

Abstract
H syndrome is a recently described autosomal recessive genodermatosis with cutaneous phenotypes of varying severity and multi-system involvement. Patients suffering from this disorder can be easily mistaken for sclerodermoid conditions. The radiological findings of H syndrome are typical but have been described only anecdotally. We present a case of a 29 year old male patient of H syndrome with typical radiological features.
Keywords: H syndrome, sclerodermoid conditions, subcutaneous edema
What was known?
H syndrome is a novel form of histiocytosis with autosomal recessive inheritance and characteristic cutaneous findings.
Introduction
H syndrome is an autosomal recessive genodermatosis, first described in 2008.[1] It has multisystemic involvement and is characterized by numerous clinical features, including cutaneous hyperpigmentation and hypertrichosis, hepatosplenomegaly, hearing loss, heart anomalies, hypogonadism, low height (short stature), hyperglycemia (insulin-dependent diabetes mellitus), and hallux valgus/flexion contractures.[1,2] Cutaneous hyperpigmentation, commonly accompanied by hypertrichosis and progressive sclerodermatous induration, typically affecting initially the medial thighs with sparing of the knees, is the hallmark of the disorder and is considered pathognomonic.[2]
Case Report
A 29-year-old male patient presented with a 6-year history of asymptomatic progressive cutaneous sclerosis, hyperpigmentation, and hypertrichosis over both thighs and trunk.
He complained of skin tightness around the abdomen while walking. He wandered to and fro between different specialists but without much help. He was diagnosed as Type 1 diabetic 6 years back and was receiving insulin since then.
Cutaneous examination revealed large ill-defined hyperpigmented indurated plaques extending symmetrically from mid-truncal area to both thighs characteristically sparing the knees and medial aspect of buttocks [Figure 1a and b]. A prominent constriction band was noted around the abdomen [Figure 2]. The lesions over thighs were associated with hypertrichosis and were warm to touch.
Figure 1.
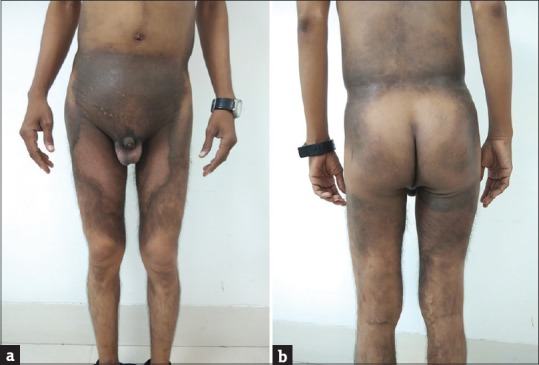
(a and b) Hyperpigmented indurated plaques extending symmetrically from mid-truncal area to both thighs characteristically sparing the knees and medial aspect of buttocks
Figure 2.
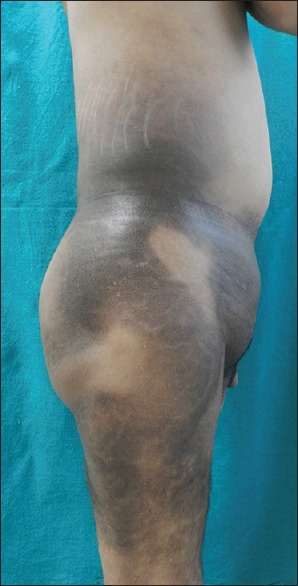
Prominent constriction band around the abdomen
Examination also revealed short stature (145 cm), hypospadias, micropenis, scrotal swelling, and mild hepatomegaly and inguinal lymphadenopathy. Axillary and pubic hairs were scanty. Other secondary sexual characters such as facial hair were sparse and voice was normal. Nails and teeth were normal. Ophthalmologic and auditory examinations were normal. On the basis of cutaneous findings, we kept differential diagnosis of morphea profunda, pseudoscleroderma, POEMS syndrome, Rosai–Dorfman syndrome, pigmented hypertrichotic dermatosis, and scleredema as the possibilities.
A deep skin biopsy was done from the indurated plaque on the lateral part of the left thigh and sent for histopathology. Skin biopsy revealed marked fibrosis of the dermis and subcutaneous tissue [Figure 3a]. Dermal appendages pushed upward, and a perivascular infiltrate of lymphocytes and histiocytes with foamy cytoplasm were seen intermingled with the bundles of dermal collagen. Septal panniculitis with plasma cell infiltration was noted [Figure 3b]. Histopathology report suggested sclerodermatous changes with immunostain CD68+ve histiocytes [Figure 3c].
Figure 3.
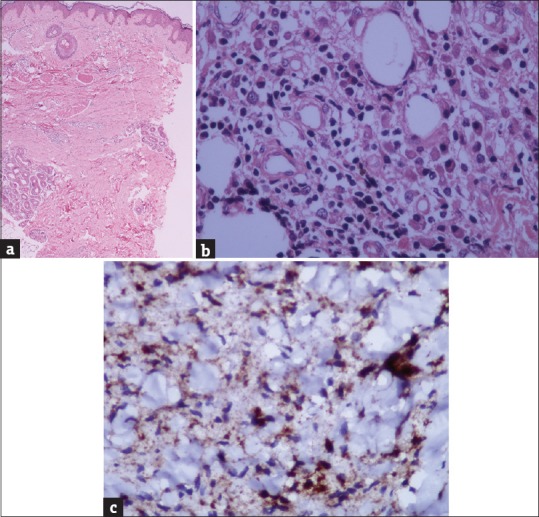
(a) Marked fibrosis of the dermis and subcutaneous tissue (H and E, x40). (b) mononuclear cell infiltrate of lymphocytes, plasma cells, and histiocytes with foamy cytoplasm (H and E, x400). (c) CD 68 +ve histiocytes (Immunostain CD68, x400)
Routine laboratory investigations revealed a fasting blood glucose level of 250 mg/dL and random blood sugar of 338 mg/dL with glycated hemoglobin 10.2%. Thyroid profile, liver function, kidney function, serum testosterone, follicle stimulating hormone and luteinizing hormone levels were normal. Antinuclear antibody was negative. Ultrasound abdomen revealed mild hepatosplenomegaly and abdominal lymphadenopathy. Color Doppler study of the scrotal region revealed small penis, normal echotexture, no calcific plaques, and small left testis. Chest radiography and nerve conduction studies were normal.
Computed tomography (CT) demonstrated marked symmetrical thickening of the skin with infiltration of subcutaneous tissue at the lower half of the body from infraumbilical region till scrotum and into bilateral gluteal region down to the knees [Figure 4a and b]. Para-aortic and inguinal lymphadenopathy [Figure 5] and mild pericardial effusion [Figure 6] were noted. Marked thickening of the scrotum was seen [Figure 7]. A diagnosis of “H syndrome” was made. Genetic testing was not possible due to resource constraints.
Figure 4.
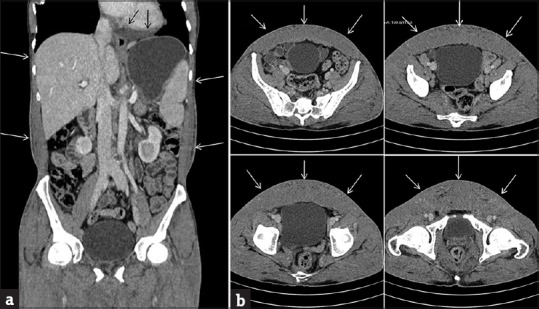
(a and b) Computed tomography scan images showing diffuse subcutaneous edema from infraumbilical region till scrotum and into bilateral gluteal region
Figure 5.
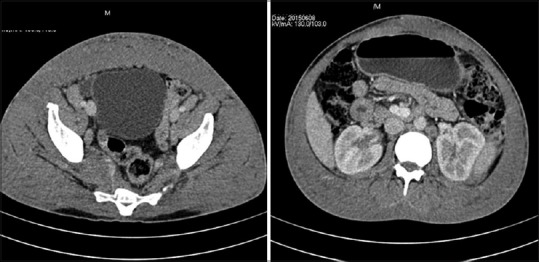
Computed tomography scan images showing para-aortic and inguinal lymph node enlargement
Figure 6.
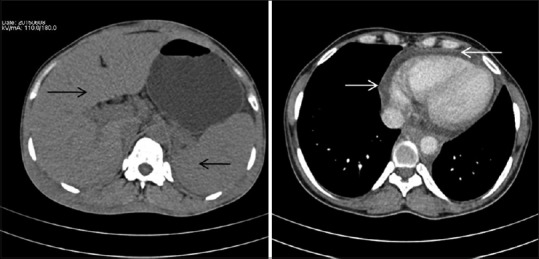
Computed tomography scan images showing hepatosplenomegaly and mild pericardial effusion
Figure 7.
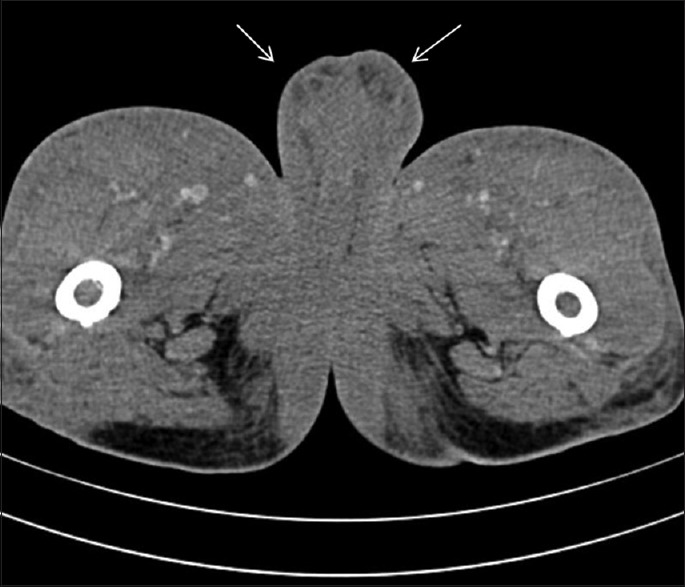
Marked thickening of the scrotum noted
Discussion
The name H syndrome was first coined by Molho-Pessach et al.[1] It is recently described as an autosomal recessive disorder characterized by unique progressive symmetrical cutaneous induration with hyperpigmentation and hypertrichosis, localized mainly to the lower half of the body, hepatosplenomegaly, hearing loss, low height, hyperglycemia/diabetes mellitus, heart anomalies, hypogonadism, and hallux valgus/flexion contractures.[2] Other features include massive lymphadenopathy, pancreatic exocrine insufficiency, renal abnormalities, bone sclerosis, lytic bone lesions, swollen respiratory mucosa, infiltrated cheeks, and recurrent fever.[1,2,3]
It occurs due to biallelic mutations in SLC29A3 gene encoding the human equilibrative nucleoside transporter 3. SLC29A3 gene is widely expressed in various organs and regulates the inflammatory cascade.[2] A defect in this protein causes disordered immune regulation inflammatory infiltrates in the skin and internal organs. Failure to suppress the inflammation finally results in fibrosis.[4]
The pathognomonic finding is the cutaneous feature seen in 68% of patients and tends to appear during the first or second decade of life, initially over the inner thighs, spreading later to involve other areas.[1,2,5] Histopathological examination of affected skin demonstrates widespread dermal and subcutaneous fibrosis with a mononuclear infiltrate composed mainly of CD68+ histiocytes and plasma cells.[6] This feature implies that H syndrome is a novel form of inherited histiocytosis.
The radiographic findings of H syndrome are typical. Hiller N et al. described the spectrum of radiological findings in 12 patients with H syndrome in a recent study.[7] In majority of them, a typical and unique pattern of diffuse symmetric infiltration of the subcutaneous fat in the lower half of the body, with varying degrees of severity, was demonstrated. Changes in subcutaneous fat were accompanied by thickening of the skin, and in severe cases, cutaneous and subcutaneous changes lead to retraction of the underdeveloped external genitalia (micropenis) as were noticed in our case. The degree of subcutaneous thickening is related to patient's age, with older patients showing more severe infiltration of the subcutaneous tissue.[7]
Several conditions are characterized by radiological findings of dermal and subcutaneous changes similar to those seen in H syndrome.[8] The major disorder considered in the differential diagnosis is morphea and plasma cell panniculitis. Morphea is not associated with systemic symptoms, tends to be distributed in a nonsymmetric pattern, and as opposed to H syndrome, has no associated hypertrichosis, while in plasma cell panniculitis, the most typical clinical finding is tender, erythematous subcutaneous nodules, as opposed to the more diffuse cutaneous lesions that are typical of H syndrome.[8,9]
Skin thickening and infiltration of the subcutaneous fat are nonspecific findings that may be demonstrated by imaging in various disorders, including myxedema, eosinophilic fasciitis, nephrogenic systemic fibrosis, lymphedema, lipodermatosclerosis, and sclerodermoid graft versus host disease.[8] However, most of these disorders can be easily differentiated from H syndrome based on localization, distribution, and extent of findings as well as by the associated clinical constellation.
Near about 100 cases of H syndrome have been reported till date, with a total of 10 cases being reported from the Indian subcontinent.[9,10] This is a remarkable number of patients for a relatively novel autosomal recessive disorder, implying that the disease is not rare. Since the disorder has been fully described only recently, it is still misdiagnosed not only due to lack of awareness but also due to the wide spectrum of possible clinical manifestations. It is possible that cases go undiagnosed or are not diagnosed properly because of its overlapping features with other syndromes.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for images and other clinical information to be reported in the journal. The patient understands that name and initial will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
What is new?
H syndrome is a rarely reported entity with characteristic radiological features.
References
- 1.Molho-Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: A genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol. 2008;59:79–85. doi: 10.1016/j.jaad.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu Libdeh A, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet. 2008;83:529–34. doi: 10.1016/j.ajhg.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. H syndrome: The first 79 patients. J Am Acad Dermatol. 2014;70:80–8. doi: 10.1016/j.jaad.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 4.Prendiville J, Rogers M, Kan A, de Castro F, Wong M, Junker A, et al. Pigmented hypertrichotic dermatosis and insulin dependent diabetes: Manifestations of a unique genetic disorder? Pediatr Dermatol. 2007;24:101–7. doi: 10.1111/j.1525-1470.2007.00352.x. [DOI] [PubMed] [Google Scholar]
- 5.Avitan-Hersh E, Mandel H, Indelman M, Bar-Joseph G, Zlotogorski A, Bergman R, et al. A case of H syndrome showing immunophenotype similarities to Rosai-Dorfman disease. Am J Dermatopathol. 2011;33:47–51. doi: 10.1097/DAD.0b013e3181ee547c. [DOI] [PubMed] [Google Scholar]
- 6.Doviner V, Maly A, Ne'eman Z, Qawasmi R, Aamar S, Sultan M, et al. H syndrome: Recently defined genodermatosis with distinct histologic features. A morphological, histochemical, immunohistochemical, and ultrastructural study of 10 cases. Am J Dermatopathol. 2010;32:118–28. doi: 10.1097/DAD.0b013e3181b28572. [DOI] [PubMed] [Google Scholar]
- 7.Hiller N, Zlotogorski A, Simanovsky N, Ingber A, Ramot Y, Molho-Pessach V, et al. The spectrum of radiological findings in H syndrome. Clin Imaging. 2013;37:313–9. doi: 10.1016/j.clinimag.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Rambhia KD, Dongre AM, Khopkar US. Sclerodermoid plaques: A riddle of 'H'. Indian J Dermatol Venereol Leprol. 2015;81:327. doi: 10.4103/0378-6323.152748. [DOI] [PubMed] [Google Scholar]
- 9.Molho-Pessach V, Varma M, Godbole K, Kamath N, Zlotogorski A. H syndrome – Four new patients from India. Indian J Dermatol Venereol Leprol. 2014;80:579. doi: 10.4103/0378-6323.144229. [DOI] [PubMed] [Google Scholar]
- 10.Mohanan S, Chandrashekar L, Semple RK, Thappa DM, Rajesh NG, Negi VS, et al. H syndrome with a novel homozygous R134C mutation in SLC29A3 gene. Int J Dermatol. 2013;52:820–3. doi: 10.1111/j.1365-4632.2012.05838.x. [DOI] [PubMed] [Google Scholar]