

Abstract
Objectives
Oncogenic potential of Notch signaling and its cooperation with other factors to affect proliferation are widely established. Notch exhibits a cooperative effect with loss of a cell polarity gene, scribble to induce neoplastic overgrowth. Oncogenic Ras also show cooperative effect with loss of cell polarity genes such as scribble (scrib), lethal giant larvae (lgl) and discs large to induce neoplastic overgrowth and invasion. Our study aims at assessing the cooperation of activated Notch with loss of function of lgl in tumor overgrowth, and the mode of JNK signaling activation in this context.
Results
In the present study, we use Drosophila as an in vivo model to show the synergy between activated Notch (Nact) and loss of function of lgl (lgl-IR) in tumor progression. Coexpression of Nact and lgl-IR results in massive tumor overgrowth and displays hallmarks of cancer, such as MMP1 upregulation and loss of epithelial integrity. We further show activation of JNK signaling and upregulation of its receptor, Grindelwald in Nact/lgl-IR tumor. In contrast to previously described Notchact/scrib−/− tumor, our experiments in Nact/lgl-IR tumor showed the presence of dying cells along with tumorous overgrowth.
Electronic supplementary material
The online version of this article (10.1186/s13104-018-3350-5) contains supplementary material, which is available to authorized users.
Keywords: Notch, lgl, Drosophila, Tumor overgrowth, JNK signaling, Cell death
Introduction
In the past decade, a keen interest has been shown to explore the oncogenic cooperation with loss of cell polarity in tumor progression and malignancy. Studies in Drosophila have revealed that the oncogenic form of Ras cooperates with loss of tumor suppressors, namely scrib, lgl and dlg to cause tumor cell invasion [1, 2]. The oncogenic form of Notch has also shown to cooperate with scrib−/− to induce neoplastic overgrowth [2]. The loss-of-function mutation of Scribble complex genes (scrib, lgl and dlg) results in disruption of epithelial integrity followed by neoplastic tissue hyperproliferation [3–5]. However, the tumor formation caused by loss of scrib, lgl and dlg has been found to be restricted by the compensatory JNK mediated apoptosis [2, 6–8]. Among the Scrib complex genes, lgl was the first neoplastic tumor suppressor gene described in Drosophila [9]. The phenotypes of lgl mutant tissues show close similarity with that of the human epithelial cancers [10–12]. Although it has been shown that Notch cooperates with scrib−/− to induce neoplastic growth, it is still unknown whether Notch works in the same way with loss-of-function of lgl also. Recently, Lgl has been shown to regulate Notch signaling via endocytosis [13]. However, it gives no substantial evidence on coupling of lgl-Notch effect on tumorigenesis. In the present study, we checked the effect of a tumor suppressor gene mutation, lgl, in activated Notch background, and found that lgl downregulation synergizes with activated Notch to induce overgrowth and migratory behavior. Here, we show that Nact/lgl-IR tissues display the hallmarks of tumor overgrowth. Moreover, our study revealed that the effect of Nact/lgl-IR tumor is mediated by the activation of JNK signaling through the upregulation of its receptor, Grindelwald.
Main text
Methods
Detailed description of methods used in this study is provided in Additional file 1.
Results
Oncogenic Notch synergizes with RNAi mediated downregulation of lgl to promote tissue overgrowth
Coexpression of both lgl-IR and Notchact in the Drosophila eye discs using ey-GAL4 dramatically induced overgrowth (Fig. 1d, d″) as compared to that of only Nact overexpressed (Fig. 1b, b″) or only lgl-IR overexpressed (Fig. 1c, c″) eye discs. To further describe the phenotype of Nact/lgl-IR tumor, expression of Matrix metalloproteinase 1 (MMP1) was monitored. MMPs are enzymes with clear association to tumor cell invasion and cancer progression [14, 15]. Coexpression of Notchact and lgl-IR resulted in massive upregulation of MMP1 expression throughout the entire eye disc (Fig. 1d′) as compared to that of only Nact or only lgl-IR (Fig. 1b′, c′). Further, we extended our observation into the brain since ey-GAL is mildly expressed in the brain also. Except endogenous expression, no MMP1 activation was observed in the ey-GAL4 driven lgl-IR (Fig. 1g′) and Nact larval brain (Fig. 1f′). In case of Notchact/lgl-IR larval brain, excessive amount of GFP marked cells with enhanced MMP1 expression was observed in the optic lobes (Fig. 1h, h′). The increment in GFP and MMP1 expression was also found in the ventral nerve cord (VNC) of Notchact/lgl-IR larval brain (Fig. 1h, h′ marked with arrows). This indicates that the weak expression of ey-GAL4 in VNC is also inducing MMP1 expression in Notchact/lgl-IR tissue. When we quantified the amount of GFP in upper region of VNC, a significant increment in the amount of GFP in Notchact/lgl-IR was found as compared to that of the controls (Additional file 2: Figure S1a). We also quantified the presence of MMP1 in the VNC of Notchact/lgl-IR (Additional file 2: Figure S1b), which clearly shows a significant increase as compared to that of the controls. Moreover, transcript levels of mmp1 in the cephalic complex were also found to be upregulated in Notchact/lgl-IR tumor as compared to that of the controls (Additional file 2: Figure S1c).
Fig. 1.
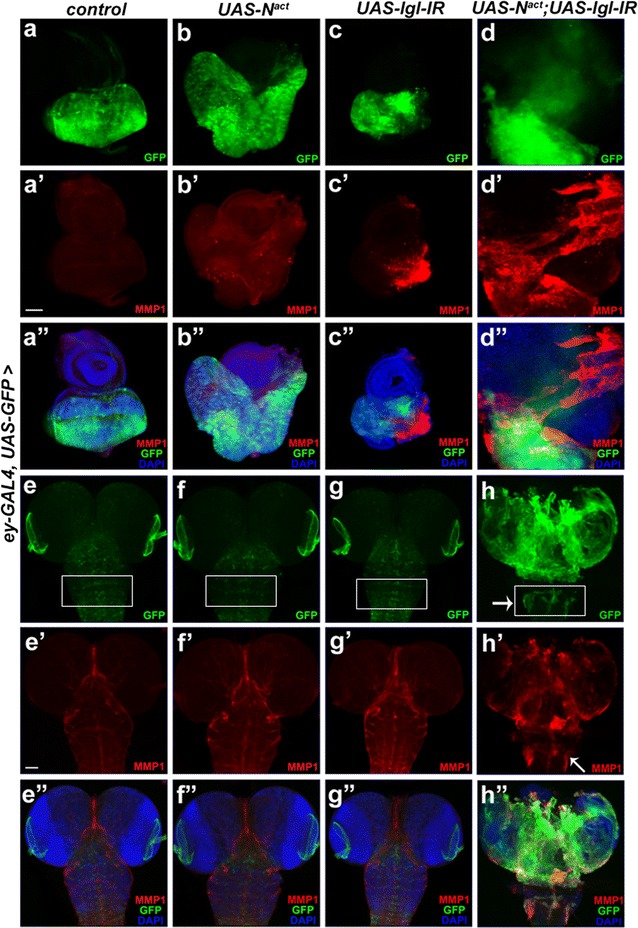
Oncogenic Notch synergizes with lgl-IR to induce tissue overgrowth. Fluorescent micrographs of eye imaginal discs and larval brains are shown. ey-GAL4/+ (control) eye imaginal disc shows expression of GFP (a) and MMP1 (a′). ey-GAL4 driven UAS-Nact results in enlarged disc size (b) and shows slight enhancement in MMP1 expression (b′). UAS-lgl-IR eye disc driven by ey-GAL4 (c) induces MMP1 expression (c′). UAS-Nact coexpressed with UAS-lgl-IR results in massively overgrown eye discs (d) and significant enhancement in MMP1 expression throughout the tissue (d′) compared to that of only lgl-IR (c′) or only Notchact (b′) overexpressed eye discs. Images a″–d″ are merges of those in a–a′, b–b′, c–c′ and d–d′ with DAPI, respectively. Expression of GFP and MMP1 in brains of ey-GAL4 driven UAS-Nact (f, f′) and UAS-lgl-IR (g, g′) remain similar as of wild-type brain (e, e′). h UAS-Nact and UAS-lgl-IR coexpressed brain shows massive expression of GFP in the optic lobes and in ventral nerve cord (arrowhead marks the expression of GFP in VNC). h′ The optic lobes and VNC of Nact/lgl-IR brain shows extensive expression of MMP1 (arrow marks the expression of MMP1 in VNC). Images e″–h″ are merges of those in e–e′, f–f′, g–g′ and h–h′ with DAPI, respectively. All eye discs are oriented with dorsal to the left and anterior to the top position. Dorsal view of the brains is shown. Scale bars, 50 µm (a–d, a′–d′, a″–d″) and 100 µm (e–h, e′–h′, e″–h″)
In order to examine the cytoskeleton network and cell–cell adhesion, we marked the tissues with phalloidin and adherens junction marker proteins, Armadillo (Arm) and Cadherin (DE-Cad). The F-actin network marked by phalloidin revealed a defective actin cytoskeleton network in Nact/lgl-IR tumor tissues compared to that of controls (Additional file 3: Figure S2). In the same way, the localization of DE-Cad and Arm were also deregulated in Nact/lgl-IR tumorous eye discs (Additional file 4: Figure S3a–d, e–h). We, next, determined if neuronal differentiation was defective in Nact/lgl-IR tumor using a neuronal marker, Elav that marks the differentiated neurons in eye disc and brain. Remarkably, coexpression of Nact and lgl-IR led to severe loss of Elav positive cells in the eye disc and abnormal expression of Elav in the optic lobes indicating an impaired neuronal differentiation (Additional file 4: Figure S3i–l, m–p).
In parallel, we also used dominant-negative version of Notch to see the effect of depletion of Notch signaling on lgl-IR tumors. Previously, expression of mamDN in lgl− tissues partially rescued the lgl− mosaic adult eye phenotype [13]. Our analysis also found that reduction of Notch signaling partially rescued the phenotypes of lgl loss-of-function flies (Additional file 5: Figure S4). Thus, our analysis support the notion that the lgl loss-of-function wing phenotype is dependent on elevated Notch signaling, consistent with the previous study [13].
Involvement of JNK pathway in Nact/lgl-IR tumor
Previous studies in Drosophila have revealed that oncogenic Ras along with loss of lgl or scrib or dlg induces JNK signaling, which is crucial for tumor invasion [7, 16]. This prompted us to check the expression of Puckered (puc), a transcriptional target of JNK signaling and widely used to check the activation of JNK signaling. An enhancer trap allele, puc-LacZ [17] was used to monitor the activation of JNK signaling. Coexpression of both Nact and lgl-IR resulted in intense upregulation of puc throughout the wing disc (Fig. 2d), indicating the activation of JNK signaling in Nact/lgl-IR tumor. We also observed a significant increase in size of the wing disc in Nact and lgl-IR coexpressed condition compared to that of the wild-type, only Nact, and only lgl-IR wing discs (Fig. 2i).
Fig. 2.

Activation of JNK signaling in Nact/lgl-IR tumor. Fluorescent micrographs of wing imaginal discs are shown. a Endogenous expression of puc in Drosophila wing imaginal disc is shown. b Only Nact overexpression shows slight increment in puc expression. c Downregulation of lgl in wing disc induces puc expression. d A significant increase in puc expression was found, when Nact is coexpressed with lgl-IR. a′–d′ are the merge images of DAPI along with a–d, respectively. Images (e–h) show the expression of Egr in wild-type, Nact, lgl-IR, and Nact/lgl-IR wing discs, respectively. The expression of Egr remains unchanged in all genotypes. e′–h′ are the merge images of DAPI along with e–h. i Analyses of differences in wing disc size show that there is a significant increase in Nact/lgl-IR wing imaginal disc size as compared to the wild-type, Nact, and lgl-IR wing disc sizes. j Real-time PCR analysis was performed to estimate the transcript levels of JNK pathway ligand egr and its receptors wgn and grnd in Nact/lgl-IR tumor. The transcript levels of egr and wgn in Nact/lgl-IR were significantly reduced, when compared to that of wild-type; however no significant change in transcript levels were seen, when compared to that of only Nact and lgl-IR. Interestingly, grnd transcript levels were significantly upregulated in Nact/lgl-IR tumor as compared to that of the wild-type, Nact and lgl-IR wing discs. qPCR was normalized with rps17 and repeated for three times. Analysis of data was done using two-way ANOVA with Tukey’s multiple comparison test; data represents mean ± SEM (***p < 0.001; **p < 0.01; *p < 0.05 and ns p > 0.05). All wing discs are oriented with dorsal to the top and posterior to the right. Scale bars: 100 µm (a–d, a′–d′, e–h, e′–h′)
To check the mode of activation of JNK signaling, we examined the transcript level expression of ligand eiger (egr), and its receptor wengen (wgn), in Nact/lgl-IR tumor. egr and wgn transcript levels were found to be depleted in case of Nact/lgl-IR tumor as compared to that of the controls (Fig. 2j). Recently, another member in tumor necrosis factor receptor superfamily, Grindelwald (Grnd), found to be associated with loss of cell polarity and neoplastic growth [18]. Interestingly, a significant upregulation of grnd transcripts in Nact/lgl-IR tumor was found, when compared to that of the wild-type, only Nact and only lgl-IR tissues (Fig. 2j). We went on to check the protein level expression of Egr in Nact/lgl-IR tumors. Immunostaining with anti-Egr antibody [19] revealed that there is no change in the level of Egr protein expression in Nact/lgl-IR tumor (Fig. 2h) as compared to that of the wild-type, only Nact and only lgl-IR tissues (Fig. 2e–g). As Egr is known to be also expressed by the tumor-associated hemocytes, leading to signaling activation [20], these immune cells may be in this case responsible for Grnd activation, but their poor adhesion to the tumor tissue may make them escape Immunofluorescence detection.
To further confirm the involvement of JNK signaling as a downstream event of Nact/lgl-IR cooperation, we blocked JNK signaling in Nact/lgl-IR tumor, and checked whether blocking JNK could affect the Nact/lgl-IR tumor. The massive upregulation of MMP1 in Nact/lgl-IR tumor (Additional file 6: Figure S5a) was drastically suppressed, when bsk-DN (a dominant negative allele of Drosophila JNK gene, basket) was expressed in the background (Additional file 6: Figure S5b). In addition, coexpression of bsk-DN with Nact; lgl-IR resulted in a reduced wing disc size as compared to Nact/lgl-IR overexpressed wing disc (Additional file 6: Figure S5c). These results indicate that JNK signaling may be involved in the tumorous overgrowth of Nact/lgl-IR tissues.
Nact/lgl-IR tumor induces cell death
Eluding apoptosis is considered as one of the acquired capabilities of many types of cancer; however, studies also explain that elevated oncogenic signaling induces apoptosis or senescence [21]. When we checked the status of cell death in Nact/lgl-IR tumor, we observed a significant amount of acridine orange (Compare Fig. 3d with a–c) and caspase positive cells (Compare Fig. 3i with f–h) indicating severe cell death. Since loss of lgl in a tissue known to induce cell competition to remove the unfit cells [22], dying cells in Nact/lgl-IR tissue could be an indication of cell competition. To check the effect of cell death on overgrowth and MMP1 expression, we blocked cell death by expressing a caspase inhibitor, p35 (Fig. 3e, j). It was found that blocking cell death in Nact/lgl-IR overexpressed condition did not obstruct MMP1 expression (Fig. 3o). Coexpression of p35 with Nact/lgl-IR resulted in an increased wing disc size as compared to Nact/lgl-IR overexpressed wing disc (Fig. 3r). As the caspase inhibitor, p35 is known to block cell death [23], the increase in the tissue size is expected since blocking cell death in Nact/lgl-IR tumor allowed more cells to overgrow that, in turn, increased the disc size.
Fig. 3.
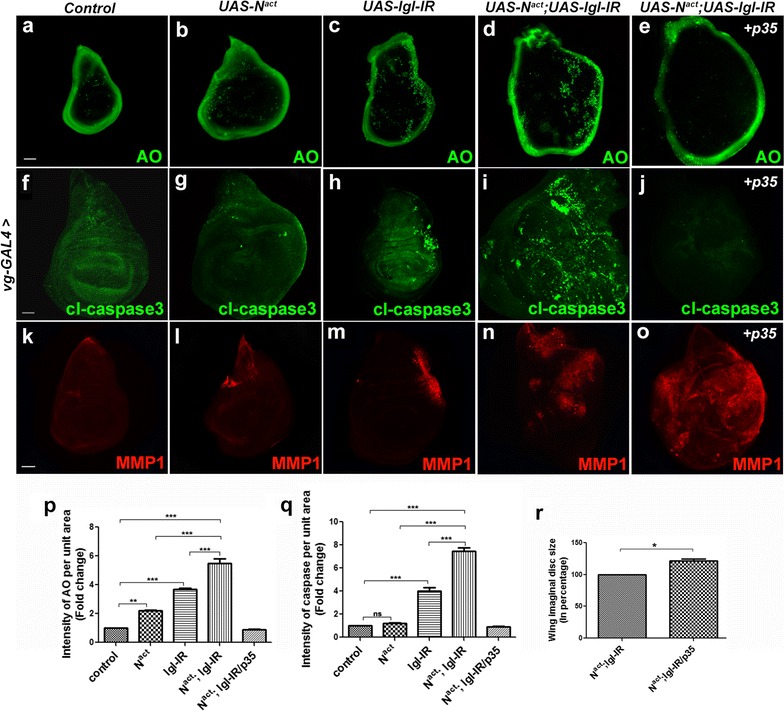
Nact/lgl-IR tumor induces cell death. Fluorescent micrographs of wing imaginal discs are shown. Acridine orange-staining in wild-type (a), Nact over-expressed (b), lgl-IR over-expressed (c) and both Nact/lgl-IR coexpressed (d) wing discs are shown. Expression of cleaved caspase 3 in wild-type (f), Nact over-expressed (g), lgl-IR over-expressed (h) are shown. i Nact/lgl-IR coexpressed wing disc shows upregulation in cleaved caspase 3 expression. MMP1 expression in wild-type (k), Nact overexpressed (l), lgl-IR overexpressed (m) are shown. n Nact/lgl-IR wing disc shows massive upregulation in MMP1 expression. Blocking cell death in Nact/lgl-IR tissues by expressing UAS-p35 leads to absence of acridine orange positive cells (e) and cleaved caspase-3 marked cells (j); whereas the expression of MMP1 is found to be unaltered (o). p Intensity per unit area for acridine orange shows that there is significant amount of upregulation of acridine orange-positive cells in Nact/lgl-IR wing disc. q Intensity per unit area for caspase staining shows that the Nact/lgl-IR wing disc contains significantly upregulated level of caspase activity than that of wild-type, lgl-IR and Nact wing discs. r Wing disc size analyses show that the disc size is increased when p35 is expressed in the background of Nact/lgl-IR, as compared to Nact/lgl-IR. Analysis of data for intensity profiling was done using two-way ANOVA with Tukey’s multiple comparison test; data represents mean ± SEM (***p < 0.001; **p < 0.01 and ns p > 0.05. Analysis of data for wing disc size was done using Unpaired t test with Welch’s correction; data represents mean ± SEM *p < 0.05). All wing discs are oriented with dorsal to the top and posterior to the right. Scale bars: 100 µm for (a–e), and 50 µm for (f–j, k–o)
Discussion
In the present study, we unveil a cooperation of Notch with RNAi-mediated downregulation of a polarity cum tumor suppressor gene, lgl to promote tumor overgrowth. Our data, presented here, illustrate that coexpression of Nact and lgl-IR in Drosophila eye disc results in overgrowth, loss of positional clues and upregulation of MMP1 expression, which is less prevalent in only Nact overexpression or only lgl-IR overexpression. Earlier the loss of polarity gene scribble found to cooperate with Notch signaling to promote neoplastic overgrowth [2]. Another two independent studies of similar context show that oncogenic Ras cooperates with loss of cell polarity genes (lgl, scrib, dlg) to induce metastasis and secondary tumor formation at distant sites [7, 14]. Interestingly, we found that Notch synergizes with loss of lgl to promote tumorous overgrowth and elevated expression of MMP1, and inhibiting Notch signaling rescues the defects caused by loss of lgl. It indicates the potential function of Notch signaling during lgl mediated tumor development. Our data also show distorted epithelial integrity in Nact/lgl-IR tumor that point towards epithelial to mesenchymal transition, where tightly joined epithelial cells with regularly spaced cell–cell junctions convert to mesenchymal cells which are of irregular shape without tight intracellular adhesion [24].
Further, we found upregulation of JNK signaling and its receptor Grindelwald in Nact/lgl-IR tumor. Two previous studies have shown that Notch cooperates with two different proteins to induce proliferation and metastasis by the activation of JNK signaling in ligand-dependent and -independent manner [25, 26]. In case of Nact/lgl-IR tumor, we show that the transcript levels of egr (ligand) and wgn (receptor) were not upregulated, whereas a significant upregulation of grnd transcripts in the Nact/lgl-IR tumor was observed. Earlier the active form of Grnd has shown to activate JNK signaling in vivo [18]. Thus, in case of Nact/lgl-IR tumor, JNK signaling might get activated through Grindelwald. Previously, it has been shown that JNK signaling can initiate tumor initiation and growth in Eiger-independent manner also [27].
Another most important hallmark of almost all types of cancer is the ability to evade apoptosis that, in turn, helps tumor cell population to increase in number [21]. In other similar tumor models such as Rasv12/dlg−/−, dying cells of dlg−/− clones evade apoptosis in presence of oncogenic Ras, where JNK signaling switches its role from proapoptotic to progrowth [7]. In contrast, Ras/scrib−/− and Ras/lgl−/−tumors were reported to show apoptosis [22, 28]. However, Notch/scrib−/− tumor did not show the presence of apoptosis [29]. In our case, Nact/lgl-IR tumor resulted in severe apoptosis along with strong overgrowth and MMP1 expression. These dying cells in Nact/lgl-IR tumor might be the indication of cell competition as there is a strong proliferation and overgrowth. In case of Nact/scrib−/− tumor, Notch is giving growth advantage to scrib−/− tissues by preventing cell death. However, in case of Nact/lgl-IR wing discs, activation of Notch failed to restrict the cell death caused by loss of lgl; rather its activation induces further cell death. These differences indicate that although oncogenic cooperation with loss of cell polarity results in similar tumor cell migration but certain property like cell death occurs depending on the context.
Limitations
The present study is not the first one to show the cooperation between Notch and loss of cell polarity genes. Activated Notch is known to cooperate with another cell polarity gene, scribble, to induce neoplastic overgrowth.
In the present study, experiments were performed using RNAi line of lgl, but not with the lgl loss-of-function mutants.
Additional files
Additional file 1. Materials and methods.
Additional file 2: Figure S1. Quantification of GFP and MMP1 in the VNC of Nact/lgl-IR tumor (a) GFP quantification in VNC shows a four-fold increment in the amount of GFP positive cells in Nact/lgl-IR as compared to that of the wild-type, only Nact and lgl-IR overexpressed tissues. b MMP1 quantification in VNC shows around four-fold increase in Nact/lgl-IR, whereas only Nact and lgl-IR overexpressed tissues show almost same level of MMP1 in VNC as of wild-type. c Real-Time PCR analysis shows significant increase in mmp1 transcripts in the cephalic complex of Nact/lgl-IR as compared to that of wild-type, only Nact or only lgl-IR tissues. Data was normalized to rps17. Analysis of data was done using One-way ANOVA with Tukey’s multiple comparison test; data represents mean ± SEM (***p < 0.001 and ns p > 0.05).
Additional file 3: Figure S2. Nact/lgl-IR tumor leads to distorted actin cytoskeleton. Coexpression of Nact and lgl-IR causes distorted actin cytoskeleton organization (d) compared to that of wild-type (a), only Nact overexpressed (b) and only lgl-IR overexpressed condition (c). F-actin was marked using phalloidin. Scale bars: 10 µm (a-d).
Additional file 4: Figure S3. Nact/lgl-IR shows hallmarks of migratory tumor. Fluorescent micrographs of eye imaginal discs and larval brains are shown. a, a′ Endogenous Cadherin and (e, e′) Armadillo localize at the adherens junctions and marks the photoreceptors in the ey-GAL4/+ eye imaginal discs. Morphogenetic furrow in a and e is marked with an arrow. Overexpression of Nact leads to overgrown discs and the localization pattern of Cadherin (b, b′) and Armadillo (f, f′) have been modified. Overexpression of lgl-IR results in distorted localization of Cadherin (c, c′) and Armadillo (g, g′). Coexpression of Nact and lgl-IR in eye imaginal disc causes complete deformation of Cadherin (d, d′) and Armadillo (h, h′) localization pattern. Images a′–d′, e′–h′ are higher magnification of the square region from a–d, e–h. i Expression of Elav, a marker for differentiated neurons in wild-type eye discs is shown. j Overexpression of Nact in eye disc shows increased expression of Elav, probably due to overproliferation of the disc. k lgl-IR over-expressed eye disc shows comparatively less Elav-positive cells. l Interestingly, Nact and lgl-IR coexpressed eye disc shows hardly any Elav-positive cells. Images i′, j′, k′ and l′ are merges of GFP along with i, j, k and l, respectively. Elav expression in the brains of Nact (n) and lgl-IR (o) driven by ey-GAL4 is found to be similar to that of the wild-type brain (m). p Coexpression of Nact and lgl-IR resulted in an abnormal expression pattern of Elav, where clump like distribution is found in the optic lobes (marked with arrow). Images m′, n′, o′ and p′ are merges of GFP along with m, n, o and p, respectively. Scale bars: 50 µm (a–d, e–h, i–l, i′–l′), 5 µm (a′–d′, e′–h′) and 100 µm (m–p, m′–p′). All eye discs are oriented with dorsal to the left and anterior to the top. Ventral view of the brains is shown.
Additional file 5: Figure S4. Lowering the dose of Notch partially rescues lgl-IR-induced MMP1 expression and restores the adult wing. a MMP1 expression in wild-type wing disc is shown. b Overexpression of only Notch-DN did not induce expression of MMP1. c Overexpression of lgl-IR induces MMP1 expression in the wing disc. d Coexpression of Notch-DN in lgl-IR background partially rescues the expression of MMP1 caused by lgl-IR overexpression. a′, b′, c′ and d′ are merges of DAPI along with a, b, c and d, respectively. Moreover, Coexpression of Notch-DN with lgl-IR resulted in reduced wing disc size as compared to that of only overexpression of lgl-IR (i). e GFP marked vestigial domain in wing disc is shown. e′ is the merge image of DAPI along with (e). f Overexpression of Notch-DN resulted in held out wings with wing nicking phenotype. g Overexpression of lgl-IR using vg-GAL4 led to necrotic lesions followed by deformation of adult wings. h Coexpression of Notch-DN with lgl-IR partially restored deformed adult wings. j Phenotype penetrance in adult flies is shown for each genotype; the phenotype observed in Notch-DN show 100% penetrance and around 70% lgl-IR flies showed deformed wings. In case of Notch-DN; lgl-IR flies, around 60% flies showed the depicted phenotype and, the rest of the flies showed less developed wings but they were not of the lgl-IR category. Analysis of data was done using One-way ANOVA with Tukey’s multiple comparison test; data represents mean ± SEM (***p < 0.001 and ns p > 0.05). All wing discs are oriented with dorsal to the top and posterior to the right. Scale bar: 50 µm (a–d, a′–d′).
Additional file 6: Figure S5. Inhibition of JNK pathway suppresses the Nact/lgl-IR tumor growth and MMP1 expression. Fluorescent micrographs of wing imaginal discs are shown. a Overexpression of both Nact and lgl-IR in wing imaginal disc using vg-GAL4 resulted in massive upregulation of MMP1. b Coexpression of bskDN in the background of Nact and lgl-IR resulted in the suppression of MMP1 expression. a″–b″ is the merge images of a–a′ and b–b″. c The Nact/lgl-IR wing disc size was significantly reduced, when bskDN was expressed in the background. Analysis of data was done using Unpaired t test with Welch’s correction; data represents mean ± SEM **p < 0.01). All wing discs are oriented with dorsal to the top and posterior to the left. Scale bar: 50 µm (a–a″, b–b″).
Authors’ contributions
AM and MSP involved in conception and design of the study. MSP performed the experiments, analyzed the data and drafted the manuscript. DD involved in critically revising the original draft. AS and MM were involved in analysis and interpretation of the data. All authors read and approved the final manuscript.
Acknowledgements
The authors wish to thank Spyros Artavanis-Tsakonas, Estee Kurant, Konrad Basler and the Bloomington Stock Center for fly stocks. Some of the antibodies used in this work were obtained from DSHB. Confocal microscopy and Real-time PCR facility at DBT-BHU-ISLS are duly acknowledged.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Data available on request from the corresponding author.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by funds from DBT, India (BT/PR14082/BRB/10/806/2010 and BT/PR14080/BRB/10/805/2010) and UGC-UPE, BHU to AM and MM. MSP was supported by the fellowship from JNMF while AS and DD were supported by CSIR, Government of India.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- scrib
scribble
- lgl
lethal giant larvae
- dlg
discs large
- Nact
activated Notch
- lgl-IR
lgl-RNAi
- MMP1
matrix metalloproteinase
- VNC
ventran nerve cord
- ey-GAL4
eyeless GAL4
- Arm
armadillo
- DE-Cad
Drosophila E-Cadherin
- Notch-DN
Notch dominant negative
- puc
puckered
- egr
eiger
- wgn
wengen
- grnd
grindelwald
- bsk-DN
basket-dominant negative
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13104-018-3350-5) contains supplementary material, which is available to authorized users.
Contributor Information
Maimuna Sali Paul, Email: maimunasid.sp@gmail.com.
Ankita Singh, Email: ankita.mhg.bhu10@gmail.com.
Debdeep Dutta, Email: debdeep.brc@gmail.com.
Mousumi Mutsuddi, Phone: 91-542-6702492, Email: mousumi@bhu.ac.in, Email: mousumi_mutsuddi@yahoo.com.
Ashim Mukherjee, Phone: 91-542-6702490, Email: amukherjee@bhu.ac.in, Email: ashim04@gmail.com.
References
- 1.Pagliarini AR, Xu T. A genetic screen in Drosophila for metastatic behavior. Science. 2003;302:1227–1231. doi: 10.1126/science.1088474. [DOI] [PubMed] [Google Scholar]
- 2.Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–5779. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gateff E, Schneiderman HA. Developmental studies of a new mutant of Drosophila melanogaster: lethal malignant brain tumor (l(2)gl 4) Am Zool. 1967;7:760. [Google Scholar]
- 4.Stewart M, Murphy C, Fristrom JW. The recovery and preliminary characterization of X chromosome mutants affecting imaginal discs of Drosophila melanogaster. Dev Bio. 1972;1927:71–83. doi: 10.1016/0012-1606(72)90113-3. [DOI] [PubMed] [Google Scholar]
- 5.Bilder D, Perrimon N. Localization of apical epithelial determinants by the basolateral PDZ protein Scribble. Nature. 2000;403:676–680. doi: 10.1038/35001108. [DOI] [PubMed] [Google Scholar]
- 6.Uhlirova M, Jasper H, Bohmann D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci USA. 2005;102:13123–13128. doi: 10.1073/pnas.0504170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol. 2006;16:1139–1146. doi: 10.1016/j.cub.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 8.Froldi F, Ziosi M, Garoia F, Pession A, Grzeschik AN, Bellosta P, et al. Therlethal giant larvae tumour suppressor mutation requires dMyc oncoprotein to promote clonal malignancy. BMC Biol. 2010;8:1–16. doi: 10.1186/1741-7007-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gateff E. The genetics and epigenetics of neoplasms in Drosophila. Biol Rev. 1978;53:123–168. doi: 10.1111/j.1469-185X.1978.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 10.Froldi F, Ziosi M, Tomba G, Parisi F, Garoia F, Pession A, et al. Drosophila lethal giant larvae neoplastic mutant as a genetic tool for cancer modeling. Curr Genom. 2008;9:147–154. doi: 10.2174/138920208784340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grifoni D, Sollazzo M, Fontana E, Froldi F, Pession A. Multiple strategies of oxygen supplies in Drosophila malignancies identify trichogenesis as a novel cancer hallmark. Sci Rep. 2015;5:9061. doi: 10.1038/srep09061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calleja M, Morata G, Casanova J. Tumorogenic properties of Drosophila epithelial cells mutant for lethal giant larvae. Dev Dyn. 2016;245:834–843. doi: 10.1002/dvdy.24420. [DOI] [PubMed] [Google Scholar]
- 13.Parsons LM, Portela M, Grzeschik NA, Richardson HE. Lgl regulates Notch signaling via endocytosis, independently of the apical aPKC-Par6-Baz polarity complex. Curr Biol. 2014;24:2073–2084. doi: 10.1016/j.cub.2014.07.075. [DOI] [PubMed] [Google Scholar]
- 14.Uhlirova M, Bohmann D. JNK and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006;25:5294–5304. doi: 10.1038/sj.emboj.7601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCawley LJ, Matrisian LM. Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol Med Today. 2000;6:149–156. doi: 10.1016/S1357-4310(00)01686-5. [DOI] [PubMed] [Google Scholar]
- 16.Leong GR, Goulding KR, Amin N, Richardson HE, Brumby AE. scribble mutants promote aPKC and JNK-dependent epithelial neoplasia independently of Crumbs. BMC Biol. 2009 doi: 10.1186/1741-7007-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shklover J, Mishnaevski K, Levy-Adam F, Kurant E. JNK pathway activation is able to synchronize neuronal death and glial phagocytosis in Drosophila. Cell Death Dis. 2015;6:e1649. doi: 10.1038/cddis.2015.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersen SD, Colombani J, Palmerini V, Chakrabandhu K, Boone E, Rothlisberger M, et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic overgrowth. Nature. 2015;522:482–486. doi: 10.1038/nature14298. [DOI] [PubMed] [Google Scholar]
- 19.Moreno E, Yan M, Basler K. Evolution of TNF signaling mechanisms: JNK dependent apoptosis triggered by Eiger, the Drosophila homologue of the TNF superfamily. Curr Biol. 2002;12:1263–1268. doi: 10.1016/S0960-9822(02)00954-5. [DOI] [PubMed] [Google Scholar]
- 20.Cordero BJ, Macagno PJ, Stefanatos KR, Strathdee EK, Cagan LR, Vidal M. Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell. 2010;8:999–1011. doi: 10.1016/j.devcel.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Menéndez J, Pérez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci USA. 2010;107:14651–14656. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meier P, Silke J, Leevers SJ, Evan GI. The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 2000;19:598–611. doi: 10.1093/emboj/19.4.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee MJ, Dedhar S, Kalluri R, Thompson WE. The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pallavi SK, Ho MD, Hicks C, Miele L, Artavanis-Tsakonas S. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 2012;31:2895–2907. doi: 10.1038/emboj.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ho MD, Pallavi SK, Artavanis-Tsakonas S. The Notch-mediated hyperplasia circuitry in Drosophila reveals a Src-JNK signaling axis. eLife. 2015;4:e05996. doi: 10.7554/eLife.05996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muzzopappa M, Murcia L, Milan M. Feedback amplification loop drives malignant growth in epithelial tissues. Proc Natl Acad Sci USA. 2017;114:E7291–E7300. doi: 10.1073/pnas.1701791114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulshammer E, Uhlirova M. The actin cross-linker filamin/cheerio mediates tumor malignancy downstream of JNK signaling. J Cell Sci. 2013;126:927–938. doi: 10.1242/jcs.114462. [DOI] [PubMed] [Google Scholar]
- 29.Doggett K, Turkel N, Willoughby LF, Ellul J, Murray MJ, Richardson HE, et al. BTB-zinc finger oncogenes are required for Ras and Notch-driven tumorigenesis in Drosophila. PLoS ONE. 2015;10:e0132987. doi: 10.1371/journal.pone.0132987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Materials and methods.
Additional file 2: Figure S1. Quantification of GFP and MMP1 in the VNC of Nact/lgl-IR tumor (a) GFP quantification in VNC shows a four-fold increment in the amount of GFP positive cells in Nact/lgl-IR as compared to that of the wild-type, only Nact and lgl-IR overexpressed tissues. b MMP1 quantification in VNC shows around four-fold increase in Nact/lgl-IR, whereas only Nact and lgl-IR overexpressed tissues show almost same level of MMP1 in VNC as of wild-type. c Real-Time PCR analysis shows significant increase in mmp1 transcripts in the cephalic complex of Nact/lgl-IR as compared to that of wild-type, only Nact or only lgl-IR tissues. Data was normalized to rps17. Analysis of data was done using One-way ANOVA with Tukey’s multiple comparison test; data represents mean ± SEM (***p < 0.001 and ns p > 0.05).
Additional file 3: Figure S2. Nact/lgl-IR tumor leads to distorted actin cytoskeleton. Coexpression of Nact and lgl-IR causes distorted actin cytoskeleton organization (d) compared to that of wild-type (a), only Nact overexpressed (b) and only lgl-IR overexpressed condition (c). F-actin was marked using phalloidin. Scale bars: 10 µm (a-d).
Additional file 4: Figure S3. Nact/lgl-IR shows hallmarks of migratory tumor. Fluorescent micrographs of eye imaginal discs and larval brains are shown. a, a′ Endogenous Cadherin and (e, e′) Armadillo localize at the adherens junctions and marks the photoreceptors in the ey-GAL4/+ eye imaginal discs. Morphogenetic furrow in a and e is marked with an arrow. Overexpression of Nact leads to overgrown discs and the localization pattern of Cadherin (b, b′) and Armadillo (f, f′) have been modified. Overexpression of lgl-IR results in distorted localization of Cadherin (c, c′) and Armadillo (g, g′). Coexpression of Nact and lgl-IR in eye imaginal disc causes complete deformation of Cadherin (d, d′) and Armadillo (h, h′) localization pattern. Images a′–d′, e′–h′ are higher magnification of the square region from a–d, e–h. i Expression of Elav, a marker for differentiated neurons in wild-type eye discs is shown. j Overexpression of Nact in eye disc shows increased expression of Elav, probably due to overproliferation of the disc. k lgl-IR over-expressed eye disc shows comparatively less Elav-positive cells. l Interestingly, Nact and lgl-IR coexpressed eye disc shows hardly any Elav-positive cells. Images i′, j′, k′ and l′ are merges of GFP along with i, j, k and l, respectively. Elav expression in the brains of Nact (n) and lgl-IR (o) driven by ey-GAL4 is found to be similar to that of the wild-type brain (m). p Coexpression of Nact and lgl-IR resulted in an abnormal expression pattern of Elav, where clump like distribution is found in the optic lobes (marked with arrow). Images m′, n′, o′ and p′ are merges of GFP along with m, n, o and p, respectively. Scale bars: 50 µm (a–d, e–h, i–l, i′–l′), 5 µm (a′–d′, e′–h′) and 100 µm (m–p, m′–p′). All eye discs are oriented with dorsal to the left and anterior to the top. Ventral view of the brains is shown.
Additional file 5: Figure S4. Lowering the dose of Notch partially rescues lgl-IR-induced MMP1 expression and restores the adult wing. a MMP1 expression in wild-type wing disc is shown. b Overexpression of only Notch-DN did not induce expression of MMP1. c Overexpression of lgl-IR induces MMP1 expression in the wing disc. d Coexpression of Notch-DN in lgl-IR background partially rescues the expression of MMP1 caused by lgl-IR overexpression. a′, b′, c′ and d′ are merges of DAPI along with a, b, c and d, respectively. Moreover, Coexpression of Notch-DN with lgl-IR resulted in reduced wing disc size as compared to that of only overexpression of lgl-IR (i). e GFP marked vestigial domain in wing disc is shown. e′ is the merge image of DAPI along with (e). f Overexpression of Notch-DN resulted in held out wings with wing nicking phenotype. g Overexpression of lgl-IR using vg-GAL4 led to necrotic lesions followed by deformation of adult wings. h Coexpression of Notch-DN with lgl-IR partially restored deformed adult wings. j Phenotype penetrance in adult flies is shown for each genotype; the phenotype observed in Notch-DN show 100% penetrance and around 70% lgl-IR flies showed deformed wings. In case of Notch-DN; lgl-IR flies, around 60% flies showed the depicted phenotype and, the rest of the flies showed less developed wings but they were not of the lgl-IR category. Analysis of data was done using One-way ANOVA with Tukey’s multiple comparison test; data represents mean ± SEM (***p < 0.001 and ns p > 0.05). All wing discs are oriented with dorsal to the top and posterior to the right. Scale bar: 50 µm (a–d, a′–d′).
Additional file 6: Figure S5. Inhibition of JNK pathway suppresses the Nact/lgl-IR tumor growth and MMP1 expression. Fluorescent micrographs of wing imaginal discs are shown. a Overexpression of both Nact and lgl-IR in wing imaginal disc using vg-GAL4 resulted in massive upregulation of MMP1. b Coexpression of bskDN in the background of Nact and lgl-IR resulted in the suppression of MMP1 expression. a″–b″ is the merge images of a–a′ and b–b″. c The Nact/lgl-IR wing disc size was significantly reduced, when bskDN was expressed in the background. Analysis of data was done using Unpaired t test with Welch’s correction; data represents mean ± SEM **p < 0.01). All wing discs are oriented with dorsal to the top and posterior to the left. Scale bar: 50 µm (a–a″, b–b″).
Data Availability Statement
Data available on request from the corresponding author.