 0 | − 6.42 | Homeostasis |
| 1 | 51524116 | A | G | 3 | WA | TMPRSS6 | 688 | E/G | 0 | − 5.771 | Reproductive, growth/size/body, endocrine/exocrine, liver/biliary, immune, homeostasis, mortality/aging, integument, hematopoietic, digestive/alimentary |
| 1 | 118357796 | A | C | 7 | W1 | PRDX4 | 81 | F/V | 0.02 | − 5.65 | Reproductive, cellular, endocrine/exocrine |
| 1 | 122963712 | T | C | 18 | W1 | OFD1 | 19 | Y/C | 0 | − 7.42 | Embryo, nervous, system, skeleton, craniofacial, limbs/digits/tail, renal/urinary, respiratory, cellular, mortality/aging, cardiovascular, growth/size/body, digestive/alimentary |
| 2 | 66538263 | T | A | 9 | WA | BPHL | 70 | D/V | 0 | − 8.815 | Hearing/vestibular/ear, homeostasis |
| 3 | 108278046 | C | T | 3 | WD | PKHD1 | 3397 | R/W | 0.03 | − 5.066 | Respiratory, growth/size/body, endocrine/exocrine, liver/biliary, renal/urinary, cellular, mortality/aging, cardiovascular, nervous system, hematopoietic, digestive/alimentary |
| 4 | 51931175 | G | T | 4 | W1 | CENPC | 729 | P/Q | 0 | − 7.783 | Embryo, mortality/aging, growth/size/body, cellular |
| 4 | 62191743 | T | C | 3 | WD | FAT1 | 796 | Y/C | 0.01 | − 7.534 | Nervous system, craniofacial, renal/urinary, vision/eye, mortality/aging, pigmentation, growth/size/body, homeostasis |
| 4 | 70191405 | G | A | 15 | WD | TBC1D1 | 182 | R/C | 0 | − 6.635 | Growth/size/body, adipose, cellular, no abnormal phenotype observed, muscle, homeostasis |
| 5 | 58052925 | G | A | 7 | W1 | NIN | 1206 | R/C | 0.02 | − 6.045 | Hearing/vestibular/ear, nervous system, behaviour, cardiovascular |
| 5 | 58235842 | C | T | 8 | W1 | NID2 | 944 | G/S | 0 | − 5.151 | Immune, skeleton |
| 9 | 17236801 | C | A | 3 | WA | CCDC39 | 857 | P/H | 0 | − 5.599 | Respiratory, skeleton, craniofacial, liver/biliary, immune, renal/urinary, homeostasis, cellular, mortality/aging, digestive/alimentary, growth/size/body, hematopoietic, cardiovascular |
| 9 | 17571682 | C | A | 3 | WD | MFN1 | 439 | G/W | 0 | − 7.551 | Embryo, mortality/aging, growth/size/body, cellular |
| 19 | 4443604 | A | T | 11 | WD | UNC45B | 225 | I/N | 0 | − 5.4 | Mortality/aging |
| 19 | 6266804 | C | T | 9 | WD | CPD | 994 | P/L | 0.01 | − 6.728 | Respiratory, behaviour, reproductive, craniofacial, endocrine/exocrine, liver/biliary, immune, digestive/alimentary, homeostasis, cellular, vision/eye, integument, nervous system, skeleton, growth/size/body, hematopoietic, cardiovascular |
| 24 | 4359027 | G | A | 3 | WA | KMT2A | 1941 | P/L | 0 | − 9.253 | Embryo, liver/biliary, muscle, cellular, reproductive, immune, craniofacial, limbs/digits/tail, hearing/vestibular/ear, renal/urinary, neoplasm, homeostasis, behaviour, cardiovascular, mortality/aging, integument, nervous system, growth/size/body, hematopoietic, skeleton |
| 27 | 3474503 | C | A | 4 | WA | MPP3 | 206 | S/Y | 0 | − 5.649 | Nervous system, vision/eye, cellular |
Fixed evolutionary-intolerant variants include potential selection candidates
We identified 473 predicted deleterious alleles that were fixed (247) or nearly fixed (allele frequency > 90%) in the three white layer lines (WA, WD, and W1) (see Additional file 5: Table S1). Gene-set enrichment analysis showed that the corresponding genes are involved in energy metabolism (e.g. ATP-binding, calmodium-binding) and muscle and motor activity (see Additional file 5: Table S2). Several of these variants were strongly selected in domesticated chicken. For example, variant (G558R) in the TSHR gene was completely fixed in all three lines and this mutant allele is associated with the absence of strict regulation of seasonal reproduction found in natural populations [16]. A deleterious inframe deletion (108delE) was also found in the P2RY2 gene, which is an ATP receptor. In addition, 12 fixed deleterious variants were identified in seven myosin-related genes (MYH7B, MYCBPAP, MYO1G, MYH9, MYLK3, MYO9B, and MYLK2) that are involved in skeletal muscle development [44]. Other gene families that contained fixed deleterious variants were the protein-tyrosine-phosphatases (PTPN7, PTPRJ, TNS3, PTPRE, PTPRF, and DUSP28), the centrosome proteins (CEP97, CEP162, CEP89, and CEP164), which are potentially involved in essential developmental processes, based on evidence of early lethality in knockout model organisms (notably CEP97 and CEP164, [31]), and collagen-like genes (e.g. C1QTNF8, C1QTNF6, EMILIN2). Forty variants in 37 genes were predicted to have a severe impact on the protein produced by these genes (PROVEAN score − 5), including a variant in the TSHR gene (see Additional file 5: Table S3).
Selection candidates
To distinguish between true selection candidates and effects of genetic drift, we examined the populations for regions under selection. Genome-wide Z-scores of heterozygosity (zHp) were calculated per 20-kb windows. We considered bins with a zHp less than − 2.7 as potential regions of selective sweeps in the genome (representing the extreme end of the distribution) (see Additional file 2: Figure S1) and found 27 fixed evolutionary intolerant variants in these regions (see Additional file 2: Figure S12 and Additional file 5: Table S4), which overlap with the TSHR (see Additional file 2: Figure S13) and FOXI1 genes, previously described as being under domestication selection [16, 17].
We focussed on predicted evolutionary-intolerant variants in smaller regions of selective sweeps to identify possible functional variation that has been under selection. We identified a splice donor variant in the CPE gene (see Additional file 2: Figure S14), which is involved in the energy metabolism of cells and insulin processing. In addition, we identified a strong selection signal in two bins that overlapped with a missense variant in the CCDC93 gene (T389 M) (see Additional file 2: Figure S15). This gene is involved in protein transport, but, although various quantitative trait loci (QTL) related to egg production and egg quality overlap with this gene [45], its exact function remains unknown. A splice acceptor variant in the PSMC6 gene, a start lost variant in the GLCCI1 gene, and an inframe insertion in the RUNXT1 gene were identified as potential additional functional target mutations (see Additional file 2: Figures S16, S17 and S18). PSMC6 and GLCCI1 are both involved in energy metabolism, and overlap respectively with an egg shell thickness QTL and a QTL for haugh unit (a measure of egg protein quality based on the height of its egg white) and growth [45]. The RUNX1T1 gene is a transcription factor involved in the generation of precursor metabolites (substances from which energy is derived). All these variants are likely functional, and while they are identified as being damaging in a natural or wild context, they may have been favourably selected for because they positively affect desired traits in egg-laying hens.
Line-specific high-frequency deleterious variation
WA breeding line
We found 26 high-frequency (allele frequency > 0.7) deleterious missense variants, one frameshift and three splice variants specific to the WA breeding line. Interestingly, the ASPM gene contains three deleterious missense variants (see Additional file 6: Table S1). This gene encodes a mitotic spindle protein and is expressed in proliferating tissues and is associated with a range of phenotypes, including decreased body weight, microcephaly, and reduced fertility in both sexes. Two variants were predicted to have a severe impact on CIB1 (R112C) and PCSK6 (R87 W) proteins (PROVEAN score < − 5), which are both involved in mammalian fertility. CIB1 is related to abnormal spermatogenesis, decreased testis weight and male infertility, while PCSK6 showed a role in female fertility (ovary cysts, increased ovary tumour incidence) [31].
WD breeding line
We annotated 77 high-frequency deleterious variants specific to the WD breeding line (see Additional file 6: Table S2), which included 59 deleterious missense variants, one inframe deletion (ENSGALG00000030853), 14 splice acceptor/donor variants, one start-loss variant (PCBD2), and two stop-gained variants (BRIC5 and NCOR1). Interestingly, the FYCO1 gene, which is associated with cataract phenotypes in mammals [31], harbours two highly deleterious missense variants. Moreover, six missense variants are predicted to be highly deleterious by PROVEAN (PIGX, CARMIL2, LPAR6, ENSGALG00000015226, LIMK2, RIC3). Three of these genes were demonstrated to have severe effects in null-mutant mice (CARMIL2, LPAR6, and LIMK2) [31].
W1 breeding line
We identified 35 high-frequency variants specific to the W1 breeding line (see Additional file 6: Table S3), which included 31 deleterious missense variants, three splice-donor variants, and one stop-gained variant (NOLC1). Three missense variants in three different genes (TAAR1: Y290 N, VWA1: P251S, MCM10: P39L) were predicted to be highly deleterious. TAAR1, a trace amine associated receptor gene, and VWA1 are both associated with various behavioural traits, including increased hyperactivity (TAAR1) and abnormal motor coordination/balance (VWA1). Null-mutants for the MCM10 gene are embryonic lethal in mammals, resulting in abnormal growth prior to termination of development [31]. Interestingly, the CSPG4 gene harbours three deleterious missense variants in the W1 line, which are associated with abnormal muscle cell physiology and increased body weight [46].
Discussion
Combining a systematic genomic survey for missing homozygosity and whole-genome sequence (WGS) data opens new opportunities to directly infer functional variants. We have presented a first full genomic catalogue of variants that provides a perspective on the deleterious and functional variation in fairly closed, and relatively inbred, purebred layer lines. We not only confirmed previous “domestic” or selective variants but also assessed the impact of deleterious variation in these lines. Taken together, this genomic framework can be used to further improve and understand the genomic elements that are selected or purged in current breeding programs. Finally, a better understanding of the variants with functional implications will provide a useful resource for further selection programs to help distinguish true deleterious variants from those with positive functional implications.
Domesticated populations are expected to be under artificial selection against inbreeding depression. Indeed, in this paper, we show that putatively highly deleterious (i.e. lethal) variants are rare in the commercial chicken populations studied here, in spite of the small effective size of these populations. However, we found several examples of putative lethal variants with allele frequencies up to 10% (e.g. OFD1 and Y19C) and showed that, although under strong selection, the purging of these variants is not always very effective, even in modern poultry breeding programs. Artificial selection in these populations may be ‘strong’, but is based on an index of a large number of phenotypic traits. Balancing selection may also be acting on these populations (e.g. heterozygote advantage), which causes deleterious variants to remain in the population.
In order to capture deleterious variants using haplotypes of SNPs that exhibit missing homozygosity, the low-frequency haplotype has to be in complete LD with the causal variant. However, most deleterious variants (EL) reside on common haplotypes that cannot be detected with medium-density SNP chip data. However, absence of specific homozygous allele states can now be inferred directly because animals can be routinely genotyped for these variants, such that they can be added to the currently used genomic selection framework. A similar study in cattle showed that 15% of the LoF and 6% of the tested missense variants are likely true EL [15]. Although predicting EL variation from sequence can be sensitive to induce false positives, we tried to reduce the number of false positives by manually examining the predicted EL variants. Moreover, the distinct allele frequency spectrum for our predicted deleterious mutations compared to neutral mutations confirms that they are subject to purifying selection.
One limitation of our study is that we focused on coding variation, however, a large proportion of the non-coding genome is also subject to purifying selection because of their biological function [47]. As a result, we may have missed a large proportion of potential deleterious or functional variants. In addition, livestock genomes still lack proper annotation of many functional elements but currently there are many efforts to improve this aspect [48].
We found no evidence of a higher load of deleterious variants in our studied chicken lines compared to other livestock species [15, 49]. However, although the impact of individual variants on the population may be limited, a recent study showed that negative selection involves synergistic epistasis, which means that the combined effect of mutations is greater than the sum of the individual effects. This supports the hypothesis that the overall effect of the deleterious mutations on population fitness might be substantial [50]. As a consequence, the number of deleterious variants found in the chicken populations studied here might represent a universal level for ‘healthy populations’, i.e. lower levels deleterious mutations are not attained because selection against low-frequency alleles is ineffective, but higher levels of deleterious mutations could occur, which then rapidly leads to disproportionately large inbreeding depression effects. This study also demonstrates the value of domesticated populations to provide insight in the genomic architecture of inbreeding depression and can be useful for future studies on inbreeding in both wild and domesticated populations.
The observed spectrum of allele frequencies for predicted deleterious and tolerated variants corroborates the hypothesis that the predicted deleterious variants (especially deleterious missense and stop-gained variants) have been under purifying selection. Conversely, the predicted tolerated missense variants followed the same distribution of allele frequencies as synonymous variants (usually considered to be neutral), which indicates that the large majority of these predicted missense variants are indeed evolutionary tolerated. Within coding regions, we also found an enrichment of indels that are multiples of three nucleotides, which was not the case for non-coding indels. Indels that alter the frame of translation in coding regions can be highly disruptive, for instance by introducing a premature stop codon and, therefore, such indels are often under purifying selection. Conversely, indels that are multiples of three nucleotides will result in losses or gains of one or multiple amino acid residues, which have a higher likelihood of being tolerated. We also observed an enrichment of frameshift and stop-gained variants at the N- and C terminal ends of the protein, which suggests that, in general, these types of variants have a stronger impact on the function of the protein when they are located in the middle part of the protein compared to the distal parts of the protein. Namely, if they are located at the N-terminal part of the protein, a functional protein product might still be generated by an alternate start codon that can “rescue” a large part of the protein (N-terminal part), as described previously [51]. In contrast, a frameshift or stop-gained variant at the C-terminal end may be tolerated since an almost complete protein is often generated. Together these genomic signatures of purifying selection support our predictions on deleterious alleles within the populations.
Evidence that the frequency of recombination in a genomic region is negatively correlated with the ratio of deleterious to tolerated mutations suggests more effective purging in regions with higher recombination rate, potentially because deleterious variants that hitchhike along with selected variants are more easily physically disconnected from variants that are under selection in regions with high recombination rates. Similar results have been reported in other species, although always with weaker correlations [2, 3, 52]. We shed light on the role of recombination (i.e. more effective selection in regions of high recombination) in genomic purging within the avian clade, which is known for its highly diverse recombination rates between chromosomes, with notably extremely high recombination rates on microchromosomes [53].
In addition to predicted deleterious variants with low frequencies, several high-frequency predicted deleterious variants were identified that likely have high functional relevance. We focussed on predicted evolutionary-intolerant, but high-frequency, variants in selective sweep regions. This study confirmed several predicted deleterious variants that were previously identified as being positively selected in domesticated chicken populations, e.g. variants in the TSHR and FOXI1L genes [16, 17]. However, we find several novel predicted deleterious variants in strong selective sweep regions (e.g. variants in the CCDC93, PSMC6 and GLCCI1 genes), that should be further investigated for phenotypic effects. In spite of a paucity of functional annotation, there is evidence that the majority of these genes have a role in cellular energy metabolism and likely cause increased metabolic activity [16, 33].
The use of genomic selection has increased the rate of genetic improvement in breeding populations substantially over the past years [6]. However, genomic selection remains a “black-box” approach and the genomic architecture that underlies selection remains unknown. Without additional prior information on the functional effects of low-frequency variants, effective selection for or against desired or unwanted variation remains challenging. Leveraging low-frequency functional variants for selection requires functional annotation, which can then be translated into statistical priors in enhanced genomic selection programs [54–56]. This study contributes to this by the identification of specific variants that can be incorporated in breeding programs to enhance genetic improvement.
Conclusions
In this study, we applied several methods to infer deleterious variation in three commercial white-layer lines. We confirmed that missing homozygosity can result from lethal variants that reside on low-frequency SNP haplotypes. We were able to capture even very low-frequency deleterious variation, including 152 likely EL variants, by exploiting WGS data of dozens of sequenced individuals within single populations. Results provided clear evidence for purifying selection, based on a distinct spectrum of allele frequencies of deleterious variants compared to that of variants that have a higher likelihood of being neutral. In spite of their low-frequency nature, the identified putative deleterious alleles generally occurred more often in regions with low recombination, which suggests that purging of these alleles is less effective in such regions. Also, frameshift and stop-gained variants were more frequent at the protein N- and C-termini, which confirms that these are likely evolutionary-tolerated, which also applies to in-frame indels. In addition, multiple predicted evolutionary intolerant coding variants were discovered in selective sweep regions, which are likely under positive selection. A comprehensive genomic catalogue of putative deleterious variants was developed for white-egg layer breeding lines, which can enhance current genomic breeding practices to lower the frequency of undesirable variants in the population.
Additional files
Additional file 1: Table S1. Statistics per breed. Table S2. Candidate mutations in regions of missing homozygosity. Table S3. Single nucleotide variants (SNV) and indels called. Table S4. Summary of identified variants for each line. Table S5. Call rate and TS/TV for all variants after filtering. Table S6. Functional annotation of variants (VEP). Table S7. Final list of deleterious variants identified in nine different classes. Table S8. Fertility phenotypes for three CXC matings that carry the OFD1 (Y19C) missense mutation.
Additional file 2: Figure S1. (A) zHp value distribution; (B) Genome-wide zHp values for the three white-layer lines. Figure S2. Pipeline overview for the detection of deleterious and functional variants using population WGS data. Figure S3. Chromosome distribution of transition/transversion (TS/TV) ratio across all sequenced animals. Figure S4. PCA analysis for the sequenced samples in WA, WD, and W1 lines. Figure S5. Population F-statistics. Figure S6. Pearson correlation of SIFT and PROVEAN scores on 13,065 missense mutations. Figure S7. Allele frequency distribution of different classes of variants. Figure S8. Relative position of tolerated and deleterious missense variants (SIFT). Figure S9. Relative position of synonymous, tolerated, and deleterious missense variants (SIFT). Figure S10. Distribution of the ratio of deleterious to tolerated alleles in two classes of recombination rates (<=2, >2). Figure S11. Missense variants predicted to be highly deleterious (PROVEAN <= -4.0) with no homozygotes observed. Figure S12. Fixed evolutionary-intolerant variants in selective sweep regions. Figure S13. Selective sweep region comprising the TSHR gene. Figure S14. Selective sweep region comprising the CPE gene. Figure S15. Selective sweep region comprising the CCDC93 gene. Figure S16. Selective sweep region comprising the PSMC6 gene. Figure S17. Selective sweep region comprising the GLCC1 gene. Figure S18. Selective sweep region comprising the RUNX1T1 gene.
Additional file 3: Table S1. Haplotypes exhibiting missing or depleted homozygosity in the WA line. Table S2. Haplotypes exhibiting missing or depleted homozygosity in CB lines. Table S3. Haplotypes exhibiting missing or depleted homozygosity in the W1 line. Table S4. Haplotypes exhibiting missing or depleted homozygosity in the B1-B2 line.
Additional file 4: Table S1. Candidate EL variants.
Additional file 5: Table S1. Evolutionary-intolerant fixed variants (AF > 90%). Table S2. DAVID gene-set enrichment analysis for evolutionary-intolerant fixed variation. Table S3. Evolutionary-intolerant variants predicted to have a severe impact on the protein (PROVEAN score <= -5). Table S4. Fixed evolutionary-intolerant variants in selective sweep regions.
Additional file 6: Table S1. High-frequency deleterious variation in the WA line. Table S2. High-frequency deleterious variation in the WD line. Table S3. High-frequency deleterious variation in the W1 line.
Authors’ contributions
MAMG and HJM conceived and designed the study. MFLD performed the data analysis and wrote the manuscript. HJM, MAMG, MB, AV, MCAMB, KP, JV, CG, DdR, and MJTR provided useful comments and suggestions and helped to draft the manuscript. AV, MCAMB, KP, and JV provided raw data. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to thank Chiara Bortoluzzi, and Vinicius da Silva for useful input on this work.
Competing interests
MCAMB, JV, KP, and AV are employees of Hendrix-Genetics, one of the funders of this study. All authors declare that the results are presented in full and as such present no conflict of interest. The other Breed4Food partners Cobb Europe, CRV, Topigs Norsvin, declare to have no competing interests for this study.
Availability of data and materials
The datasets analysed during the current study are not publicly available, due to data restriction from Hendrix-Genetics.
Consent for publication
Not applicable
Ethics approval and consent to participate
The genotype data used in this study were obtained as part of routine data collection from Hendrix-Genetics breeding programs, and not specifically for the purpose of this project. Therefore, approval of an ethics committee was not mandatory. Sample collection and data recording were conducted strictly according to the Dutch law on animal protection and welfare (Gezondheids- en welzijnswet voor dieren).
Funding
This research was funded by the STW-Breed4Food Partnership, project number 14283: From sequence to phenotype: detecting deleterious variation by prediction of functionality. This study was financially supported by NWO-TTW and the Breed4Food partners Cobb Europe, CRV, Hendrix Genetics and Topigs-Norsvin. In addition, this study was supported by the IMAGE project (Horizon 2020, No. 677353). The funders had no role in the design of the study, data collection and analysis, decision to publish, or preparation of the manuscript. The use of the HPC cluster was made possible by CAT-AgroFood (Shared Research Facilities Wageningen UR).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12711-018-0390-1) contains supplementary material, which is available to authorized users.
Contributor Information
Martijn F. L. Derks, Email: martijn.derks@wur.nl
Hendrik-Jan Megens, Email: hendrik-jan.megens@wur.nl.
Mirte Bosse, Email: mirte.bosse@wur.nl.
Jeroen Visscher, Email: jeroen.visscher@hendrix-genetics.com.
Katrijn Peeters, Email: katrijn.peeters@hendrix-genetics.com.
Marco C. A. M. Bink, Email: marco.bink@hendrix-genetics.com
Addie Vereijken, Email: addie.vereijken@hendrix-genetics.com.
Christian Gross, Email: c.gross@tudelft.nl.
Dick de Ridder, Email: dick.deridder@wur.nl.
Marcel J. T. Reinders, Email: m.j.t.reinders@tudelft.nl
Martien A. M. Groenen, Email: martien.groenen@wur.nl
References
- 1.Charlesworth D, Willis JH. Fundamental concepts in genetics. The genetics of inbreeding depression. Nat Rev Genet. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- 2.Zhang M, Zhou L, Bawa R, Suren H, Holliday JA. Recombination rate variation, hitchhiking, and demographic history shape deleterious load in poplar. Mol Biol Evol. 2016;33:2899–2910. doi: 10.1093/molbev/msw169. [DOI] [PubMed] [Google Scholar]
- 3.Chun S, Fay JC. Evidence for hitchhiking of deleterious mutations within the human genome. PLoS Genet. 2011;7:e1002240. doi: 10.1371/journal.pgen.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.VanRaden PM, Olson KM, Null DJ, Hutchison JL. Harmful recessive effects on fertility detected by absence of homozygous haplotypes. J Dairy Sci. 2011;94:6153–6161. doi: 10.3168/jds.2011-4624. [DOI] [PubMed] [Google Scholar]
- 5.Derks MFL, Megens HJ, Bosse M, Lopes MS, Harlizius B, Groenen MAM. A systematic survey to identify lethal recessive variation in highly managed pig populations. BMC Genomics. 2017;18:858. doi: 10.1186/s12864-017-4278-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sitzenstock F, Ytournel F, Sharifi AR, Cavero D, Taubert H, Preisinger R, et al. Efficiency of genomic selection in an established commercial layer breeding program. Genet Sel Evol. 2013;45:29. doi: 10.1186/1297-9686-45-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kearney JF, Navarro P, Haley CS, Villanueva B. Consequences of selection for improving production traits on the frequency of deleterious alleles for fitness. J Anim Sci. 2009;87:850–859. doi: 10.2527/jas.2008-1099. [DOI] [PubMed] [Google Scholar]
- 8.Rakonjac S, Bogosavljevic-Boskovic S, Pavlovski Z, Skrbic Z, Doskovic V, Petrovic MD, et al. Laying hen rearing systems: a review of major production results and egg quality traits. Worlds Poult Sci J. 2014;70:93–104. doi: 10.1017/S0043933914000087. [DOI] [Google Scholar]
- 9.Ellen ED, Visscher J, van Arendonk JA, Bijma P. Survival of laying hens: genetic parameters for direct and associative effects in three purebred layer lines. Poult Sci. 2008;87:233–239. doi: 10.1080/00071660802094206. [DOI] [PubMed] [Google Scholar]
- 10.Savory CJ. Feather pecking and cannibalism. Worlds Poult Sci J. 1995;51:215–219. doi: 10.1079/WPS19950016. [DOI] [Google Scholar]
- 11.Johnson PA, Stephens CS, Giles JR. The domestic chicken: causes and consequences of an egg a day. Poult Sci. 2015;94:816–820. doi: 10.3382/ps/peu083. [DOI] [PubMed] [Google Scholar]
- 12.Webster AB. Welfare implications of avian osteoporosis. Poult Sci. 2004;83:184–192. doi: 10.1093/ps/83.2.184. [DOI] [PubMed] [Google Scholar]
- 13.Habier D, Fernando RL, Garrick DJ. Genomic BLUP decoded: a look into the black box of genomic prediction. Genetics. 2013;194:597–607. doi: 10.1534/genetics.113.152207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meuwissen TH, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charlier C, Li W, Harland C, Littlejohn M, Coppieters W, Creagh F, et al. NGS-based reverse genetic screen for common embryonic lethal mutations compromising fertility in livestock. Genome Res. 2016;26:1333–1341. doi: 10.1101/gr.207076.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464:587–591. doi: 10.1038/nature08832. [DOI] [PubMed] [Google Scholar]
- 17.Gheyas AA, Boschiero C, Eory L, Ralph H, Kuo R, Woolliams JA, et al. Functional classification of 15 million SNPs detected from diverse chicken populations. DNA Res. 2015;22:205–217. doi: 10.1093/dnares/dsv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid M, Smith J, Burt DW, Aken BL, Antin PB, Archibald AL, et al. Third report on chicken genes and chromosomes 2015. Cytogenet Genome Res. 2015;145:78–179. doi: 10.1159/000430927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fritz S, Capitan A, Djari A, Rodriguez SC, Barbat A, Baur A, et al. Detection of haplotypes associated with prenatal death in dairy cattle and identification of deleterious mutations in GART, SHBG and SLC37A2. PLoS One. 2013;8:e65550. doi: 10.1371/journal.pone.0065550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files. https://github.com/najoshi/sickle. Accessed 20 Feb 2018.
- 24.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv 2012:1207.3907 [q-bio.GN].
- 28.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 30.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ, et al. Mouse genome database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res. 2017;45:D723–D729. doi: 10.1093/nar/gkw1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elferink MG, Megens HJ, Vereijken A, Hu X, Crooijmans RPMA, Groenen MAM. Signatures of selection in the genomes of commercial and non-commercial chicken breeds. PLoS One. 2012;7:e32720. doi: 10.1371/journal.pone.0032720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 35.Elferink MG, van As P, Veenendaal T, Crooijmans RP, Groenen MA. Regional differences in recombination hotspots between two chicken populations. BMC Genet. 2010;11:11. doi: 10.1186/1471-2156-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Megens HJ, Crooijmans RP, Bastiaansen JW, Kerstens HH, Coster A, Jalving R, et al. Comparison of linkage disequilibrium and haplotype diversity on macro- and microchromosomes in chicken. BMC Genet. 2009;10:86. doi: 10.1186/1471-2156-10-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amuzu-Aweh EN, Bovenhuis H, de Koning DJ, Bijma P. Predicting heterosis for egg production traits in crossbred offspring of individual White Leghorn sires using genome-wide SNP data. Genet Sel Evol. 2015;47:27. doi: 10.1186/s12711-015-0088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang Y, Ding Q, Xie X, Libby RT, Lefebvre V, Gan L. Transcription factors SOX4 and SOX11 function redundantly to regulate the development of mouse retinal ganglion cells. J Biol Chem. 2013;288:18429–18438. doi: 10.1074/jbc.M113.478503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, et al. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res. 2003;144:83–90. doi: 10.1016/S0165-3806(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 40.Aslam ML, Bastiaansen JW, Elferink MG, Megens HJ, Crooijmans RP, Blomberg A, et al. Whole genome SNP discovery and analysis of genetic diversity in Turkey (Meleagris gallopavo) BMC Genomics. 2012;13:391. doi: 10.1186/1471-2164-13-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smeds L, Qvarnström A, Ellegren H. Direct estimate of the rate of germline mutation in a bird. Genome Res. 2016;26:1211–1218. doi: 10.1101/gr.204669.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denholm L, Marron B, Parnell P, Teseling C, Beever J. Pleiotropic embryonic malformation associated with cranial and caudal neural tube defects from a single amino-acid substitution (V311A) at a conserved locus in the Nhlrc2 protein. In: Proceedings of the 9th international conference on neural tube defects: 26–29 October 2015; Austin. 2015.
- 43.Adams HA, Sonstegard TS, VanRaden PM, Null DJ, Van Tassell CP, Larkin DM, et al. Identification of a nonsense mutation in APAF1 that is likely causal for a decrease in reproductive efficiency in Holstein dairy cattle. J Dairy Sci. 2016;99:6693–6701. doi: 10.3168/jds.2015-10517. [DOI] [PubMed] [Google Scholar]
- 44.Lagrutta AA, McCarthy JG, Scherczinger CA, Heywood SM. Identification and developmental expression of a novel embryonic myosin heavy-chain gene in chicken. DNA. 1989;8:39–50. doi: 10.1089/dna.1.1989.8.39. [DOI] [PubMed] [Google Scholar]
- 45.Hu ZL, Park CA, Reecy JM. Developmental progress and current status of the animal QTLdb. Nucleic Acids Res. 2016;44:D827–D833. doi: 10.1093/nar/gkv1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grako KA, Ochiya T, Barritt D, Nishiyama A, Stallcup WB. PDGF (alpha)-receptor is unresponsive to PDGF-AA in aortic smooth muscle cells from the NG2 knockout mouse. J Cell Sci. 1999;112:905–915. doi: 10.1242/jcs.112.6.905. [DOI] [PubMed] [Google Scholar]
- 47.Ponting CP, Hardison RC. What fraction of the human genome is functional? Genome Res. 2011;21:1769–1776. doi: 10.1101/gr.116814.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andersson L, Archibald AL, Bottema CD, Brauning R, Burgess SC, Burt DW, et al. Coordinated international action to accelerate genome-to-phenome with FAANG, the functional annotation of animal genomes project. Genome Biol. 2015;16:57. doi: 10.1186/s13059-015-0622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bosse M, Megens HJ, Madsen O, Crooijmans RPMA, Ryder OA, Austerlitz F, et al. Using genome-wide measures of coancestry to maintain diversity and fitness in endangered and domestic pig populations. Genome Res. 2015;257:970–981. doi: 10.1101/gr.187039.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sohail M, Vakhrusheva OA, Sul JH, Pulit SL, Francioli LC, Genome of the Netherlands Consortium, et al Negative selection in humans and fruit flies involves synergistic epistasis. Science. 2017;356:539–542. doi: 10.1126/science.aah5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, et al. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramu P, Esuma W, Kawuki R, Rabbi IY, Egesi C, Bredeson JV, et al. Cassava haplotype map highlights fixation of deleterious mutations during clonal propagation. Nat Genet. 2017;48:959–963. doi: 10.1038/ng.3845. [DOI] [PubMed] [Google Scholar]
- 53.Backström N, Forstmeier W, Schielzeth H, Mellenius H, Nam K, Bolund E, et al. The recombination landscape of the zebra finch Taeniopygia guttata genome. Genome Res. 2010;20:485–495. doi: 10.1101/gr.101410.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez-Enciso M, Rincon JC, Legarra A. Sequence—versus chip-assisted genomic selection: accurate biological information is advised. Genet Sel Evol. 2015;47:43. doi: 10.1186/s12711-015-0117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perez-Enciso M, Forneris N, de los Campos G, Legarra A. Evaluating sequence-based genomic prediction with an efficient new simulator. Genetics. 2017;205:939–953. doi: 10.1534/genetics.116.194878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacLeod IM, Bowman PJ, Vander Jagt CJ, Haile-Mariam M, Kemper KE, Chamberlain AJ, et al. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics. 2016;17:144. doi: 10.1186/s12864-016-2443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Statistics per breed. Table S2. Candidate mutations in regions of missing homozygosity. Table S3. Single nucleotide variants (SNV) and indels called. Table S4. Summary of identified variants for each line. Table S5. Call rate and TS/TV for all variants after filtering. Table S6. Functional annotation of variants (VEP). Table S7. Final list of deleterious variants identified in nine different classes. Table S8. Fertility phenotypes for three CXC matings that carry the OFD1 (Y19C) missense mutation.
Additional file 2: Figure S1. (A) zHp value distribution; (B) Genome-wide zHp values for the three white-layer lines. Figure S2. Pipeline overview for the detection of deleterious and functional variants using population WGS data. Figure S3. Chromosome distribution of transition/transversion (TS/TV) ratio across all sequenced animals. Figure S4. PCA analysis for the sequenced samples in WA, WD, and W1 lines. Figure S5. Population F-statistics. Figure S6. Pearson correlation of SIFT and PROVEAN scores on 13,065 missense mutations. Figure S7. Allele frequency distribution of different classes of variants. Figure S8. Relative position of tolerated and deleterious missense variants (SIFT). Figure S9. Relative position of synonymous, tolerated, and deleterious missense variants (SIFT). Figure S10. Distribution of the ratio of deleterious to tolerated alleles in two classes of recombination rates (<=2, >2). Figure S11. Missense variants predicted to be highly deleterious (PROVEAN <= -4.0) with no homozygotes observed. Figure S12. Fixed evolutionary-intolerant variants in selective sweep regions. Figure S13. Selective sweep region comprising the TSHR gene. Figure S14. Selective sweep region comprising the CPE gene. Figure S15. Selective sweep region comprising the CCDC93 gene. Figure S16. Selective sweep region comprising the PSMC6 gene. Figure S17. Selective sweep region comprising the GLCC1 gene. Figure S18. Selective sweep region comprising the RUNX1T1 gene.
Additional file 3: Table S1. Haplotypes exhibiting missing or depleted homozygosity in the WA line. Table S2. Haplotypes exhibiting missing or depleted homozygosity in CB lines. Table S3. Haplotypes exhibiting missing or depleted homozygosity in the W1 line. Table S4. Haplotypes exhibiting missing or depleted homozygosity in the B1-B2 line.
Additional file 4: Table S1. Candidate EL variants.
Additional file 5: Table S1. Evolutionary-intolerant fixed variants (AF > 90%). Table S2. DAVID gene-set enrichment analysis for evolutionary-intolerant fixed variation. Table S3. Evolutionary-intolerant variants predicted to have a severe impact on the protein (PROVEAN score <= -5). Table S4. Fixed evolutionary-intolerant variants in selective sweep regions.
Additional file 6: Table S1. High-frequency deleterious variation in the WA line. Table S2. High-frequency deleterious variation in the WD line. Table S3. High-frequency deleterious variation in the W1 line.
Data Availability Statement
The datasets analysed during the current study are not publicly available, due to data restriction from Hendrix-Genetics.