

Abstract
Background
Brassica crops are cultivated widely for human consumption and animal feed purposes, and oilseed rape/canola (Brassica napus and rapa) is the second most important oilseed worldwide. Because of its natural diversity and genetic complexity, genomics studies on oilseed rape will be a useful resource base to modify the quantity and quality of biomass in various crops, and therefore, should have a positive impact on lignocellulosic biofuel production. The objective of this study was to perform microarray analysis on two variable lignin containing oilseed rape cultivars to target novel genes and transcription factors of importance in Brassica lignin regulation for applied research.
Results
To gain insight into the molecular networks controlling cell wall biosynthetic and regulatory events, we conducted lignin and microarray analysis of top and basal stem sections of brown seeded Brassica napus DH12075 and yellow seeded YN01–429 cultivars. A total of 9500 genes were differentially expressed 2-fold or higher in the stem between the cultivars, with a higher number of expressed genes in the basal section. Of the upregulated genes, many were transcription factors and a considerable number of these were associated with secondary wall synthesis and lignification in B. napus and other plant species. The three largest groups of transcription factors with differential expression were C2H2 and C3HC4 zinc fingers and bHLH. A significant number of genes related to lignin and carbohydrate metabolism also showed differential expression patterns between the stem sections of the two cultivars. Within the same cultivar, the number of upregulated genes was higher in the top section relative to the basal one.
Conclusion
In this study, we identified and established expression patterns of many new genes likely involved in cell wall biosynthesis and regulation. Some genes with known roles in other biochemical pathways were also identified to have a potential role in cell wall biosynthesis. This stem transcriptome profiling will allow for selecting novel regulatory and structural genes for functional characterization, a strategy which may provide tools for modifying cell wall composition to facilitate fermentation for biofuel production.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4645-6) contains supplementary material, which is available to authorized users.
Keywords: Biomass, Brassica, Cell wall, Stem lignin, Microarray, Transcription factors
Background
Limiting sources of fossil energy resources and potential climate change underline increasing worldwide economic interest and scientific focus on renewable biofuel. Lignocellulosic biomass is abundant in nature and can be used as sustainable and renewable feedstocks for biofuel production. Grasses, such as maize, sorghum, switchgrass and Miscanthus, and fast-growing trees, such as poplar and willow are potential sources of lignocellulosic biomass. Crop residues collected from the production of major food crops such as canola, maize, rice and wheat can also be used as raw material for biofuel. Brassica crops are among the oldest cultivated plants, and oilseed rape/canola (Brassica napus and rapa) is currently the second most important oilseed worldwide [1]. Because of its wide spread cultivation, natural diversity and genetic complexity [2, 3], genomic studies on oilseed rape are a useful resource base from which to modify the quantity and quality of biomass in various crops, and hence, should have a positive impact on lignocellulosic biofuel production.
Crop residues are a rich source of plant biomass, which is mainly composed of plant cell walls. The major components of plant cell walls are cellulose, hemicellulose, pectin and lignin. Cellulose is the world’s most abundant biopolymer [4] and can be used as a sustainable and renewable feedstock for biofuels [5, 6]. However, this process is inhibited by the presence of lignin [7], a major component of secondary cell walls, which provides structural support to plants but also acts as an obstacle to glucose fermentation by increasing the recalcitrance of cell wall digestibility [8]. Moreover, lignin can adsorb hydrolytic enzymes that are used to generate monosaccharides from lignocellulose, and some lignin degradation products inhibit subsequent fermentation steps [9]. Novel strategies to reduce lignin and/or modify its composition in the plant cell wall are therefore needed, and a clear understanding of cell wall biogenesis-related gene expression with emphasis on lignin biosynthesis and factors regulating its accumulation and polymerization is essential to addressing cell wall recalcitrance.
Although many of the structural genes in lignin biosynthesis have been characterized, additional knowledge on transcription factors controlling this pathway is still needed. Studies have shown developmental regulation of the phenylpropanoid pathway genes by various classes of trans-acting factors [10]. Although, most of the genes encoding lignin biosynthetic enzymes have been targets for modification of lignin biosynthesis, only a few studies have taken advantage of the possibility of modulating the trans-acting factors involved in phenylpropanoid metabolism. Some promising findings on the role of transcription factors with phenylpropanoid end products, including lignin biosynthesis, have been reported through mutant analysis [11–15], but additional work is still needed in this area. Presumably, altering the expression level of appropriate transcription factors would coordinately affect a group of genes in the pathway. By doing so, it might be possible to avoid deleterious phenotypes associated with the accumulation of pathway intermediates due to single gene/enzyme manipulations [16].
Microarrays have been an important technology for the global analysis of gene expression in plants, including in cell walls, although more recently RNA sequencing has risen in importance. Hertzberg et al. [17] used poplar cDNA arrays to profile changes in gene expression at various stages of secondary xylem differentiation. Ko et al. [18] used short oligonucleotide arrays to identify genes that display preferred expression in secondary xylem and during the transition from primary to secondary growth in Arabidopsis stems. Ehlting et al. [19] studied the metabolic, developmental and regulatory events at different stages of vascular and interfascicular fiber differentiation in inflorescence stems of Arabidopsis using oligo arrays.
The yellow seeded B. napus YN01–429 cultivar was recently developed through classical breeding approaches. It was found to contain significantly reduced seed lignin content relative to the more conventional brown seeded cultivar DH12075 [20, 21]. The yellow-colored trait of Brassica seeds is valuable to Brassica breeders since it is associated with a thinner seed coat and reduced dietary fiber content [21]. Light seed color and low fiber content are believed to share precursors and biochemical pathways leading to lignin and pigment synthesis [22, 23]. Since plant stems represent the major contribution to Brassica plant residue, we analyzed the total lignin content of stem sections of these two unique cultivars. On the basis of differential lignin content, we performed microarray experiments to determine variations in the transcription profiles of different sections of the stem. Analysis of transcript profiles allowed us to categorize genes into specific sets involved in various regulatory, developmental and metabolic processes including transcriptional regulation, carbohydrate metabolism, and lignin and cellulose biosynthesis. By correlating changes in gene expression with changes in the levels of lignin, we hypothesized that it should be possible to derive insights into the regulatory mechanisms of cell wall biosynthesis and to find target transcription factors of importance in Brassica lignin regulation for practical purposes.
Methods
Plant materials and growth conditions
Brown seeded B. napus DH12075 (designated DH), yellow seeded B. napus YN01–429 (designated YN), alfalfa (Medicago sativa L.) and Arabidopsis thaliana ecotypes and mutant lines were grown in soil in a controlled greenhouse environment (16 h light/8 h dark, 20 °C/17 °C). For B. napus stem samples, plants with well-developed siliques were harvested and 40 cm of the stem was selected (excluding the lower most 10 cm). Collected stem samples were divided into 4 segments, 10 cm each, and apical (section 1) and basal sections (section 4) were used for lignin, microarray and qPCR analyses. For Arabidopsis lignin analysis, Arabidopsis plants (42-day-old) were cut at the base of the inflorescence stem and leaves and thin branches were removed, keeping the stem and main/robust branches for analysis.
Lignin analysis
Lignin content of stem material was determined by thioglycolic acid (TGA) method [24]. Fifty mg of stem tissue ground in liquid N2 from 3 different plants per cultivar (i.e., 3 biological replicates) was used for lignin content determination.
DNA microarray
Total RNA from stem sections of both DH and YN were extracted as described in Carpenter and Simon [25] followed by clean-up using the commercial RNeasy mini kit (Qiagen, Valencia, CA, USA). Two biological replicates were collected for each sample. RNA amplification, labeling with cy3- or cy5-dCTP dyes (GE Healthcare, Buckinghamshire, UK), and probe fragmentation was carried out using Ambion AminoAllyl MessageAmp II RNA amplification kit according to the manufacturer’s instructions (Ambion, Austin, TX, USA). A spotted 15 K 50-mer B. napus oligo array, previously developed at the Saskatoon Research and Development Centre, Agriculture and Agri-Food Canada [26], was hybridized with the cy5- and cy3-labelled probe pairs. Cy3 dye was used for labeling the YN RNA and Cy5 for DH RNA. A dye swap (cy3/cy5) labelling experiment was performed for each biological replicate. Labeling, hybridization and post- hybridization washing were conducted according to the protocol for Corning epoxide slides (Corning Inc., Lowell, MA, USA). Hybridization was carried out at 37 °C for 17 h in a MAUI hybridization station (BioMicro Systems, Salt Lake City, UT, USA). Following the post-hybridization washes, slides were scanned with a VersArray ChipReader laser scanner (Bio-Rad Laboratories, Hercules, CA, USA).
Image files were transferred to ArrayPro Analyzer software (Media Cybernetics Inc., Bethesda, MD, USA) for image analysis, spot verification, normalization, filtering and feature extractions. A standard statistical program GeneSpring GX (Agilent Technologies, Santa Clara, CA, USA) was used to determine that the data set would fit a normal distribution (log 10) and to check the validity of expression data by assessing the variation between control spots. Data files for the duplicates on individual slides and the dye swap data files for each experiment were merged and average spot intensities used to reduce experimental bias. Normalization was performed on merged data using the Lowess (sub-grid) method, and the local background was subtracted from the values of each spot. The intensity of each spot at λ549 nm (Cy5) and λ647 nm (Cy3) was analyzed using a BASE plug-in and finally transformed into a DH/YN ratio value that denoted the most upper (section 1) and lowest stem sections (section 4), including DH1:YN1, DH4:YN4, DH1:DH4 and YN1:YN4. Additional data processing was performed using tools available in BASE (http://base.thep.lu.se). Gene ontology (GO) analysis was conducted using the TAIR database [27]. Microarray ratios with transcript changes > 2-fold upregulated or downregulated between two different B. napus stem sections were considered for all ratios where p ≤ 0.05 (Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12: Table S1A and B to S6A and B). Data in these 12 Additional files were screened using either Excel or Microsoft Access to identify individual or groups of B. napus genes expressed in multiple DH and YN ratios in the main categories of cell wall, other carbohydrate genes and transcription factors, and in additional subcategories. Ten major transcription factor families, Arabidopsis homologue IDs and corresponding mutant lines for selected B. napus genes were retrieved from http://www.arabidopsis.org/browse/genefamily/index.jsp [28].
Quantitative real-time RT-PCR
For quantitative real time reverse transcription PCR (qRT-PCR) experiments, total RNA was extracted from stem sections using a TRIzol reagent (http://www.invitrogen.com) following the manufacturer’s instructions. Three biological replicates (independent RNA preparations) were collected and used with three technical reps for qRT-PCR analysis. RNA concentration was determined using a nanodrop spectrophotometer (Fisher Scientific, Canada). Twenty micrograms of total RNA (per sample in each replicate) was treated with Turbo DNase (www.ambion.com) as per the manufacturer’s instructions to eliminate trace amounts of genomic DNA. Reverse transcription reactions were performed with Superscript™ III Reverse Transcriptase (http://www.invitrogen.com) according to the manufacturer’s instructions using 1.5 μg RNA per reaction. Two RT reactions per sample were done and reactions were pooled after 10-fold dilution with nuclease-free water. Polymerase chain reactions were carried out using 96-well plates in a LightCycler® 480 II (http://www.roche-applied-science.com/lightcycler-online) using SYBR® Green. Reactions contained 10 μl of 2× SYBR® Green Master Mix (Roche), 5 μl of diluted cDNA and 200 nM of gene/EST-specific primer (Additional file 13: Table S7) in a final volume of 20 μl. For data normalization, five reference genes (actin, adenine phosphorybosyl transferase, β-tubulin, cyclophilin and elongation factor 1-α) were included in the experiment and two genes with stable expression, namely β-tubulin and actin, were selected using geNorm software [29]. PCR efficiency, in the range of 85 to 100%, was determined from amplification plots using the program LinRegPCR [30].
Plant transformation
RNAi and over-expression constructs were developed using either the closest Arabidopsis homologue according to Hossain et al. [31, 32] or the closest alfalfa stem cDNA according to Li et al. [33]. Alfalfa primers were identified by Laberge (personal communication). Floral bud transformation was conducted in Arabidopsis according to Hossain et al. [31, 32]. Alfalfa transformation was conducted using Agrobacterium tumefaciens according to Aung et al. [34].
Results
DH and YN stems differ in lignin and exhibit different gene expression profiles
To validate our introductory hypothesis, we first determined the stem lignin content of DH and YN plants. Total lignin content showed higher levels in both apical (section 1) (DH1) and basal sections (section 4) (DH4) of DH compared to their counterparts in YN cultivar sections (YN1 and YN4) (Fig. 1a). Within stem sections of each cultivar, we found the highest lignin content to be in the basal sections and the lowest in the apical sections. Increased lignin accumulation is the result of increased enzyme activity and/or increased gene expression. Hence, we compared global gene expression levels between the two cultivars and also between these sections using a 15 K B. napus microarray that was used earlier to investigate gene expression in developing B. napus seed [35] and diseased stem sections [26]. In detail, we related lignin levels with transcriptome changes between DH and YN (both apical and basal sections for each cultivar (Fig. 1b; Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12: Tables S1A and B to S6A and B). The greater upregulation of genes in the apical stem sections compared with basal stem section expression pattern is reasonable because the apical region of the plant is developmentally more active compared to the basal region.
Fig. 1.
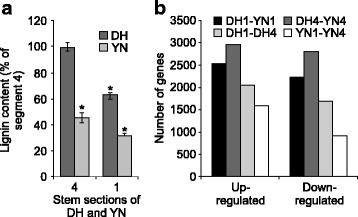
Top and basal section of stem from B. napus DH12075 (DH) and YN01–429 (YN) used for lignin analysis. a Lignin in top section (1) and basal section (4). b Total number of changed gene transcripts in microarray analysis of four pairwise comparisons of the transcriptome in sections 1 and 4. For A, each value is the mean ± SD of 3 biological experiments each replicated 3 times. Values with asterisks varied significantly where p ≤ 0.05
Functional classification of genes up-regulated in high lignin stems of B. napus
Gene annotation of sequences differentially expressed using the 15 K B. napus array was conducted using The Arabidopsis Information Resource (http://www.arabidopsis.org). Annotated genes were grouped into three major functional categories: cellular component, molecular function and biological process, and then divided into subcategories (Fig. 2; Table 1). For ease of presentation, data was expressed as the average of genes in each subcategory in DH vs. the average in YN. Within the category of biological processes in DH4 vs. YN4, genes involved in defense responses were 7.6% of the total, whereas stress responsive genes were 7.4%; genes involved in developmental processes were 4.9 and 4.7% were involved in transport; 2.35% of genes were involved in transcription and 0.6% in DNA or RNA metabolism. In the cellular component category, 6.4% genes were related to plasma membrane, and nucleus genes accounted for 6.02%; cell wall genes were 2.12 and 1.6% genes were related to cytoplasm functions. In the molecular function category, 7.4% genes were involved in protein binding, and 5.7% genes accounted for DNA or RNA binding activity; 4.8% genes were involved in transporter activity, and 3.4% genes showed transcription factor activity (Fig. 2; Table 1).
Fig. 2.
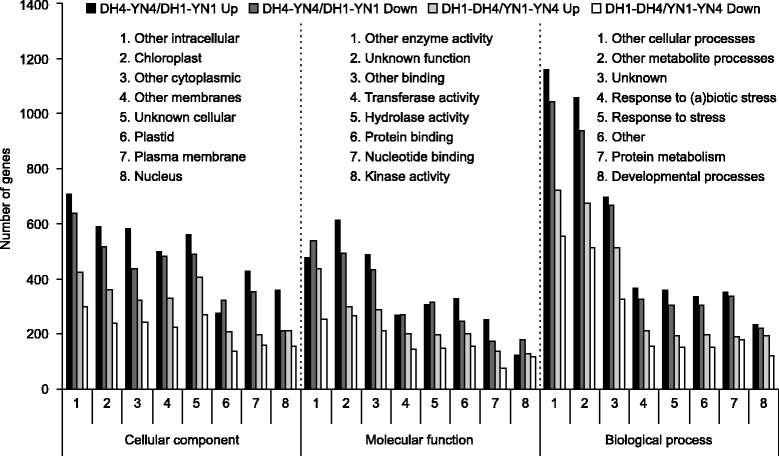
Functional classification of differentially expressed genes in pairwise microarray comparisons of stem section transcriptomes from B. napus DH12075 (DH) and YN01–429 (YN). 1, section 1. 4, section 4. Up, upregulated. Down, down regulated. Genes were first compiled into three large biochemical classes and then subdivided into as many as 16 sub-categories. The eight largest sub-categories are shown
Table 1.
Total number of changed stem transcripts in three major functional categories of B. napus DH12075 (DH) and YN01–429 (YN)
| DH4:YN4 | DH1:YN1 | DH1:DH4 | YN1:YN4 | |||||
|---|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | Up | Down | |
| Cellular Component | ||||||||
| Other intracellular components | 754 | 693 | 668 | 580 | 475 | 396 | 377 | 205 |
| Chloroplast | 602 | 567 | 581 | 462 | 413 | 320 | 308 | 159 |
| Other cytoplasmic components | 636 | 469 | 531 | 403 | 361 | 321 | 280 | 161 |
| Other membranes | 531 | 482 | 466 | 483 | 374 | 290 | 284 | 156 |
| Unknown cellular components | 607 | 607 | 513 | 373 | 463 | 352 | 347 | 187 |
| Plastid | 291 | 343 | 260 | 303 | 174 | 151 | 246 | 122 |
| Plasma membrane | 438 | 381 | 421 | 326 | 262 | 240 | 133 | 79 |
| Nucleus | 392 | 227 | 331 | 200 | 247 | 203 | 181 | 107 |
| Other cellular components | 284 | 285 | 184 | 153 | 225 | 162 | 163 | 103 |
| Mitochondria | 198 | 191 | 210 | 188 | 125 | 94 | 97 | 55 |
| Cell wall | 127 | 123 | 115 | 88 | 75 | 66 | 64 | 40 |
| Cytosol | 121 | 102 | 106 | 80 | 66 | 66 | 56 | 31 |
| Extracellular | 110 | 82 | 82 | 62 | 66 | 62 | 44 | 33 |
| ER | 90 | 76 | 81 | 60 | 60 | 48 | 44 | 33 |
| Golgi apparatus | 49 | 43 | 40 | 37 | 31 | 26 | 18 | 12 |
| Ribosome | 23 | 23 | 23 | 18 | 16 | 13 | 10 | 8 |
| Molecular Function | ||||||||
| Other enzyme activity | 515 | 695 | 439 | 383 | 492 | 295 | 383 | 216 |
| Unknown molecular functions | 650 | 471 | 578 | 515 | 341 | 376 | 259 | 156 |
| Other binding | 525 | 469 | 455 | 393 | 327 | 274 | 248 | 150 |
| Transferase activity | 295 | 308 | 243 | 229 | 211 | 186 | 188 | 100 |
| Hydrolase activity | 341 | 328 | 275 | 299 | 240 | 199 | 151 | 98 |
| Protein binding | 351 | 260 | 306 | 230 | 223 | 199 | 176 | 115 |
| Nucleotide binding | 279 | 132 | 231 | 217 | 131 | 96 | 139 | 53 |
| Kinase activity | 137 | 245 | 114 | 110 | 175 | 143 | 85 | 88 |
| DNA or RNA binding | 257 | 182 | 160 | 183 | 83 | 125 | 121 | 73 |
| Transporter activity | 164 | 201 | 197 | 131 | 153 | 93 | 70 | 56 |
| Other molecular functions | 185 | 157 | 143 | 110 | 122 | 93 | 103 | 45 |
| Transcription factor activity | 165 | 149 | 130 | 115 | 117 | 93 | 79 | 47 |
| Nucleic acid binding | 119 | 111 | 98 | 102 | 76 | 64 | 61 | 28 |
| Structural molecule activity | 35 | 22 | 28 | 25 | 26 | 16 | 20 | 8 |
| Receptor binding or activity | 15 | 18 | 17 | 12 | 16 | 10 | 9 | 5 |
| Biological Process | ||||||||
| Other cellular processes | 1254 | 1145 | 1066 | 943 | 803 | 710 | 641 | 398 |
| Other metabolic processes | 1139 | 1016 | 974 | 860 | 754 | 664 | 597 | 364 |
| Unknown biological processes | 763 | 760 | 637 | 575 | 583 | 418 | 444 | 233 |
| Response to abiotic or biotic stimulus | 387 | 373 | 350 | 279 | 221 | 197 | 200 | 115 |
| Response to stress | 381 | 342 | 337 | 263 | 206 | 188 | 181 | 115 |
| Other biological processes | 353 | 334 | 325 | 269 | 215 | 197 | 183 | 105 |
| Protein metabolism | 375 | 359 | 333 | 317 | 247 | 229 | 130 | 127 |
| Developmental processes | 251 | 261 | 222 | 181 | 197 | 158 | 189 | 83 |
| Transport | 266 | 232 | 232 | 193 | 165 | 124 | 139 | 85 |
| Cell organization and biogenesis | 171 | 154 | 150 | 137 | 127 | 110 | 97 | 56 |
| Transcription | 159 | 146 | 130 | 119 | 104 | 88 | 73 | 42 |
| Electron transport or energy pathways | 81 | 74 | 61 | 54 | 51 | 31 | 45 | 17 |
| Signal transduction | 75 | 52 | 66 | 46 | 39 | 38 | 37 | 22 |
| DNA or RNA metabolism | 35 | 27 | 24 | 21 | 25 | 20 | 18 | 14 |
NB data was ranked in descending order using the DH4-YN4 pair wise comparison. Up., upregulated. Down, down regulated
Annotation for genes differentially expressed in DH1 vs. YN1, DH1 vs. DH4 and YN1 vs. YN4 pairwise comparisons was also carried out. In all three functional categories, genes belonging to the above three groupings showed a similar pattern of distribution to those of DH4 vs. YN4.
Validation of microarray data by quantitative RT-PCR
To validate the results of microarray experiments, transcript levels of seven genes were examined by qRT-PCR using total RNA prepared from stem sections. Emphasis was placed on functional diversity in selecting the following genes for qRT-PCR experiments; ANAC062 (Accession # ES901271), PHYTOCHOME INTERACTING FACTOR 4 (PIF4; EV181356), GLYCINE-RICH RNA BINDING PROTEIN (RBP/ CAA78513.1), DIMINUTO 1, LIPASE 1 (AY866419), FIBRILLIN (AF084554.1) and GDSL-MOTIF CONTAINING LIPASE/HYDROLASE (CN730052) (Fig. 3a and b; Additional file 13: Table S7). From the microarray data, it was determined that these selected genes were upregulated in the DH cultivar with high lignin content. The expression profiles of most of the genes tested by qRT-PCR were consistent with our microarray data, although the difference in expression was less in the latter data set. Expression of DIMINUTO 1 was significantly higher in the basal section of DH (DH4). Expression of ANAC062 was also higher in the basal section of DH4, whereas PIF4 showed higher expression in both apical (DH1) and basal sections (DH4). Expression of LIPASE1 was also higher in both sections of DH compared to those of YN. Transcripts of RBP exhibited slight numerical increase in both sections of DH, although the difference was not statistically significant. Because of their extremely low expression levels relative to other tested genes, the two genes encoding FIBRILLIN and GDSL-MOTIF PROTEIN were assessed separately (Fig. 3c).
Fig. 3.
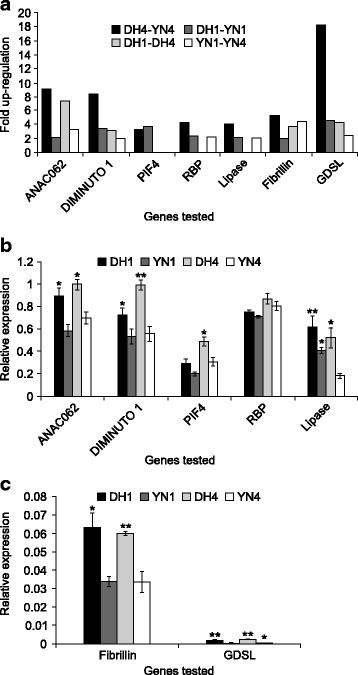
Relative expression pattern of seven regulatory and biochemical genes in stem sections DH12075 (DH) and YN01–429 (YN) using qRT-PCR. a Each value is the mean ± SD from 2 biological replicates each technically replicated 3 times. b and c. Each value is the mean ± SD from 3 biological replicates, each technically replicated 3 times. 1, stem section 1. 4, stem section 4. Values with asterisks represent significantly different data of DH relative to YN where * p ≤ 0.05 and ** p ≥ 0.01. The significance test was done separately for each gene
Both of the latter genes showed significantly higher expression in the two sections of DH relative to their counterparts in YN.
Upregulation of monolignol biosynthetic genes
Genes involved in monolignol biosynthesis were retrieved from the TAIR database (www.arabidopsis.org) and used to identify B. napus biochemical genes that were upregulated at-least 2-fold in high lignin-containing stem material using the microarray data (Table 2). Tissue comparisons showed that several genes encoding enzymes involved in monolignol biosynthesis were upregulated in the DH cultivar relative to the YN cultivar. Among these genes, caffeic acid/5-hydroxyferulic acid O-methyltransferase (OMT1) was upregulated in the top and basal sections of DH relative to the top and basal sections of YN (Table 2). In addition, S-adenosyl methionine synthase (SAMS) and CCoAOMT enzymes that methylate lignin precursors [36] were coded by strongly upregulated genes in our microarray experiments (Table 2). SAMS catalyzes the transfer of an adenosyl group from ATP to the sulphur atom of methionine, resulting in the synthesis of SAM, a common methyl group donor. In the microarray experiments, SAMS3 was one of the stronger upregulated genes in the top and basal sections of DH relative to those of YN and in the basal section relative to the top section of DH (Table 2).
Table 2.
Differential upregulation of monolignol biosynthetic genes ≥ two-fold in stem sections of B. napus DH12075 (DH) and YN01–429 (YN)
| Probe name | Upregulated expression | Corresponding Arabidopsis Loci | Description | |||
|---|---|---|---|---|---|---|
| DH4:YN4 | DH1:YN1 | DH4:DH1 | YN4:YN1 | |||
| BN18476 | 2.04 | 2.13 | AT1G09500 | Similar to Eucalyptus gunnii alcohol dehydrogenase | ||
| BN10302 | 3.64 | 4.25 | AT1G12840 | De-etiolated 3 (DET3) | ||
| BN21410 BN21411 | 6.45 | 2.18 | 2.25 | AT1G15950 | Cinnamoyl CoA reductase (CCR1) | |
| BN26995 | 2.27 | AT1G33030 | O-methyltransferase family 2 protein | |||
| BN17817 | 2.54 | AT1G67980 | Caffeoyl-CoA 3-O-methyltransferase | |||
| BN24319 BN24320 | 2.43 | AT1G77520 | O-methyltransferase family 2 protein | |||
| BN23842 | 2.5 | AT1G80820 | Cinnamoyl CoA reductase (CCR2) | |||
| BN25563 | 2.14 | 2.19 | AT2G02400 | Cinnamoyl-CoA reductase family | ||
| BN15586 | 8.73 | 2.64 | AT2G40890 | Coumarate 3-hydroxylase (C3H), a P450-dependent monooxygenase (CYP98A3) | ||
| BN26731 | 2.99 | AT2G33600 | Cinnamoyl-CoA reductase family | |||
| BN14781 BN14782 BN14783 |
8.79 | 4.72 | 2.9 | AT3G17390 | S-Adenosylmethionine synthetase 3 (SAMS3) | |
| BN22074 | 4.24 | AT3G19450 | Cinnamyl-Alcohol Dehydrogenase (CAD4) | |||
| BN18533 BN18534 | 3.09 | 3.51 | 2.48 | AT4G36220 | Ferulic acid 5-hydroxylase 1 (FAH1) | |
| BN13060 | 2.18 | AT5G14700 | Cinnamoyl-CoA reductase-related | |||
| BN19086 BN19087 | 3.0 | 3.26 | AT5G48930 | Hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase (HCT) | ||
| BN13706 | 5.22 | 5.01 | AT5G54160 | Caffeic acid/5-hydroxyferulic acid O-methyltransferase (OMT1) | ||
| BN20498 | 2.49 | AT5G58490 | Cinnamoyl-CoA reductase family | |||
Cinnamoyl-CoA reductase (CCR) and cinnamyl alcohol dehydrogenase (CAD) also play important roles in monolignol biosynthesis. The cinnamoyl-CoA esters, precursors of monolignol biosynthesis, are generated by the general phenylpropanoid pathway and then converted into monolignols by CCR and CAD [7]. CCR1 was among the most upregulated genes in the top and basal sections of DH relative to those of YN, and the same gene was upregulated in the basal section relative to the top section of YN in the array (Table 2). CCR2 was also upregulated in the top section of DH relative to the top section of YN. Another major gene change in the phenylpropanoid pathway was coded by p-coumarate 3-hydroxylase (C3H), also known as REDUCED EPIDERMAL FLUORESCENCE (REF8) was upregulated in the top and basal sections of DH relative to those of YN (Table 2).
The DE-ETIOLATED 3 (DET3), At1g12840, was upregulated in the top and basal section of DH relative to the top and basal section of YN (Table 2). This gene encodes the C-subunit of the V-type ATPase [37] implicated in the maintenance of pH homeostasis in plants [38]. Moreover, the FAH1 gene was upregulated in the basal section of DH4 relative to the basal section of YN4, and the same gene is upregulated in the basal section relative to the top section of both cultivars (Table 2). The basal section is more mature and richer in lignin content relative to the top section (Fig. 1a), suggesting the involvement of this gene with lignin accumulation in Brassica. The ferulate-5-hydroxylase (F5H) enzyme is a key regulatory point in the determination of lignin monomer composition [39] by catalyzing the conversion of ferulic acid to sinapic acid and syringyl lignin.
Differential expression of glucosyltransferase, peroxidase and laccase genes
It is well established that monolignols are synthesized in the cytoplasm and then exported into the cell wall for polymerization [7]. Cellular homeostasis of these monomers is regulated through glucosylation [40]. This modification step increases the solubility and stability of monomers and provides access to the membrane transport systems for transportation and storage. Glucosyltransferases play an important role in these processes [7]. After transport of the monolignols to the cell wall, lignin is formed through dehydrogenative polymerization of the monolignols [7]. The dehydrogenation to monolignol radicals has been attributed to different classes of oxidative proteins, such as peroxidases, laccases, polyphenol oxidases and coniferyl alcohol oxidase. However, it is still unknown which particular peroxidase and laccase genes are directly involved in this process during secondary wall formation.
To shed light on the differential expression pattern of glucosyltransferases, peroxidases and laccases in B. napus stem, we retrieved genes that belong to these groups from www.arabidopsis.org. A total of 26 glucosyltransferase genes were upregulated 2-fold or more in high lignin containing stem materials (Table 3), whereas 29 and 2 genes belonging to peroxidases and laccases, respectively, were upregulated 2-fold or more (Table 4). Most glucosyltransferases, peroxidases and laccases were upregulated two- to four-fold in more lignified tissues (Tables 3 and 4).
Table 3.
Differential upregulation of glucosyltransferase genes ≥ two-fold in stem sections of B. napus DH12075 (DH) and YN01–429 (YN)
| Probe name | DH4:YN4 | DH1:YN1 | DH4:DH1 | YN4:YN1 | Corresponding Arabidopsis loci | Description |
|---|---|---|---|---|---|---|
| BN18392 | 2.44 | AT1G05530 | UDP-GLUCOSYL TRANSFERASE 75B2 | |||
| BN25565 | 2.12 | AT1G07250 | UDP-GLUCOSYL TRANSFERASE 71C4 | |||
| BN19757 | 2.83 | AT1G23870 | Arabidopsis thaliana TREHALOSE-PHOSPHATASE/SYNTHASE 9 | |||
| BN18181 | 2.28 | AT1G55850 | CELLULOSE SYNTHASE LIKE E1 | |||
| BN20190 | 5.55 | 2.19 | 2.09 | AT1G70290 | TREHALOSE −6-PHOSPHATASE SYNTHASE S8 | |
| BN17018 | 3.95 | AT2G31750 | UDP-GLUCOSYL TRANSFERASE 74D1 | |||
| BN11938 | 2.24 | AT2G31960 | GLUCAN SYNTHASE-LIKE 3 | |||
| BN13391 | 2.12 | 2.39 | AT2G36850 | GLUCAN SYNTHASE-LIKE 8 | ||
| BN18400 | 2.05 | AT2G43820 | UDP-GLUCOSYLTRANSFERASE 74F2 | |||
| BN19396 | 2.01 | AT3G03050 | CELLULOSE SYNTHASE LIKE D3 | |||
| BN25630 | 2.35 | AT3G50740 | UDP-GLUCOSYL TRANSFERASE 72E1 | |||
| BN16340 | 2.26 | AT3G55830 | ECTOPICALLY PARTING CELLS | |||
| BN19754 | 12.95 | AT4G01070 | UDP-GLUCOSE-DEPENDENT GLUCOSYLTRANSFERASE 72 B1 | |||
| BN18305 | 2.23 | AT4G02280 | SUCROSE SYNTHASE 3 | |||
| BN17034 | 2.09 | AT4G03550 | GLUCAN SYNTHASE-LIKE 5 | |||
| BN16416 | 2.33 | AT4G10120 | ATSPS4F | |||
| BN26358 | 2.56 | AT4G15490 | UGT84A3 | |||
| BN25190 | 2.69 | AT4G17770 | TREHALOSE −6-PHOSPHATASE SYNTHASE S5 | |||
| BN20567 | 3.81 | 2.87 | At4g31590 | CELLULOSE-SYNTHASE LIKE C5 | ||
| BN12852 | 7.67 | 2.04 | At4g32410 | CELLULOSE SYNTHASE 1 | ||
| BN22038 | 4.45 | 2.19 | AT4G34135 | UDP-GLUCOSYLTRANSFERASE 73B2 | ||
| BN23469 | 3.48 | 2.43 | AT4G39350 | CELLULOSE SYNTHASE A2 | ||
| BN20784 | 2.13 | AT5G05170 | CELLULOSE SYNTHASE 3 | |||
| BN26739 | 2.21 | AT5G05870 | UDP-GLUCOSYL TRANSFERASE 76C1 | |||
| BN12647 | 4.39 | 2.08 | AT5G20830 | SUCROSE SYNTHASE 1 | ||
| BN17387 | 2.38 | 2.32 | AT5G64740 | CELLULOSE SYNTHASE 6 |
Table 4.
Differential upregulation of peroxidase and laccase genes ≥ two-fold in stem sections of B. napus DH12075 (DH) and YN01–429 (YN)
| Probe name | DH4: YN4 | DH1:YN1 | DH4:DH1 | YN4:YN1 | Corresponding Arabidopsis loci | Description |
|---|---|---|---|---|---|---|
| Peroxidase genes | ||||||
| BN24392 | 2.23 | AT1G05260 | RARE COLD INDUCIBLE GENE 3 | |||
| BN18476 | 2.04 | AT1G09500 | similar to Eucalyptus gunnii alcohol dehydrogenase | |||
| BN10302 | 4.25 | AT1G12840 | ARABIDOPSIS THALIANA VACUOLAR ATP SYNTHASE SUBUNIT C | |||
| BN21410 | 3.73 | AT1G15950 | CINNAMOYL COA REDUCTASE 1 | |||
| BN14306 | 2.16 | 2.54 | AT1G20620 | CATALASE 3 | ||
| BN18084 | 6.60 | 2.27 | AT1G20630 | CATALASE 1 | ||
| BN16003 | 2.56 | AT1G44970 | Peroxidase, putative | |||
| BN27576 | 2.14 | AT1G48130 | ARABIDOPSIS THALIANA 1-CYSTEINE PEROXIREDOXIN 1 | |||
| BN11140 | 2.02 | AT1G71695 | Peroxidase 12 | |||
| BN23842 | 2.50 | AT1G80820 | CINNAMOYL COA REDUCTASE | |||
| BN25563 | 2.41 | AT2G02400 | Cinnamoyl-CoA reductase family | |||
| BN13322 | 2.48 | AT2G25080 | GLUTATHIONE PEROXIDASE 1 | |||
| BN11700 | 2.0 | AT2G30860 | GLUTATHIONE S-TRANSFERASE PHI 9 | |||
| BN19562 | 3.68 | AT2G38380 | Peroxidase 22 (PER22) | |||
| BN15586 | 3.54 | AT2G40890 | CYP98A3, encodes coumarate 3-hydroxylase (C3H) | |||
| BN24228 | 2.77 | AT3G03670 | Peroxidase, putative | |||
| BN13555 | 2.09 | AT3G11630 | Encodes a 2-Cys peroxiredoxin (2-Cys PrxA) | |||
| BN14781 82,83,84 |
4.72 | AT3G17390 | S-ADENOSYLMETHIONINE SYNTHETASE 3 | |||
| BN19400 | 7.76 | AT3G49110 | PEROXIDASE 33 | |||
| BN16513 | 4.57 | 2.62 | 2.02 | AT4G21960 | PEROXIDASE PRXR1 | |
| BN24317 | 2.03 | AT4G26010 | Peroxidase, putative | |||
| BN23955 | 2.67 | AT4G30170 | Peroxidase, putative | |||
| BN27325 BN13535 |
4.35 | 2.17 | 2.17 | AT4G35000 | ASCORBATE PEROXIDASE 3 | |
| BN16234 | 4.13 | AT4G36430 | Peroxidase, putative | |||
| BN22581 | 3.69 | AT4G37530 | Peroxidase, putative | |||
| BN14948 | 3.30 | AT5G06290 | 2-CYSTEINE PEROXIREDOXIN B | |||
| BN19087 | 2.29 | AT5G48930 | HYDROXYCINNAMOYL-COA SHIKIMATE/QUINATE HYDROXYCINNAMOYL TRANSFERASE | |||
| BN13706 | 5.01 | AT5G54160 | O-METHYLTRANSFERASE 1 | |||
| Laccase genes | ||||||
| BN23268 | 2.17 | AT2G38080 | LACCASE-LIKE MULTICOPPER OXIDASE 4 | |||
| BN14150 | 3.23 | AT5G60020 | LACCASE 17 | |||
Genes involved in carbohydrate biosynthesis
The importance of cellulose and its integration with lignin and pectin in plant cell walls led us to determine transcript levels for genes coding for cellulose biosynthetic enzymes in the microarray experiment. Genes involved in cellulose biosynthesis in Arabidopsis were downloaded from www.arabidopsis.org and were compared with our microarray data to identify genes that were upregulated by 2-fold or more in high lignin-containing material (Table 5, Additional file 14: Table S8). Comparisons showed that several genes encoding enzymes involved in cellulose biosynthesis were upregulated in the basal section of the cultivars. CELLULOSE SYNTHASE A1 (BnCESA1; BN12852) was strongly upregulated in the basal section of DH relative to its counterpart in YN (Table 5). CESA2 (BN17389) was also upregulated in the basal section of both cultivars relative to the top sections, while CESA6 was upregulated in the top and basal sections of DH relative to the top and basal sections of YN in the array experiments. DEFECTIVE GLYCOSYLATION (DGL1) (BN19297) was upregulated in the top and basal sections of DH relative to the top and basal sections of YN (Table 5). DGL1 encodes a plant ortholog of human SOT48 or yeast WBP1 and is an essential protein subunit of the oligosaccharyltransferase (OST) complex.
Table 5.
Differential upregulation of carbohydrate biosynthetic genes by ≥2-fold in stem sections of B. napus DH12075 (DH) and YN01–429 (YN)
| Probe name | Upregulated expression | Corresponding Arabidopsis Loci |
Description | |||
|---|---|---|---|---|---|---|
| DH4: YN4 |
DH1: YN1 |
DH4: DH1 |
YN4: YN1 |
|||
| BN18181 | 2.28 | AT1G55850 | CELLULOSE SYNTHASE LIKE E1 | |||
| BN17967 | 3.43 | AT1G71100 | RADIAL SWELLING 10 (RSW10) | |||
| BN12852 | 7.67 | AT4G32410 | CELLULOSE SYNTHASE 1 (CESA1) | |||
| BN23469 | 3.48 | 2.43 | AT4G39350 | CELLULOSE SYNTHASE A2 (CESA2) | ||
| BN20787 | 2.44 | AT5g05170 | Constitutive Expression of VSP1 (CEV1) | |||
| BN18701 BN18702 |
6.43 | 2.21 | AT5G19220 | ADP GLUCOSE PYROPHOSPHORYLASE LARGE SUBUNIT 1 (APL1) | ||
| BN19160 | 2.66 | 4.97 | 2.21 | AT5G49720 | ARABIDOPSIS THALIANA GLYCOSYL HYDROLASE 9A1 (ATGH9A1) | |
| BN17389 | 3.1 | 2.22 | AT5G64740 | CELLULOSE SYNTHASE 6 (CESA6) | ||
| BN19297 | 8.49 | 5.27 | AT5G66680 | DEFECTIVE GLYCOSYLATION (DGL1) | ||
Other genes involved in carbohydrate synthesis were also upregulated in DH stems (Table 5). One of these, RADIAL SWELLING 10 (RSW10) (BN17967), was upregulated in the top section of DH relative to the top section of YN (Table 5). RSW 10 encodes a ribose 5-phosphate isomerase involved in the formation of uridine used for the synthesis of UDP-sugars.
Plant primary carbohydrate metabolism is complex, yet flexible, and is regulated at many levels. To gain molecular insights into the carbohydrate metabolic process in B. napus stem, we retrieved genes involved in cellular carbohydrate metabolic process from www.arabidopsis.org. A total of 419 loci were retrieved and compared to identify upregulated genes in the arrays. In total, 85 genes were found to be upregulated of which 52, 33, 35 and 29 belonged to DH4:YN4, DH1:YN1, DH4:DH1 and YN4:YN1, respectively (Additional file 14: Table S8).
Targets for transcriptional regulation of cell wall formation
Transcriptional regulation plays a key role in the complex series of events leading to cell wall formation [41]. To identify transcription factors (TFs) that were upregulated in the array, a search of our data was performed using 10 major transcription factors from the A. thaliana Transcription Factor Database [42]. The search revealed that a wide range of TFs were upregulated and downregulated in DH vs. YN stems. Several gene families, zinc finger (C2H2 and C3HC4), basic/helix-loop-helix (bHLH), basic region/leucine zipper motif (bZIP), AP2/EREBP and NAC (NAM/ATAF1,2/CUC2), were over-represented among the differentiated TFs (Table 6; Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12: Tables S1A and B to S6A and B).
Table 6.
Ten major transcription factors differentially expressed 2-fold or more in stem sections of B. napus DH12075 (DH) and YN01–429 (YN)
| Transcription factor | Sample groups in microarray | |||||||
|---|---|---|---|---|---|---|---|---|
| DH4-YN4 | DH1-YN1 | DH1-DH4 | YN1-YN4 | |||||
| Up | Down | Up | Down | Up | Down | Up | Down | |
| C2H2 | 16 | 22 | 14 | 11 | 4 | 7 | 6 | 5 |
| C3HC4 | 13 | 15 | 12 | 9 | 3 | 4 | 2 | 2 |
| bHLH | 13 | 11 | 11 | 7 | 7 | 3 | 3 | 1 |
| bZIP | 9 | 15 | 4 | 7 | 6 | 5 | 7 | 4 |
| NAC | 8 | 7 | 8 | 4 | 6 | 2 | 4 | 1 |
| AP2-EREBP | 8 | 8 | 4 | 6 | 4 | 3 | 1 | 1 |
| Homeobox | 6 | 8 | 6 | 3 | 4 | 4 | 3 | 4 |
| MYB | 4 | 2 | 3 | 0 | 4 | 4 | 5 | 2 |
| MADS | 4 | 4 | 1 | 3 | 3 | 3 | 3 | 3 |
| WRKY | 4 | 6 | 2 | 5 | 2 | 3 | 2 | 1 |
The largest group of transcription factors with differential expression (25 gene loci; 13 upregulated and 12 downregulated) consisted of Cys2His2-like (C2H2) zinc finger genes. C2H2 zinc fingers are well known and display a wide range of functions, from DNA and RNA binding to the involvement in protein-protein interactions. In our array data, the numbers of differentially expressed members (> 2-fold) for this family in DH4:YN4, DH1:YN1, DH1:DH4 and YN1:YN4 were 38, 25, 11 and 11, respectively (Table 6; Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12: Tables S1A and B to S6A and B). BN21811 related to At1g14580, a member of this family, was over-expressed in all 4 groups whereas the other 3 family members were upregulated in 3 groups. Another gene, BN15654 related to At5g04240, was upregulated in the basal section of DH relative to the basal section of YN. This latter gene is a member of the jumonji group of C2H2 transcription factors (Table 6; Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12: Tables S1A and B to S6A and B).
C3HC4 zinc finger genes formed the next largest gene category (Table 6). One of these genes BN22477 (related to At5g05830) was strongly upregulated in three of the four tissue comparisons and another in two comparisons. In contrast, BN17974 (related to At3g09770) was strongly down regulated in the DH4:YN4 and DH4:DH1 comparisons. B helix loop helix transcription factors were the third most differentially expressed genes in DH1:YN1, DH1:DH4 and YN1:YN4, but to a lesser extent than in DH4:YN4 (Table 6). In more detail, BN155636 related to At2g18300 (AtbHLH64) and BN12702 related to At1g05805 (AtbHLH128) were over-expressed in three groups of our array (Additional files 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12: Tables S1A and B to S6A and B). AtbHLH64 is known to play a role in Arabidopsis cytokinin signaling [43]. Though the role of bHLH TFs in lignin regulation is correlative rather than firmly established, we cannot rule out their possible involvement considering the common precursors of the lignin and flavonoid pathways and the known influence of bHLH on the flavonoid pathway genes, as well as the known interaction between bHLHs and MYB TFs in other physiological and developmental systems such as Brassica trichome development [44].
Plant basic region/leucine zipper motif (bZIP) transcription factors regulate processes including pathogen defense, light and stress signaling, seed maturation and flower development in Arabidopsis [45]. The numbers of bZIP family members differentially expressed more than 2-fold in DH4-YN4, DH1-YN1, DH1-DH4 and YN1-YN4 were 24, 11, 11 and 11, respectively in the array data (Table 6). BN15088 related to At2g18160 (ATBZIP2) were upregulated in 3 groups.
AP2 (APETALA2) and EREBPs (ethylene responsive element binding proteins) are a family of transcription factors unique to plants and share a highly conserved region of about 60 to 70 amino acids (the so-called AP2 domain) with no apparent similarity outside this domain. The numbers of differentially expressed members (> 2-fold) for this family in DH4-YN4, DH1-YN1, DH1-DH4 and YN1-YN4 were 16, 10, 7 and 2, respectively, in the array data (Table 6). In detail, BN16694 related to At1g68550 (AtERF118), was upregulated in 3 groups in our microarray experiments. Another of these genes, BN16341 related to At5g67190 (DREB2) was upregulated in the basal section relative to the top section of DH relative to YN. DREB2 contains the conserved DNA binding domain found in ERF (ethylene response factor) and AP2 (APETALA2).
NAC proteins constitute one of the largest families of plant-specific transcription factors, and are expressed in various developmental stages and tissues [46]. The NAC domain was originally characterized from consensus sequences from petunia NAM and from Arabidopsis ATAF1, ATAF2 and CUC2. In our array data, the number of differentially expressed members (> 2-fold) for this family in DH4-YN4, DH1-YN1, DH1-DH4 and YN1-YN4 were 15, 12, 8 and 5, respectively (Table 6). One member of this family, BN13387 related to At1g61110, was over-expressed in all 4 groups of our array and was previously reported to be expressed in stamens [47]. Other NAC transcription factors were only upregulated in 3 groups (Table 6). BN24136 was highly upregulated and is related to At3g49530 (ANAC062), a gene that plays an integrative role in plant responses to stress [48]. BN17366 related to At1g01720 is another Arabidopsis NAC-domain containing protein 1 (ATAF1) upregulated in the basal section relative to the top section of DH stems.
In addition to the six classes of over-represented transcription factors, MYB factors were more than 2-fold upregulated in our microarray comparisons of lignified and less lignified tissues. MYB factors represent a family of proteins with a conserved R2R3 (stimulating) or R3 (inhibiting) MYB DNA-binding domain. They often bind to bHLHs. In our array data, the numbers of overexpressed members of this family in DH4-YN4, DH1-YN1, DH1-DH4 and YN1-YN4 were 6, 3, 8 and 7, respectively (Table 6). BN17384 related to At5g60890 (AtMYB34) was expressed in all 4 groups of the array data while 2 other members of this family were upregulated in 3 groups (Table 6). BN26939 related to At2g47460 (AtMYB12) was also strongly upregulated in the array data.
In order to highlight the promising genes for future functional characterization from these elaborated sets, 25 TFs with representation from families mentioned above, plus six biochemical or physiological genes, were selected and their Arabidopsis knockdown mutants retrieved from TAIR (Additional files 14, 15, 16 and 17: Tables S8 to S11). In our analysis on a subsample of these lines, 4 TF lines and 4 non-TF lines showed lower lignin content than the Col-0 control line, while 2 TF lines and 3 non-TF lines showed enhanced lignin content (Additional files 16 and 17: Tables S10 and 11).
Discussion
The yellow seeded B. napus YN01–429 cultivar was recently developed through classical breeding, and was found to contain significantly reduced seed lignin content relative to seeds of the more conventional brown seeded cultivar DH12075 [20, 21]. Here, we determined that the stems of the brown-seeded cultivar were more lignified than those of the yellow-seeded cultivar and that basal stem sections were more lignified than apical stem sections for both cultivars.
Biochemical genes
Biochemical genes specifying monolignol formation, such as COMT [10], CCoAOMT 1 [49], SAMS [50], CAD [51], REF8 (C3H) [52] and F5H (FAH5) [53], were more highly expressed in the more lignified stem tissues we examined. Upregulation in more lignified tissue also occurred for genes specifying monolignol glucosylation, and monolignol polymerization, including peroxidases and laccases, when pairs of tissue sections were compared. Cellulose biosynthesis, the entire cell wall and plant development appeared to be affected by some of these differentially expressed Brassica genes. For example, we found that Brassica CES genes [54, 55] and peroxidases [56] were also more upregulated in more lignified tissues. Passardi et al. [56] showed that two highly homologous Arabidopsis peroxidases, AtPrx33 and Atprx34, were involved in cell elongation, and the knockout mutant, cesa2, also had severe defects in cell wall formation and microtubule orientation [57]. However, the numbers of cellulose biosynthetic genes upregulated in DH were fewer than lignin biosynthetic genes. Hence, our comparative transcriptome analyses of lignified and less lignified material could serve as a source of differentially expressed biochemical genes that impact on cell wall recalcitrance and could be useful to enable plant molecular breeders to fine tune crop residue composition for industrial applications.
Targets for transcription regulation
Microarray analysis of differentially lignified tissues resulted in six transcription factor families being more prominent in expression changes than other major TFs. Transcription factors have recently received attention from plant molecular breeders because of their ability to modify entire biochemical pathways or developmental families of genes. For example, overexpression of the Arabidopsis GLABRA3 (AtGL3) bHLH transcription factor in B. napus enhanced trichome coverage and insect resistance [44, 58].
Here, we screened our microarray data for the top 10 TF families present in lignified B. napus stems with the intent of beginning the process of characterizing the function of a subset of regulatory genes using Arabidopsis knockdown mutants. The most modified TF family in our stem microarrays was the zinc finger family. Several zinc finger proteins (ZFPs) in plants, e.g. Arabidopsis and petunia, have already been found to be involved in a variety of processes such as the regulation of floral organogenesis, leaf initiation, lateral shoot initiation, gametogenesis and stress response [59]. The C2H2 group was the most highly upregulated set of zinc finger families in our analyses. This latter group is known to be involved in the regulation of Arabidopsis flowering time [60]. Prigge and Wagner [61] reported that SERRATE (At2g27100), another member of the C2H2 family, plays a role in embryogenesis and is transcribed in shoot meristem and in emerging organ primordia throughout development. Dong and co-workers [62] also showed that SERRATE is required for the accurate processing of microRNA (miRNA) precursors in the plant cell nucleus. Moreover, some zinc finger genes highly expressed in trichomes have been reported to play a role in cell wall biosynthesis [63]. Thus, selections from among these differentially expressed zinc finger genes could target B. napus regulatory genes that can modify entire developmental programs.
BHLH TFs are one of the largest TF families in Arabidopsis. Phylogenetic analysis, divergence in binding site specificity and their varied ability to engage in homodimerization and heterodimerization events strongly support their roles in a multiplicity of transcriptional programs [64, 65]. BHLH TFs are known to play important roles in the flavonoid branch of the phenylpropanoid pathway; hence it makes sense that we would find them so plentiful in the closely related lignified tissue. In Arabidopsis seeds, the MYB transcription factor TRANSPARENT TESTA 2 (TT2) forms a complex together with the bHLH transcription factor TT8 and a WD40 scaffold protein to control the expression of BANYULS, a proanthocyanidin biosynthetic gene [66]. In a leaf study, TT8 and its closest homolog GL3 and ENHANCED GLABRA (EGL3) regulate flavonoid biosynthesis through interaction with two homologous MYB proteins PRODUCTION OF ANTHOCYANIN PIGMENTS 1 (PAP1, MYB75) and PAP2 (MYB90) [67]. In our stem microarrays, several bHLH genes were also differentially expressed along with a number of MYB genes. These genes should be paired and tested to determine whether any are binding partners and whether their modification affects lignin in transgenic plants or mutants. Already, the alfalfa bHLH TT8 homologue has been shown in RNAi studies to downregulate carbohydrate and dry matter accumulation, but not lignin, in alfalfa forage [68].
AP2/EREBP genes represent the fifth most differentially expressed genes in the B. napus stem microarrays. These genes play a variety of roles throughout the plant life cycle; from being key regulators of several developmental processes, like floral organ identity determination or control of leaf epidermal cell identity, to forming part of the mechanisms used by plants to respond to various types of biotic and environmental stresses [69]. NAC genes ranked sixth among our most differentially expressed TFs in the stem microarrays. Mitsuda et al. [70] showed that NAC SECONDARY WALL THICKENING PROMOTING FACTOR1 (NST1) and NST3 are key regulators of the formation of secondary walls in woody tissues of Arabidopsis. Lu et al. [71] showed that ATAF1 mRNA expression is strongly induced by dehydration and abscisic acid (ABA) treatment and they suggested a general role of this protein as a repressor in drought stress responses. Wu et al. [72] also implicated ATAF1 in diverse biotic and abiotic stress responses including drought, high-salinity, ABA, methyl jasmonate, mechanical wounding and Botrytis cinerea infection. Considering the expression pattern of NAC transcription factors in the array data and their role in lignin biosynthesis as well as in diverse biotic and abiotic stress responses, this family is a promising target for lignin modulation of the plant cell wall.
Although not among the most differentially expressed B. napus stem TFs, MYB-related transcription factors are an important group since they regulate different branches of secondary metabolism, in addition to the identity and fate of plant cells [73]. Studies on MYB transcription factors in Arabidopsis, pine and eucalyptus confirmed regulatory roles that MYB TFs play in lignin biosynthesis [14, 15, 74, 75]. MYB58 and MYB63 are transcriptional regulators specifically activating lignin biosynthetic genes during secondary wall formation in Arabidopsis [75]. Dominant repression of their functions led to a reduction in secondary wall thickening and lignin content, while overexpression of MYB58 and MYB63 resulted in specific activation of lignin biosynthetic genes and concomitant ectopic deposition of lignin in cells that are normally unlignified [75]. Newman and co-workers [13] suggested a role for another Arabidopsis MYB gene, AtMYB61, in cell wall deposition in normal vascular development [76]. However, transcription of these three MYB genes was unaltered in our array experiments. Instead, MYB12 and MYB34 were differentially regulated in the arrays. MYB34 plays a key role in the regulation of indole glucosinolate homeostasis in Arabidopsis [77], and MYB12 acts as a flavonol-specific activator of flavonoid biosynthesis [78]. Luo et al. [79] showed that MYB12 can also activate the caffeoyl quinic acid (CQA) biosynthetic pathway when expressed in a tissue-specific manner in tomato. The role of these two MYB TFs in secondary metabolism pathways and the correlation of their expression patterns with stem lignification establish this family as an important target in the modulation of lignin biosynthesis.
Analysis of Arabidopsis mutants for lignin, eight mutant lines showed lower lignin content than the Col-0 control line, while five showed enhanced lignin content. These verified lignin genes can now be used to modify Brassica lignin using our microarray oligonucleotides. Moreover, the Brassica and Arabidopsis sequences can be used to find homologues in other species. In fact, DIM1 and eEF-1Bβ1 have already been shown to modify lignin and carbohydrate in Arabidopsis [31, 32] and TT8 and HB12 have been shown to modify carbohydrate structure in alfalfa [34, 68]. In addition, several other genes (HB5, DIM1, ZFP2, eEF-1Bβ1, ANAC62) have also been used to modify alfalfa (Amyot, personal communication).
Conclusions
Most of the structural genes in the lignin biosynthetic pathway in plants have been characterized; however, more investigation is needed in the areas of understanding transcriptional regulation and unraveling the mechanism of monolignol polymerization. Here, we examined the expression pattern of genes in stem sections of two Brassica cultivars with differential lignin content. In doing so, we identified and established expression pattern of many new genes likely involved in cell wall biosynthesis and regulation. This transcript profiling allowed for the identification of novel gene candidates with strong potential at playing key roles in cell wall construction during stem development. The genes represent valuable tools to make cell walls more amenable to hydrolysis. More important, we were able to identify the 10 most important transcription factor families and listed 25 promising TF genes for future characterization. Elucidation of their precise mechanisms of transcriptional activation or repression of the cell wall biosynthetic process will not only shed light on the effect of these transcriptional regulators in several plant species, but may also make it possible for the molecular breeding community to modify entire biosynthetic pathways of cellulose or lignin by altering one or a few transcription factors.
Additional files
Table S1A. Differential expression of DH1 vs DH4 less than point 5 (microarray). (XLSX 360 kb)
Table S1B. Differential expression of DH1 vs DH4 greater than 2 (microarray). (XLSX 442 kb)
Table S2A. Differential expression of DH1 vs YN1 less than point 5 (microarray). (XLSX 496 kb)
Table S2B. Differential expression of DH1 vs YN1 greater than 2 (microarray). (XLSX 565 kb)
Table S3A. Differential expression of YN1 vs YN4 less than point 5 (microarray). (XLSX 226 kb)
Table S3B. Differential expression of YN1 vs YN4 greater than 2 (microarray). (XLSX 417 kb)
Table S4A. Differential expression of DH4 vs YN4 less than point 5 (microarray). (XLSX 728 kb)
Table S4B. Differential expression of DH4 vs YN4 greater than 2 (microarray). (XLSX 718 kb)
Table S5A. Differential expression of DH2 vs YN2 less than point 5 (microarray). (XLSX 567 kb)
Table S5B. Differential expression of DH2 vs YN2 greater than 2 (microarray). (XLSX 585 kb)
Table S6A. Differential expression of DH3 vs YN3 less than point 5 (microarray). (XLSX 512 kb)
Table S6B. Differential expression of DH3 vs YN3 greater than 2 (microarray). (XLSX 556 kb)
Table S7. qRT-Primers used in this study. (DOC 47 kb)
Table S8. Differential upregulation of genes involved in cellular carbohydrate metabolic processing. (DOC 143 kb)
Table S9. Ten categories of TFs with BN oligonucleotides retrieved from stem section comparisons of B. napus DH12075 (DH) and YN01–429 (YN). (XLSX 100 kb)
Table S10. Targeted transcription factors selected for lignin validation using Arabidopsis mutants. (DOCX 25 kb)
Table S11. Selected non-TF genes for lignin validation using Arabidopsis mutants. (DOCX 17 kb)
Acknowledgements
ZH and BVSP were recipients of Visiting Fellowships in Canadian Government Laboratories. We thank Dr. Serge Laberge for confirming the alfalfa transcription factors in lignified and non-lignified tissues.
Funding
This research was partially funded through the Cellulosic Biofuel Network as a part of the Agricultural Innovation Program of Agriculture and Agri-Food Canada. The funding body had no role in study design, collection, analysis, and interpretation of the data, preparation of the manuscript or decision to publish.
Availability of data and materials
The data sets supporting the results of this article are included within the article and as Additional files.
Abbreviations
- ABA
Abscisic acid
- ADP-Glc PPase
ADP-glucose pyrophosphorylase
- AP2
APETALA2
- bHLH
Basic/helix-loop-helix
- bZIP
Basic region/leucine zipper
- C3H
p-coumarate 3-hydroxylase
- CAD
Cinnamyl alcohol dehydrogenase
- CCoAOMT
Caffeoyl-coenzyme A O-methyltransferase
- CCR
Cinnamoyl-CoA reductase
- cDNA
Complementary DNA
- CESA
CELLULOSE SYNTHASE
- DET3
DE-ETIOLATED 3
- DGL
DEFECTIVE GLYCOSYLATION
- DIM1
DIMINUTO 1
- DNA
Deoxyribonucleic acid
- EGL
ENHANCED GLABRA
- EREBP
Ethylene responsive element binding protein;
- F5H
Ferulate-5-hydroxylase
- GO
Gene ontology
- miRNA
microRNA
- NST
NAC SECONDARY WALL THICKENING PROMOTING FACTOR
- OMT1
Caffeic acid/5-hydroxyferulic acid O-methyltransferase
- OST
Oligosaccharyltransferase
- PAP
PRODUCTION OF ANTHOCYANIN PIGMENTS
- qRT-PCR
Quantitative real time reverse transcription PCR
- REF
REDUCED EPIDERMAL FLUORESCENCE
- RNA
Ribonucleic acid
- RSW
RADIAL SWELLING
- SAMS
S-adenosyl methionine synthase
- TAIR
The Arabidopsis Information Resource
- TF
Transcription factor
- TGA
Thioglycolic acid
- TT
TRANSPARENT TESTA
- ZFP
Zinc finger protein
Authors’ contributions
ZH, BVSP, MY, and LA conducted experiments, analyzed data and drafted the manuscript. AH and MYG planned the project, secured funding, analyzed data, and edited the manuscript. All authors have read and approved the manuscript.
Ethics approval and consent to participate
Plant materials such as Brassica napus and alfalfa germplasm/cultivars were obtained from an on-going research program within Agriculture and Agri-Food Canada. Arabidopsis ecotypes were procured from the Arabidopsis Biological Resource Center (ABRC) located at the Ohio State University, Columbus OH, USA for academic research. Agriculture and Agri-Food Canada is a department of the Government of Canada and all experimental research (laboratory, growth chamber/greenhouse) were conducted according to the national guidelines.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4645-6) contains supplementary material, which is available to authorized users.
Contributor Information
Zakir Hossain, Email: Zakir.Hossain@canada.ca.
Bhinu V.-S. Pillai, Email: bhinu.pillai@canada.ca
Margaret Y. Gruber, Email: margie.gruber22@gmail.com
Min Yu, Email: Min.Yu@agr.gc.ca.
Lisa Amyot, Email: Lisa.Amyot@agr.gc.ca.
Abdelali Hannoufa, Email: Abdelali.Hannoufa@agr.gc.ca.
References
- 1.Wittkop B, Snowdon JR, Friedt W. Status and perspectives of breeding for enhanced yield and quality of oilseed crops for Europe. Euphytica. 2009;170:131–140. doi: 10.1007/s10681-009-9940-5. [DOI] [Google Scholar]
- 2.Li M, Zhang C, Liu L, Yu L. Development of relationship between A, B and C genomes in Brassica genera. Hereditas (Beijing). 2005;27:671–6. [PubMed]
- 3.Yang T-J, Kim JS, Kwon S-J, Lim K-B, Choi B-S, Kim J-A, et al. Sequence-level analysis of the diploidization process in the triplicated flowering locus C region of Brassica rapa. Plant Cell. 2006;18:1339–1347. doi: 10.1105/tpc.105.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arioli T, Peng L, Betzner AS, Burn J, Wittke W, Herth W, et al. Molecular analysis of cellulose biosynthesis in Arabidopsis. Science. 1998;279:717–720. doi: 10.1126/science.279.5351.717. [DOI] [PubMed] [Google Scholar]
- 5.Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, et al. The path forward for biofuels and biomaterials. Science. 2006;311:484–489. doi: 10.1126/science.1114736. [DOI] [PubMed] [Google Scholar]
- 6.Solomon BD, Barnes JR, Halvorsen KE. Grain and cellulosic ethanol: history, economics, and energy policy. Biomass Bioenergy. 2007;31:416–425. doi: 10.1016/j.biombioe.2007.01.023. [DOI] [Google Scholar]
- 7.Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annu Rev Plant Physiol Plant Mol Biol. 2003;54:519–546. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- 8.Boerjan W. Biotechnology and the domestication of forest trees. Curr Opin Biotechnol. 2005;16:159–166. doi: 10.1016/j.copbio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Keating JD, Panganiban C, Mansfield SD. Tolerance and adaptation of ethanologenic yeasts to lignocellulosic inhibitory compounds. Biotechnol Bioeng. 2006;93:1196–1206. doi: 10.1002/bit.20838. [DOI] [PubMed] [Google Scholar]
- 10.Raes J, Rohde A, Christensen JH, Van de Peer Y, Boerjan W. Genome-wide characterization of the lignification toolbox in Arabidopsis. Plant Physiol. 2003;133:1051–1071. doi: 10.1104/pp.103.026484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamagnone L, Merida A, Parr A, Mackay S, Culianez-Macia FA, Roberts K, et al. The AmMYB308 and AmMYB330 transcription factors from Antirrhinum regulate phenylpropanoid and lignin biosynthesis in transgenic tobacco. Plant Cell. 1998;10:135–154. doi: 10.1105/tpc.10.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin H, Cominelli E, Bailey P, Parr A, Mehrtens F, Jones J, et al. Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. EMBO J. 2000;19:6150–6161. doi: 10.1093/emboj/19.22.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman LJ, Perazza DE, Juda L, Campbell MM. Involvement of the R2R3-MYB, AtMYB61, in the ectopic lignification and dark-photomorphogenic components of the det3 mutant phenotype. Plant J. 2004;37:239–250. doi: 10.1046/j.1365-313X.2003.01953.x. [DOI] [PubMed] [Google Scholar]
- 14.Goicoechea M, Lacombe E, Legay S, Mihaljevic S, Rech R, Jauneau A, et al. EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant J. 2005;43:553–567. doi: 10.1111/j.1365-313X.2005.02480.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhong R, Lee C, Zhou J, McCarthy RL, Ye Z-H. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 2008;20:2763–2782. doi: 10.1105/tpc.108.061325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Besseau S, Hoffmann L, Geoffroy P, Lapierre C, Pollet B, Legrand M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell. 2007;19:148–162. doi: 10.1105/tpc.106.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hertzberg M, Aspeborg H, Schrader J, Andersson A, Erlandsson R, Blomqvist K, et al. A transcriptional roadmap to wood formation. Proc Natl Acad Sci U S A. 2001;98:14732–14737. doi: 10.1073/pnas.261293398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko JH, Han KH, Park S, Yang J. Plant body weight-induced secondary growth in Arabidopsis and its transcription phenotype revealed by whole-transcriptome profiling. Plant Physiol. 2004;135:1069–1083. doi: 10.1104/pp.104.038844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehlting J, Mattheus N, Aeschliman DS, Li E, Hamberger B, Cullis IF, et al. Global transcript profiling of primary stems from Arabidopsis thaliana identifies candidate genes for missing links in lignin biosynthesis and transcriptional regulators of fiber differentiation. Plant J. 2005;42:618–640. doi: 10.1111/j.1365-313X.2005.02403.x. [DOI] [PubMed] [Google Scholar]
- 20.Relf-Eckstein J, Rakow G, Raney JP. Yellow-seeded Brassica napus – a new generation of high quality canola for Canada. Proc 11th Int Rapeseed Cong., Copenhagen, Denmark, July 6–10, 2003. Vol. 2: p 458–460.
- 21.Relf-Eckstein J, Raney JP, Rakow G. Meal quality improvement in Brassica napus canola through the development of yellow-seeded germplasm. Proc 12th Int Rapeseed Cong., Wuhan, China, March 26–30, 2007. V5. p 289–291.
- 22.Theander O, Aman P, Miksche GE, Yasuda S. Carbohydrates, polyphenols, and lignin in seed hulls of different colors from turnip rapeseed. J Agric Food Chem. 1977;25:270–273. doi: 10.1021/jf60210a042. [DOI] [Google Scholar]
- 23.Whetten RW, Mackay JJ, Sederoff R. Recent advances in understanding lignin biosynthesis. Annu Rev Plant Physiol Plant Mol Biol. 1998;49:585–609. doi: 10.1146/annurev.arplant.49.1.585. [DOI] [PubMed] [Google Scholar]
- 24.Bhinu VS, Li R, Huang J, Kaminskyj S, Sharpe A, Hannoufa A. Perturbation of lignin biosynthesis pathway in Brassica napus (canola) plants using RNAi. Can J Plant Sci. 2009;89:441–453. doi: 10.4141/CJPS08164. [DOI] [Google Scholar]
- 25.Carpenter CD, Simon AE. Preparation of RNA. In: Martinez-Zapater JM, Salinas J, editors. Methods in molecular biology 82: Arabidopsis protocols. Totowa: Humana Press; 1998. pp. 85–89. [DOI] [PubMed] [Google Scholar]
- 26.Zhao J, Buchwaldt L, Rimmer SR, Sharpe A, McGregor L, Bekkaoui D, et al. Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol Plant Pathol. 2009;10:635–649. doi: 10.1111/j.1364-3703.2009.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The Arabidopsis Information Resource. [http://www.arabidopsis.org/tools/bulk/go/index.jsp] Accessed 15 Jan 2010.
- 28.The Arabidopsis Information Resource. [http://www.arabidopsis.org/browse/genefamily/index.jsp] Accessed 25 Jan 2017.
- 29.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed]
- 30.Ramakers C, Ruijter JM, Deprez RH, Moorman AF. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–66. doi: 10.1016/S0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 31.Hossain Z, McGarvey BD, Amyot L, Gruber MY, Jung J, Hannoufa A. DIMINUTO 1 affects the lignin profile and secondary cell wall formation in Arabidopsis. Planta. 2011;235:485–498. doi: 10.1007/s00425-011-1519-4. [DOI] [PubMed] [Google Scholar]
- 32.Hossain Z, Amyot L, McGarvey BD, Gruber MY, Jung J, Annoufa A. The translation elongation factor eEF-1Bß1 is involved in cell wall biosynthesis and plant development in Arabidopsis thaliana. PLoS One. 2012;7:1. doi: 10.1371/journal.pone.0030425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Hannoufa A, Zhang Y, Yu P. Transformation of TT8 and HB12 RNAi constructs in model forage (Medicago sativa, alfalfa) affects carbohydrate structure and metabolic characteristics in ruminant livestock systems. J Agric Food Chem. 2015;63:9590–9600. doi: 10.1021/acs.jafc.5b03717. [DOI] [PubMed] [Google Scholar]
- 34.Aung B, Gruber MY, Amyot L, Omari K, Bertrand A, Hannoufa A. 2015. MicroRNA156 as a promising tool for alfalfa improvement. Plant Biotech J. 2014;13:779–790. doi: 10.1111/pbi.12308. [DOI] [PubMed] [Google Scholar]
- 35.Yu B, Gruber M, Khachatourians GG, Hegedus DD, Hannoufa A. Gene expression profiling of developing Brassica napus seed in relation to changes in the content of major storage compounds. Plant Sci. 2010;178:381–389. doi: 10.1016/j.plantsci.2010.02.007. [DOI] [Google Scholar]
- 36.Zhong R, Morrison IIIWH, Negrel J, Ye Z. Dual methylation pathways in lignin biosynthesis. Plant Cell. 1998;10:2033–2046. doi: 10.1105/tpc.10.12.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schumacher K, Vafeados D, McCarthy M, Sze H, Wilkins T, Chory J. The Arabidopsis det3 mutant reveals a central role for the vacuolar H(+)-ATPase in plant growth and development. Genes Dev. 1999;13:3259–3270. doi: 10.1101/gad.13.24.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taiz L. The plant vacuole. J Exp Biol. 1992;172:113–122. doi: 10.1242/jeb.172.1.113. [DOI] [PubMed] [Google Scholar]
- 39.Meyer K, Shirley AM, Cusumano JC, Bell-Lelong DA, Chapple C. Lignin monomer composition is determined by the expression of a cytochrome P450-dependent monooxygenase in Arabidopsis. Proc Natl Acad Sci U S A. 1998;95:6619–6623. doi: 10.1073/pnas.95.12.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Chantreau M, Sibout R, Hawkins S. Plant cell wall lignification and monolignol metabolism. Front Plant Sci. 2013;4:220. doi: 10.3389/fpls.2013.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong R, Ye Z-H. Regulation of cell wall biosynthesis. Curr Opin Plant Biol. 2007;10:564–572. doi: 10.1016/j.pbi.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 42.AGRIS AtTFDB. [http://arabidopsis.med.ohio-state.edu/AtTFDB] Accessed 26 Jan 2017.
- 43.Brenner WG, Romanov GA, Kӧllmer I, Bürkle L, Schmülling T. Immediate-early and delayed cytokinin response genes of Arabidopsis thaliana identified by genome-wide expression profiling reveal novel cytokinin-sensitive processes and suggest cytokinin action through transcriptional cascades. Plant J. 2005;44:314–333. doi: 10.1111/j.1365-313X.2005.02530.x. [DOI] [PubMed] [Google Scholar]
- 44.Gruber MY, Wang S, Ethier S, Holowachuk J, Bonham-Smith PC. Soroka J, et al. “HAIRY CANOLA” – Arabidopsis GL3 induces a dense covering of trichomes on Brassica napus seedlings. Plant Mol Biol. 2006;60:679–698. doi: 10.1007/s11103-005-5472-0. [DOI] [PubMed] [Google Scholar]
- 45.Jakoby M, Weisshaar B, Drӧge-Laser W, Vicente-Carbajosa J, Tiedemann J, Kroj T, et al. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002:7106–11. [DOI] [PubMed]
- 46.Olsen AN, Ernst HA, Leggio LL, Skriver K. NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005;10:79–87. doi: 10.1016/j.tplants.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 47.Wellmer F, Riechmann JL, Alves-Ferreira M, Meyerowitz EM. Genome-wide analysis of spatial gene expression in Arabidopsis flowers. Plant Cell. 2004;16:1314–1326. doi: 10.1105/tpc.021741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seo PJ, Kim MJ, Park J-Y, Kim S-Y, Jeon J, Lee Y-H, et al. Cold activation of a plasma membrane-tethered NAC transcription factor induces a pathogen resistance response in Arabidopsis. Plant J. 2010;61:661–671. doi: 10.1111/j.1365-313X.2009.04091.x. [DOI] [PubMed] [Google Scholar]
- 49.Do CT, Pollet B, Thévenin J, Sibout R, Denoue D, Barrière Y, et al. Both caffeoyl coenzyme A 3-O-methyltransferase 1 and caffeic acid O-methyltransferase 1 are involved in redundant functions for lignin, flavonoids and sinapoyl malate biosynthesis in Arabidopsis. Planta. 2007;226:1117–1129. doi: 10.1007/s00425-007-0558-3. [DOI] [PubMed] [Google Scholar]
- 50.Sookyung O, Sunchung P, Kyung-Hwan H. Transcriptional regulation of secondary growth in Arabidopsis thaliana. J Exp Bot. 2003;54:2709–2722. doi: 10.1093/jxb/erg304. [DOI] [PubMed] [Google Scholar]
- 51.Sattler SE, Saathoff AJ, Haas ES, Palmer NA, Funnell-Harris DL, Sarath G, et al. A nonsense mutation in a cinnamyl alcohol dehydrogenase gene is responsible for the sorghum brown midrib6 phenotype. Plant Physiol. 2009;150:584–595. doi: 10.1104/pp.109.136408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franke R, Hemm MR, Denault JW, Ruegger MO, Humphreys JM, Chapple C. Changes in secondary metabolism and deposition of an unusual lignin in the ref8 mutant of Arabidopsis. Plant J. 2002;30:47–59. doi: 10.1046/j.1365-313X.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- 53.Marita JM, Ralph J, Hatfield RD, Chapple C. NMR characterization of lignins in Arabidopsis altered in the activity of ferulate 5-hydroxylase. Proc Natl Acad Sci U S A. 1999;96:12328–12332. doi: 10.1073/pnas.96.22.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fagard M, Desnos T, Desprez T, Goubet F, Refregier G, Mouille G, et al. PROCUSTE1 encodes a cellulose synthase required for normal cell elongation specifically in roots and dark-grown hypocotyls of Arabidopsis. Plant Cell. 2000;12:2409–2424. doi: 10.1105/tpc.12.12.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, Elliott JE, Williamson RE. Features of the primary wall CESA complex in wild type and cellulose-deficient mutants of Arabidopsis thaliana. J Exp Bot. 2008;59:2627–2637. doi: 10.1093/jxb/ern125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Passardi F, Tognolli M, De Meyer M, Penel C, Dunand C. Two cell wall associated peroxidases from Arabidopsis influence root elongation. Planta. 2006;223:965–974. doi: 10.1007/s00425-005-0153-4. [DOI] [PubMed] [Google Scholar]
- 57.Chu Z, Chen H, Zhang Y, Zhang Z, Zheng N, Yin B, et al. Knockout of the AtCESA2 gene affects microtubule orientation and causes abnormal cell expansion in Arabidopsis. Plant Physiol. 2007;143:213–224. doi: 10.1104/pp.106.088393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alahakoon UI, Taheri A, Nayidu NK, Epp D. Yu M, Parkin I, etal. Hairy canola (Brasssica napus) re-visited: down-regulating TTG1 in an AtGL3- enhanced hairy leaf background improves growth, leaf trichome coverage, and metabolite gene expression diversity. BMC Plant Biol. 2016;16:12. doi: 10.1186/s12870-015-0680-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Englbrecht CC, Schoof H, Böhm S. Conservation, diversification and expansion of C2H2 zinc finger proteins in the Arabidopsis thaliana genome. BMC Genomics. 2004;5:39. doi: 10.1186/1471-2164-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noh B, Lee SH, Kim HJ, Yi G, Shin EA, Lee M, et al. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time. Plant Cell. 2004;16:2601–2613. doi: 10.1105/tpc.104.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prigge MJ, Wagner DR. The Arabidopsis SERRATE gene encodes a zinc-finger protein required for normal shoot development. Plant Cell. 2001;13:1263–1280. doi: 10.1105/tpc.13.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong Z, Han MH, Fedoroff N. The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc Natl Acad Sci U S A. 2008;105:9970–9975. doi: 10.1073/pnas.0803356105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gan Y, Liu C, Yu H, Broun P. Integration of cytokinin and gibberellin signalling by Arabidopsis transcription factors GIS, ZFP8 and GIS2 in the regulation of epidermal cell fate. Development. 2007;134:2073–2081. doi: 10.1242/dev.005017. [DOI] [PubMed] [Google Scholar]
- 64.Bailey PC, Martin C, Toledo-Ortiz G, Quail PH, Huq E, Heim MA, et al. Update on the basic helix-loop-helix transcription factor gene family in Arabidopsis thaliana. Plant Cell. 2003;15:2497–2502. doi: 10.1105/tpc.151140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duek PD, Frankhauser C. bHLH class transcription factors take centre stage in phytochrome signalling. Trends Plant Sci. 2005;10:51–4. [DOI] [PubMed]
- 66.Zimmermann IM, Heim MA, Weisshaar B, Uhrig JF. Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant J. 2004;40:22–34. doi: 10.1111/j.1365-313X.2004.02183.x. [DOI] [PubMed] [Google Scholar]
- 67.Baudry A, Heim MA, Dubreucq B, Caboche M, Weisshaar B, Lepiniec L. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004;39:366–380. doi: 10.1111/j.1365-313X.2004.02138.x. [DOI] [PubMed] [Google Scholar]
- 68.Li X, Hannoufa A, Zhang Y, Yu P. Gene-silencing-induced changes in carbohydrate conformation in relation to bioenergy value and carbohydrate sub-fractions in model plant (Medicago sativa) with down-regulation of HB12 and TT8 transcription factors. Int J Mol Sci. 2016;17:1–14. doi: 10.3390/ijms17050720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riechmann JL, Meyerowitz EM. The AP2/EREBP family of plant transcription factors. Biol Chem. 1998;379:633–654. doi: 10.1515/bchm.1998.379.6.633. [DOI] [PubMed] [Google Scholar]
- 70.Mitsuda N, Iwase A, Yamamoto H, Yoshida M, Seki M, Shinozaki K, et al. NAC transcription factors, NST1 and NST3, are key regulators of the formation of secondary walls in woody tissues of Arabidopsis. Plant Cell. 2007;19:270–280. doi: 10.1105/tpc.106.047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu P-L, Chen N-Z, An R, Su Z, Qi B-S, Ren F, et al. A novel drought-inducible gene, ATAF1, encodes a NAC family protein that negatively regulates the expression of stress-responsive genes in Arabidopsis. Plant Mol Biol. 2006;63:289–305. doi: 10.1007/s11103-006-9089-8. [DOI] [PubMed] [Google Scholar]
- 72.Wu Y, Deng Z, Lai J, Zhang Y, Yang C, Yin B, et al. Dual function of Arabidopsis ATAF1 in abiotic and biotic stress responses. Cell Res. 2009;19:1279–1290. doi: 10.1038/cr.2009.108. [DOI] [PubMed] [Google Scholar]
- 73.Stracke R, Werber M, Weisshaar B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol. 2001;4:447–456. doi: 10.1016/S1369-5266(00)00199-0. [DOI] [PubMed] [Google Scholar]
- 74.Patzlaff A, McInnis S, Courtenay A, Surman C, Newman LJ, Smith C, et al. Characterisation of a pine MYB that regulates lignification. Plant J. 2003;36:743–754. doi: 10.1046/j.1365-313X.2003.01916.x. [DOI] [PubMed] [Google Scholar]
- 75.Zhou J, Chan L, Zhong R, Ye Z-H. MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis. Plant Cell. 2009;21:248–266. doi: 10.1105/tpc.108.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian Q, Wang X, Li C, Lu W, Yang L, Jiang Y, et al. Functional characterization of the poplar R2R3-MYB transcription factor PtoMYB216 involved in the regulation of lignin biosynthesis during wood formation. PLoS One. 2013;8:10. doi: 10.1371/journal.pone.0076369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Celenza JL, Quiel JA, Smolen GA, Merrikh H, Silvestro AR, Normanly J, et al. The Arabidopsis ATR1 Myb transcription factor controls indolic glucosinolate homeostasis. Plant Physiol. 2005;137:253–262. doi: 10.1104/pp.104.054395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehrtens F, Kranz H, Bednarek P, Weisshaar B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005;138:1083–1096. doi: 10.1104/pp.104.058032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luo J, Butelli E, Hill L, Parr A, Niggeweg R, Bailey P, et al. AtMYB12 regulates caffeoyl quinic acid and flavonol synthesis in tomato: expression in fruit results in very high levels of both types of polyphenol. Plant J. 2008;56:316–326. doi: 10.1111/j.1365-313X.2008.03597.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1A. Differential expression of DH1 vs DH4 less than point 5 (microarray). (XLSX 360 kb)
Table S1B. Differential expression of DH1 vs DH4 greater than 2 (microarray). (XLSX 442 kb)
Table S2A. Differential expression of DH1 vs YN1 less than point 5 (microarray). (XLSX 496 kb)
Table S2B. Differential expression of DH1 vs YN1 greater than 2 (microarray). (XLSX 565 kb)
Table S3A. Differential expression of YN1 vs YN4 less than point 5 (microarray). (XLSX 226 kb)
Table S3B. Differential expression of YN1 vs YN4 greater than 2 (microarray). (XLSX 417 kb)
Table S4A. Differential expression of DH4 vs YN4 less than point 5 (microarray). (XLSX 728 kb)
Table S4B. Differential expression of DH4 vs YN4 greater than 2 (microarray). (XLSX 718 kb)
Table S5A. Differential expression of DH2 vs YN2 less than point 5 (microarray). (XLSX 567 kb)
Table S5B. Differential expression of DH2 vs YN2 greater than 2 (microarray). (XLSX 585 kb)
Table S6A. Differential expression of DH3 vs YN3 less than point 5 (microarray). (XLSX 512 kb)
Table S6B. Differential expression of DH3 vs YN3 greater than 2 (microarray). (XLSX 556 kb)
Table S7. qRT-Primers used in this study. (DOC 47 kb)
Table S8. Differential upregulation of genes involved in cellular carbohydrate metabolic processing. (DOC 143 kb)
Table S9. Ten categories of TFs with BN oligonucleotides retrieved from stem section comparisons of B. napus DH12075 (DH) and YN01–429 (YN). (XLSX 100 kb)
Table S10. Targeted transcription factors selected for lignin validation using Arabidopsis mutants. (DOCX 25 kb)
Table S11. Selected non-TF genes for lignin validation using Arabidopsis mutants. (DOCX 17 kb)
Data Availability Statement
The data sets supporting the results of this article are included within the article and as Additional files.