

Abstract
σ factors are single subunit general transcription factors that reversibly bind core RNA polymerase and mediate gene-specific transcription in bacteria. Previously, an atypical two-subunit σ factor was identified that activates transcription from a group of related promoters in Bacillus subtilis. Both of the subunits, named SigO and RsoA, share primary sequence similarity with the canonical σ70 family of σ factors and interact with each other and with RNA polymerase subunits. Here we show that the σ70 region 2.3-like segment of RsoA is unexpectedly sufficient for interaction with the amino-terminus of SigO and the β′ subunit. A mutational analysis of RsoA identified aromatic residues conserved amongst all RsoA homologues, and often amongst canonical σ factors, that are particularly important for the SigO–RsoA interaction. In a canonical σ factor, region 2.3 amino acids bind non-template strand DNA, trapping the promoter in a single-stranded state required for initiation of transcription. Accordingly, we speculate that RsoA region 2.3 protein-binding activity likely arose from a motif that, at least in its ancestral protein, participated in DNA-binding interactions.
Keywords: transcription, RNA polymerase, sigma factor, Bacillus subtilis, protein–protein interaction
Introduction
Bacterial σ factors are general transcription factors that reversibly bind core RNA polymerase (cRNAP) to form RNAP holoenzyme in the cell (Burgess et al., 1969; Helmann & Chamberlin, 1988; Murakami & Darst, 2003; Murakami, 2015). σ factors target the holo-enzyme to cognate gene promoters and bind single-stranded non-template promoter DNA, trapping the −10 region and transcription start site in a single-stranded state called the open complex. Stabilization of the open complex is a prerequisite for the initiation of RNA synthesis at the RNAP active site.
The most ubiquitous and widespread σ factors are members of the σ70 family (Paget & Helmann, 2003) that are often structurally characterized as having four functional amino acid segments called regions 1–4 (Gribskov & Burgess, 1986). While the primary σ factors of bacteria are essential and possess all four regions, alternative members of this family may lack either region 1 or 3 or both. For example, group IV or extracytoplasmic function (ECF) σ factors are smaller (~20 kDa) than primary σ factors and possess only regions 2 and 4 (Helmann, 2002; Missiakas & Raina, 1998). Region 2 and region 4 define the minimal σ factor as these play crucial roles in holo-enzyme formation, promoter binding and transcription activation. Region 4 amino acids interact with the flap domain of the core β subunit and form a helix–turn–helix motif that recognizes and binds the −35 region of the double-stranded promoter (Geszvain et al., 2004; Kuznedelov et al., 2002). Region 2 was previously subdivided into four contiguous sub-regions (Helmann & Chamberlin, 1988), and we follow this convention here to identify regions of sequence similarity amongst σ factors. These sub-regions are called regions 2.1, 2.2, 2.3 and 2.4 (Helmann & Chamberlin, 1988). Regions 2.1 and 2.2 form two α helices, and highly conserved residues in region 2.2 are important for the interaction of the σ factor with the N-terminus of the core β′ subunit (Arthur & Burgess, 1998; Johnston et al., 2009; Ma et al., 2013). In a transcription complex, region 2.3 amino acids participate in the formation of an interface that binds non-template promoter DNA, thus stabilizing open complex formation (Campagne et al., 2014; Feklistov & Darst, 2011; Juang & Helmann, 1994; Murakami, 2013; Panaghie et al., 2000; Zuo & Steitz, 2015). Region 2.3 possesses a high level of abundance of hydrophobic and basic residues, including seven aromatic amino acids that are almost universally conserved in primary σ factors, partially conserved amongst the alternate members of the family and some of which play an important role in open complex formation (deHaseth & Helmann, 1995).
Although σ factors are single subunit proteins, an unusual two-subunit σ factor system exists in Bacillus subtilis. The two proteins comprising this atypical σ factor are SigO (formerly YvrI) and RsoA (formerly YvrHa). The SigO–RsoA two-subunit σ factor system is widespread (MacLellan et al., 2009b) amongst diverse species in the Bacillus sensu stricto clade and in the Bacillus cereus clade of bacilli (Bhandari et al., 2013). In B. subtilis, this σ factor regulates the expression of three operons (see Fig. S1, available in the online Supplementary Material) and is induced during growth under low pH conditions (MacLellan et al., 2009b), after exposure to certain cell wall-acting antibiotics (Hachmann et al., 2009), and under conditions that lead to accumulation of cyclic-di-AMP in the cytoplasm (Gundlach et al., 2016).
SigO and RsoA are σ factor-related proteins that are co-transcribed from an auto-regulated promoter (MacLellan et al., 2008). SigO is the size (23 kDa) of a typical ECF σ factor and possesses a canonical region 4 but highly divergent region 2. Evidence suggests that it can bind cRNAP and target the complex to a cognate promoter, but it cannot stabilize open complex formation in vitro or activate transcription in vivo (MacLellan et al., 2009a). RsoA is a 10 kDa protein that interacts specifically with the N-terminus of SigO, and its co-expression with SigO is required both in vivo and in vitro for open complex formation and transcription activation. RsoA possesses a region 2.2-like sequence that is highly conserved both amongst its orthologues and in canonical σ70 family σ factors (Fig. 1). Previously (MacLellan et al., 2009a), we were able to detect an interaction between RsoA and the β′ subunit clamp helices, and another group (Sengupta et al., 2015) recently confirmed that RsoA does independently interact with cRNAP. Contiguous with region 2.2 in RsoA is a highly hydrophobic segment of approximately 24 amino acids that includes a number of aromatic residues spatially conserved with those in region 2.3 of canonical σ factors. Mechanistically, amino acids in region 2.3 play a predominant role in binding single-stranded non-template DNA and trapping the promoter in an open complex state that is required for the initiation of transcription. Although the mechanistic role of RsoA in a transcription complex is not yet clear, based on sequence similarity with canonical σ factors we provisionally designate RsoA amino acids 36–55 as region 2.2 and amino acids 56–79 as region 2.3 (see Fig. 1) following the original delineations by Lonetto et al. (1992). RsoA interacts specifically with the N-terminus of SigO (MacLellan et al., 2009a) and, in this communication, amino acids in RsoA critical for this interaction are identified. Unexpectedly, we found that the C-terminal RsoA region 2.3-like segment is sufficient for interaction with SigO and that a phenylalanine residue (F67) in RsoA region 2.3, which is spatially conserved in almost all canonical σ factors, is required either directly or indirectly for the protein–protein interaction. We also show that this same amino acid segment accounts for the previously described interaction between RsoA and the RNAP β′ subunit.
Fig. 1.

Structural features of 79 amino acid B. subtilis RsoA. Alignment of partial sequences of group I (primary), III and IV (ECF) σ factors and full sequence of RsoA (bottom) aligned with full sequences of five closely related RsoA orthologues. Region 2.1, 2.2 and 2.3 delineations (top) are based on Lonetto et al. (1992). Key residues in regions 2.2 and 2.3 broadly conserved amongst most σ factors are shaded, including the seven universally conserved aromatic amino acids in type I σ region 2.3. Asterisks below alignment indicate conservation of residues amongst RsoA orthologues only. Arrows indicate RsoA residues mutated to alanine in this study. Lobed arrows indicate residues that were mutated to alanine and were also found mutated (i.e. F67S, E69V, I71N, M74T) in screen for random mutations that impair interaction with SigO. Numbered line indicates key amino acid positions between regions of RsoA. Secondary structure prediction (SS) for RsoA made using Jnet algorithm. H indicates high level of probability of α helix formation. RsoA orthologues are as follows: Bam, Bacillus amyloliquefaciens FZB42 (YP_001422598.1); Bmo, Bacillus mojavensis (WP_010331850.1); Bat, Bacillus atrophaeus 1942 (YP_003974761.1); Bpu, Bacillus pumilus SAFR-032 (YP_001486052.1); Bli, Bacillus licheniformis. Other protein abbreviations are as follows: Bsu, B. subtilis SigA; Eco, Escherichia coli σ70; Sco, Streptomyces coelicolor RpoD; Taq, Thermus aquaticus RpoD; SigH (B. subtilis); RpoH (E. coli); SigMXYW (B. subtilis); RpoE (E. coli σE); SigR (S. coelicolor σR).
Methods
Bacterial strains and plasmids.
Bacterial strains used in this study are listed in Table S1; plasmids used are listed in Table S2. E. coli and B. subtilis strains were cultured in LB broth or in this medium solidified with 1.6 % agar. For some experiments, M9 minimal medium was used. Antibiotic concentrations used were as follows: 100 µg ampicillin ml−1, 50 µg kanamycin ml−1 and 100 µg streptomycin ml−1. Indicator media included either 40 µg (E. coli) or 80 µg (B. subtilis) X-Gal ml−1. All plasmids generated were confirmed by colony PCR, restriction digest and sequencing (McGill University and Genome Quebec Innovation Centre, Montreal, PQ). Oligonucleotides were purchased from Integrated DNA Technologies.
Bacterial two-hybrid assay.
The analysis of protein–protein interactions was conducted using an adenylate cyclase-based bacterial two-hybrid (BACTH) assay (Euromedex). Full-length or partial sigO and rsoA gene sequences were amplified using PCR and ligated into either pUT18C or pKT25 to generate C-terminal fusions to the CyaA T18 and T25 catalytic sub-domains. All possible combinations of fusions using the available BACTH plasmids have been assessed using full-length SigO and RsoA and relevant sub-regions of these proteins and all yield comparable results in terms of apparent interactions (data not shown). For some experiments, SigO and RsoA were appended with C-terminal FLAG or haemagglutinin (HA) tags (respectively) to enable immunoblot detection or pull-down assays using antibodies recognizing the epitopes. In none of the circumstances we tested did the C-terminal epitopes alter interactions between the test proteins relative to untagged proteins.
Constructed bait and prey plasmids were co-transformed into the test strain E. coli BTH101. Strains were tested for interaction on selective LB medium containing X-Gal. Quantitative measurements were conducted by growing transformants at 37 °C overnight, subculturing a 2.5 % volume into fresh LB broth and growing to an OD of 0.3–0.4 at 30 °C before subjecting cells to a β-galactosidase assay using 4 mg ONPG ml−1 as substrate.
When mutations that impair interaction are observed, it is essential to confirm that the mutant protein is expressed (and accumulates in cells in a soluble form) to the same relative extent as non-mutant protein. Owing to the nature of the BACTH system, we cannot determine the abundance of non-interacting proteins in the BACTH host strain (Battesti & Boveret, 2012). Therefore, to determine expression levels of wt and mutant RsoA from the BACTH expression plasmids, we transformed the plasmids into the CyaA+ E. coli strain DH5α and grew cultures with selection at 30 °C overnight. On the next day, a 1 % volume was used to inoculate 5 ml of fresh medium and cultures were grown at 30 °C to an OD of 0.3–0.4. Cells were collected, re-suspended in 0.5 ml of 10 mM Tris/HCl (pH 8.0) and subjected to lysis using 50 µl PopCulture cell lysis reagent (Novagen). Processing was assisted by the addition of 0.1 mg ml−1 hen egg white lysozyme and 10 U DNase I. After lysis, each sample was divided into equal volumes where one volume was subjected to centrifugation at 13 000 r.p.m. for 5 min to sediment insoluble protein. Proteins in 20 µl volumes of crude lysate and centrifuged lysate were separated on a 12 % SDS-PAGE gel, transferred to PVDF membrane and probed for total (soluble and insoluble) and soluble (sedimentation-resistant) protein using Western immunoblotting and anti-HA antibodies. Note that the site-directed alanine substitution mutants had to be re-amplified by PCR and re-cloned into the BACTH plasmid prior to immunoblotting so that an HA tag could be imparted to the fusion proteins.
Some assays were also conducted using an alternative BACTH system (Dove & Hochschild, 2004) as previously described (MacLellan et al., 2009a). This assay involves test protein fusions to the DNA-binding protein λcI and the C-terminus of the RNAP α subunit. Expression of the fusion proteins is induced with increasing concentrations of IPTG, and interaction between the test proteins activates expression of a β-galactosidase gene that is integrated into the E. coli host cell genome.
Site-directed mutagenesis.
Site-directed mutagenesis was conducted on plasmid pSM019 carrying the full-length RsoA gene sequences using a strategy based upon the QuikChange method (Stratagene). Mutagenized plasmid DNA was treated with DpnI for 2 h and transformed into E. coli DH5α for screening. After confirming point mutations by sequencing, we used the mutant plasmid DNA as substrate for PCR and we ligated amplified fragments into other relevant test plasmids (e.g. BACTH assay plasmids).
Random mutagenesis and screening protocol.
Error-prone PCR was used to randomly mutagenize rsoA gene DNA that carried a C-terminal HA epitope fusion. Using pSM019 DNA as template, we supplemented 50 µl amplification reactions with 10× Taq polymerase buffer, dNTP mixture (200 µM each), 5 pmol µl−1 of each primer, 7.5 U Taq polymerase (NEB) and 6 mM MgCl2. Extension time was 2 min. Amplified DNA was cloned into the BACTH plasmid pUT18C to screen for mutations in RsoA that impair interaction with SigO.
After mutagenizing rsoA DNA, we ligated the amplified product into BACTH plasmid pUT18C. After ligation but before transformation, the ligation reaction mixture was subjected to treatment with SmaI for 1 h to linearize any plasmid DNA not containing an insert in the multiple cloning site. After SmaI digestion, the mutant plasmid pool was transformed into BACTH test strain E. coli BTH101 carrying the other plasmid (pKT25) expressing SigO fusion protein and also pLysE. Co-transformants were plated onto selective media containing X-Gal, and colonies displaying a white or weak blue phenotype (relative to cells expressing non-mutagenized RsoA fusion protein) were selected for further screening. Multiple independent error-prone PCRs were processed in this fashion and only a small number of potential mutants were picked for further analysis from each trial, in order to avoid sibling mutants. All selected putative mutant colonies were subjected to colony PCR to screen against those not containing rsoA. We developed a quasi-high-throughput protocol for screening putative interaction-defective mutants that was designed to screen against undesirable mutations (such as frameshift and nonsense mutations) that frequently arise during error-prone PCR. Putative mutant colonies were picked from indicator plates and inoculated into wells of a 96-well microtitre plate each containing 200 µl of selective LB broth. After overnight growth at 30 °C with shaking, cells were lysed by the addition of 20 µl PopCulture cell lysis reagent and a freeze–thaw treatment (promoting cell wall disruption by T4 lysozyme expressed from pLysE). DNaseI (1 U) was added to each well to digest liberated genomic DNA. After centrifugation, clarified lysate from each well was transferred to a 96-well vacuum dot blot apparatus and proteins were adsorbed to PVDF membrane. Anti-HA rabbit antibodies and goat anti-rabbit alkaline phosphatase conjugated antibodies were used to screen lysates for the expression of full-length HA-tagged RsoA. Since RsoA was tagged at the C-terminus, a positive HA signal screened against mutations that prematurely terminated RsoA translation. Plasmid DNA from positive colonies was subjected to sequencing to identify point mutations in rsoA. All mutants carried a mutation in RsoA region 2.3 and some possessed one or two additional point mutations outside this region. In order to determine whether the mutations in region 2.3 were responsible for the observed loss of interaction with SigO, we amplified mutant region 2.3 DNA, re-cloned it into the BACTH plasmid and re-tested it for interaction against full-length SigO hybrid protein. We observed in each case that the region 2.3 mutation accounted for the loss of apparent interaction with SigO. The possible contribution of mutations outside of region 2.3 to the loss of interaction was not further examined.
Tandem SigO/RsoA expression and pull-down assays.
To directly detect interactions between SigO and RsoA, we fused the 5′ half (codons 1–105) of sigO to the T18 cyaA gene fragment in pUT18C and imparted a FLAG epitope 3′ to sigO. The rsoA gene (and mutant derivatives of the gene) was fused to the T25 cyaA gene fragment and an HA epitope was imparted 3′ to rsoA. Relevant plasmid pairs were co-transformed into E. coli BL21 carrying pLysS. Transformed cells were grown overnight at 37 °C with shaking and 100 µl of this culture was used to inoculate 5 ml of fresh medium. After growth at 30 °C to an OD of ~0.4, 4 ml of each culture was collected by centrifugation and frozen as a pellet overnight. Cell pellets were subjected to two freeze–thaw cycles and the pellets were re-suspended in 200 µl of 10 mM Tris/HCl (pH 8.0), supplemented with 1× micrococcal nuclease buffer and 10 000 U micrococcal nuclease (NEB) and incubated at room temperature for 30 min until cells were lysed and genomic DNA was digested. The cell lysate was brought to a volume of 1 ml with binding buffer [20 mM Tris/HCl (pH 7.5), 500 mM NaCl and 0.05 % Tween-20] and a 300 µl volume was retained as crude lysate. To capture SigO-FLAG from the remaining 700 µl of each sample, we used magnetic beads conjugated to anti-FLAG antibodies (Sigma-Aldrich). A 100 µl aliquot of bead suspension (50 % slurry) was washed twice in 1 ml volumes of Tris-buffered saline, re-suspended in 100 µl of binding buffer and added to the 700 µl lysate. Binding occurred at room temperature for 45 min with slow rotation, and beads were sedimented using a magnet and washed with three 1 ml volumes of binding buffer. The magnetic beads were ultimately re-suspended in 250 µl of 1× Laemmli buffer, thus liberating the antibody small chain and any bound SigO-FLAG. We ultimately separated 20 µl volumes of the crude (whole cell) lysate and bound fractions by SDS-PAGE using 12 % gels. One gel was stained with Coomassie blue to visualize total proteins, and the other two gels were transferred to PVDF membranes and separately probed with anti-FLAG and anti-HA rabbit antibodies to detect SigO and RsoA, respectively. Alkaline phosphatase conjugated goat anti-rabbit antibodies were used as the secondary antibody and the program ImageJ (National Institutes of Health) was used to quantify band intensities.
Transcription assays in B. subtilis.
To test the impact of RsoA region 2.3 mutations that impair interaction with SigO for their impact on transcription initiation dependent on SigO–RsoA in the native host (B. subtilis), we co-expressed SigO and RsoA from separate xylose-inducible ectopic locations on the B. subtilis chromosome as previously described (MacLellan et al., 2008). Briefly, SigO was expressed from pSWEET integrated at the amyE locus and RsoA proteins were expressed from a kanamycin-resistant derivative of pAX01 (pSM016) integrated at the lacA locus. The ability of these proteins to initiate transcription was monitored by expression of a lacZ gene fused to the target promoter from gene oxdC and integrated at the thrC locus. After overnight growth in selective media, a 1 % volume was used to inoculate 5 ml of fresh medium and cultures were grown to an OD of 0.3–0.4. One hour prior to harvest, cultures were supplemented with 5 % (final volume) xylose to induce protein expression. Collected cells were lysed by treatment with lysozyme and sonication. After centrifugation, lysate was subjected to β-galactosidase assays using ONPG as substrate. All assays were conducted in triplicate. Triplicate volumes were pooled, lysed with lysozyme and sonication and centrifuged in order to ensure similar expression of RsoA in each assay. Proteins from equal volumes of lysate (25 µl) were separated on a 15 % Tris-glycine gel and transferred to PVDF membrane. Proteins were detected using anti-HA antibodies and goat anti-rabbit antibodies conjugated to alkaline phosphatase.
β-Galactosidase assays.
For some experiments, assays were conducted in triplicate in a 96-well microtitre plate format. Cells were inoculated into 200 µl selective medium and grown overnight at either 30 or 37 °C with shaking. On the next day, 5 µl volumes were used to inoculate 195 µl of fresh media and cultures were grown to an OD of 0.3–0.4. Plates were centrifuged at 3700 r.p.m. for 10 min and supernatant was removed. Cells were re-suspended in 200 µl of Z buffer [60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4 and 400 nM DTT (pH 7.0)] and cell density was determined at a wavelength of 600 nm. A 96-pin replicator tool was used to patch growth onto an X-Gal indicator plate for a qualitative assessment of β-galactosidase activity. We removed 110 µl of cell suspension from each well and we supplemented the remaining suspension with 10 µl PopCulture cell lysis reagent and allowed it to incubate at 37 °C for 30 min. After equilibration to room temperature, reactions were started by the addition of 20 µl of ONPG (4 mg ml−1). Reactions were stopped by the addition of 50 µl of 1 M sodium carbonate and absorbance was read at 550 and 420 nm. All absorbance readings were conducted using a Molecular Devices Paradigm Multimodal Plate reader.
Computational techniques.
Alignments were conducted using ClustalOmega (Sievers et al., 2011) and by manual manipulation. Secondary structure predictions were conducted using the Jnet algorithm (Cole et al., 2008). Adobe Photoshop was used in the preparation of composite images and to improve presentation, without altering information content in images.
Results
Predicted primary and secondary structure of RsoA
RsoA is a 79-amino acid protein with a predicted secondary structure that resembles the two α helices that comprise regions 2.1 and 2.2 in canonical σ70 σ factors (Fig. 1). The predicted α helix coincident with region 2.2 includes a motif (EDLXQE) that is highly conserved amongst RsoA orthologues and canonical σ factors. Downstream of this segment, aromatic amino acids (e.g. F67, F70) are spatially conserved with those universally conserved in region 2.3 of primary σ factors and broadly conserved amongst the alternative σ factors (Fig. 1) (Tanaka et al., 1993). We therefore designate the segment of 24 amino acids downstream of region 2.2 as region 2.3. We also refer to the predicted α helix upstream of region 2.2 as region 2.1, merely by spatial convention (Lonetto et al., 1992), although there is limited sequence conservation in this region with canonical σ factors, or indeed amongst σ factors in general. Since we currently lack experimentally verified structural information for RsoA, these regional designations as applied to RsoA are useful for delineating regions of sequence similarity with canonical σ factor. We think it most likely that conserved motifs reflect the σ factor ancestry of RsoA (see Discussion), though it remains to be determined whether the respective σ factor activities normally associated with regions 2.2 and 2.3 are necessarily exhibited by the extant RsoA protein. Based upon the above-mentioned structural designations, we assigned mutations that impair the SigO–RsoA interaction to specific sub-regions of RsoA and genetically dissected RsoA by region to determine which segment of RsoA interacts with SigO.
Isolation of random mutations in RsoA that inhibit interaction with SigO
It was previously shown that RsoA interacts specifically with the N-terminal half of SigO in cells (BACTH assay) and in vitro (direct pull-down assays) (MacLellan et al., 2009a). To identify residues in RsoA important for its interaction with the amino-terminus of SigO, we designed a random mutagenesis screen using an adenylate cyclase-based BACTH assay (Karimova et al., 1998). rsoA (bearing a C-terminal HA) tag was amplified using error-prone PCR and fused to the T25 cyaA gene fragment to generate mutant plasmid libraries of rsoA. These libraries were transformed into an E. coli test strain containing a T18 cyaA-sigO fusion. Putative interaction-deficient mutants of rsoA were identified as pale blue or white colonies on indicator medium. These mutants were subsequently subjected to a 96-well dotblot screen to detect the C-terminal HA epitope on the T25-RsoA fusion protein, thus screening against nonsense and frameshift mutations in rsoA. Immunoblot screening of approximately 400 putative mutants followed by sequencing revealed three mutants (Fig. 2a) that expressed full-length RsoA with an apparently defective ability to interact with SigO. Mutations F67S and I71N and a double mutation (E69V/M74T) all occur in region 2.3 of RsoA (see Fig. 1, lobed arrows), suggesting that this peptide segment plays an important role in mediating the interaction between RsoA and SigO.
Fig. 2.

Random mutations in RsoA that impair interaction with SigO. (a) Mutants isolated in the BACTH assay random mutagenesis screen (F67S, I71N, double mutant E69V/M74T). (b) Qualitative and quantitative BACTH analysis of the effects of the four mutations after regeneration in T25-RsoA fusion protein using site-directed mutagenesis. Quantitative assays conducted using β-galactosidase assays and activity reported as mean and sd (n=3). Qualitative activities shown as triplicate patches on selective medium containing X-Gal. Negative control is expression of T18-SigO protein in the absence of T25-RsoA expression. (c) Expression of wt and mutant T25-RsoA proteins tested in CyaA+ strain E. coli DH5α (see Methods). Upper panel is Coomassie blue-stained loading control. Lower panel is immunoblot detection of T25-RsoA fusion proteins tagged with HA epitope. For each allele, first lane is total protein and second lane is soluble (sedimentation-resistant) fraction. Control is pKT25 in E. coli DH5α.
We re-generated these mutations using site-directed mutagenesis in T25-RsoA and re-tested the constructs for interaction with SigO. All four mutations impaired the interaction relative to wt RsoA (Fig. 2b). With respect to the double E69V/M74T mutant, it initially seemed surprising that two mutations independently impairing interaction would be obtained within one mutant gene. However, these mutations in isolation led to only a partial interaction defect and markedly blue colonies on indicator medium (Fig. 2b). Since the first component of our random mutagenesis screen involved selection of white colonies, had these two mutations occurred independently they may well have been overlooked in our colorimetric screen. Because they occurred in tandem, however, the combined effect of mutations E69V and M74T led to white colonies (Fig. 2a) and ultimately the selection of that isolate for further investigation.
To ensure that each mutant protein accumulated in cells to similar levels exhibited by non-mutated RsoA, we expressed the T25-RsoA fusion proteins in a CyaA+ genetic background (see Methods) (Fig. 2c). Mutant I71N accumulated poorly in several replicated experiments, and the observed interaction defect by this mutation may be related to poor accumulation in the cell. The other three mutant proteins (F67S, E69V and M74T) accumulated total and soluble T25-RsoA levels similar to wt T25-RsoA, indicating that impaired interaction with SigO cannot be attributed to poor accumulation of these mutant proteins.
Deletion analysis of RsoA reveals that region 2.3 is sufficient for interaction with SigO
Since the mutations identified in our random mutagenesis screen all coincided with RsoA region 2.3, we generated deletion mutants of RsoA and tested these for their ability to interact with SigO.
Based on predicted secondary structure and the regional delineations of RsoA and canonical σ factors (Fig. 1), in separate constructs, we genetically deleted from RsoA the N-terminal leader sequence (amino acids 1–14), the leader plus region 2.1 (amino acids 1–35) and the leader sequence plus regions 2.1 and 2.2 (amino acids 1–55) and we tested whether these deletion derivatives were able to interact with SigO using the BACTH assay. A quantitative and qualitative assessment (Fig. 3a) demonstrated that all of the T25-RsoA deletion derivatives interacted with SigO similarly to full-length RsoA. Thus, of the 79 amino acids in RsoA, the C-terminal 24 amino acids (comprising the region 2.3-like segment) are sufficient for the observed protein–protein interaction between RsoA and SigO.
Fig. 3.
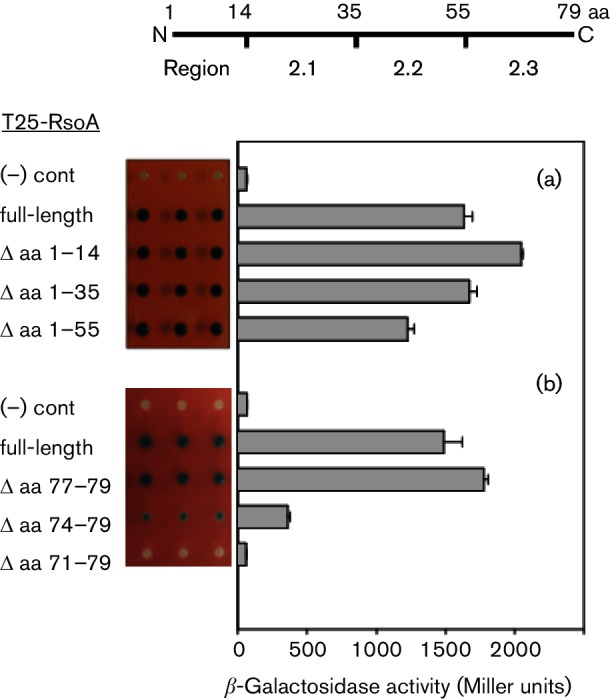
BACTH protein–protein interaction assay of T18-SigO fusion proteins co-expressed with (a) RsoA N-terminal and (b) RsoA C-terminal deletions derivatives. The full-length RsoA is 79 amino acids long and all derivatives are fused to the C-terminus of the T25 CyaA fragment. Quantitative assays conducted using β-galactosidase assays and activity reported as mean and sd (n=3). Negative control is expression of T18-SigO protein in the absence of T25-RsoA expression. Qualitative activities shown as triplicate spots on selective medium containing X-Gal. The line diagram indicates amino acid positions that delineate RsoA region 2.1, 2.2 and 2.3 sub-regions.
We next genetically deleted residues from the C-terminus of full-length RsoA in three amino acid steps. The deletion of three amino acids from the C-terminus of a T25-RsoA fusion protein had little effect on interaction relative to the full-length protein (Fig. 3b), but the deletion of six amino acids partially impaired the interaction and the deletion of nine amino acids apparently abolished the interaction, implying that RsoA region 2.3 is required for its interaction with SigO.
The RsoA deletion mutants were also tested in a substantially different BACTH system (Dove & Hochschild, 2004) that employed fusions to λcI and the RNAP α subunit. This showed (Fig. S2) interaction patterns that closely paralleled the results from the adenylate cyclase BACTH system (Fig. 3a, b). Taken together, the results from our random mutagenesis screen and gene deletion analysis indicate that region 2.3 of RsoA is sufficient to mediate its observed protein–protein interaction with SigO.
The effect of alanine substitutions in RsoA on its interaction with SigO
In order to appreciate the contribution of individual amino acid residues in RsoA to its interaction with SigO, we generated a number of region 2.2 and region 2.3 alanine substitutions, primarily at residues that are conserved amongst RsoA homologues and many canonical σ factors (Fig. 1, arrows). These were tested in the context of both full-length T25-RsoA (RsoAFL) fusion protein (Fig. 4a) and truncated T25-RsoA region 2.3 (RsoA2.3) fusion protein (Fig. 4b) expression in the BACTH assay.
Fig. 4.

BACTH protein–protein interaction assay of T18-SigO fusion proteins co-expressed with wt and alanine substitution mutants of T25-RsoA fusion proteins. (a) Mutations in full-length RsoA (T25-RsoAFL) fused to T25 CyaA fragment and (b) mutations in the 24-amino acid RsoA region 2.3 (T25-RsoA2.3) fused to CyaA fragment. Quantitative assays conducted using β-galactosidase assays and activity reported as mean and sd (n=3). Qualitative activities shown as triplicate spots on selective medium containing X-Gal. Negative control is expression of T18-SigO protein in the absence of T25-RsoA expression. For the T25-RsoA2.3 alleles, total and soluble protein expression levels were tested in a CyaA+ background (Fig. S3).
None of the alanine substitutions in region 2.2 had a pronounced effect on the apparent interaction of RsoAFL with SigO, while mutations in region 2.3 trended towards decreased apparent interaction (Fig. 5a). Mutation F67A dramatically impaired interaction between RsoAFL and SigO (Fig. 4a) to an extent observed for the F67S mutation isolated from our random screen (Fig. 2b).
Fig. 5.

BACTH assay testing interaction between N-terminus of β′ subunit (RpoC, amino acids 1–310) and deletion derivatives of RsoA. RpoC is expressed as a λcI fusion protein and RsoA derivatives are expressed as RpoA (α subunit) fusions in host strain E. coli FW Kan OL2-62 lac. Details of assay methodology are as described in Dove & Hochschild (2004) and MacLellan et al. (2009b).
The effects of region 2.3 alanine substitutions (Fig. 4b) on the SigO–RsoA2.3 interaction were largely consistent with those using full-length protein. The F67A mutation abolished the interaction with SigO in both RsoA contexts (RsoAFL and RsoA2.3). We confirmed that the T25-RsoAFL fusion proteins bearing region 2.3 alanine substitution mutants accumulated total and soluble RsoA levels similar to those of wt RsoA (Fig. S3).
Consistent with our overall analysis, mutation of conserved residues in region 2.3, but not region 2.2, impairs the SigO–RsoA interaction. Residue F67, and to a lesser extent residue F70, plays major roles, directly or indirectly, in mediating this interaction. These alanine substitution mutants have not been screened to ensure proper folding, but the data nevertheless also point to several amino acid residues, most of them highly conserved amongst RsoA orthologues, that likely do not play a role in mediating the SigO–RsoA interaction.
RsoA region 2.3 mediates weak interaction with β′ subunit
In order to test that the region 2.3 RsoA mutants yielded properly folded protein, we tested selected mutant derivatives for interaction with the β′ subunit N-terminus (β′N) [an interaction we previously reported (MacLellan et al., 2009a)]. Surprisingly, we found that, as with its interaction with SigO, the region 2.3 amino acid segment was also sufficient to account for the interaction between RsoA and the β′N amino acid segment (Fig. 5) and that the imposition of mutation F67A likewise abolished the interaction. Furthermore, mutations E69V, F70A and M74T also largely abolished the apparent interaction (Fig. S4). For this reason, we cannot identify amino acid residues in RsoA region 2.3 that act directly as interaction determinants with SigO without guaranteeing that they do not also perturb structure. Indeed, based on additional results (see the next section) it is likely that some of the key residues identified in the region 2.3-like protein–protein interaction element contribute to structure in the peptide, as well as potentially contributing to the observed interaction.
Co-sedimentation assay reveals interaction defects
To corroborate our BACTH-based interaction results, we co-expressed epitope-tagged N-terminal half of SigO (amino acids 1–105, SigONterm-FLAG) and RsoA (and RsoA-HA) as T18 and T25 (respectively) CyaA fusion proteins in E. coli cells and subsequently pulled down the SigO-FLAG fusion derivatives using anti-FLAG antibodies conjugated to magnetic beads. Samples were separated by SDS-PAGE and probed for co-sedimentation of RsoA-HA (Fig. 6).
Fig. 6.
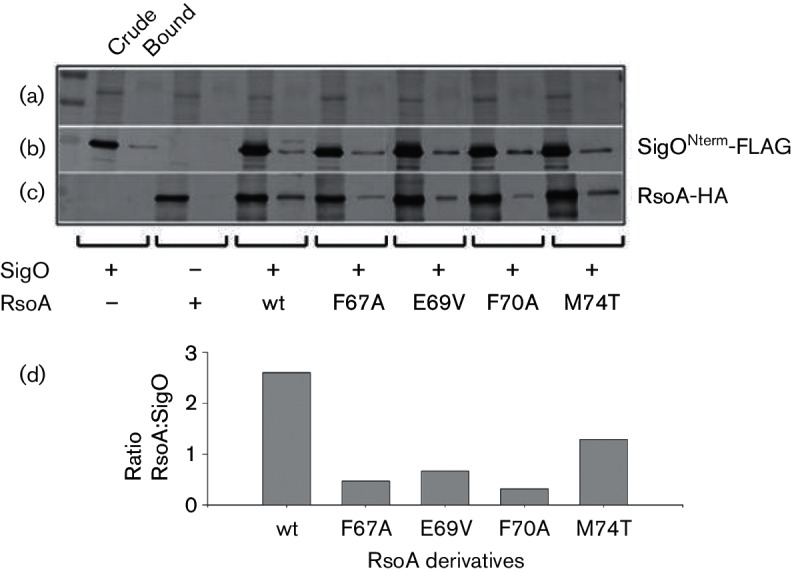
Pull-down of RsoA-HA point mutants by SigONterm-FLAG. Proteins were co-expressed as CyaA T18 and T25 fusion proteins from separate plasmids in E. coli BL21. (a) Coomassie-stained loading control. (b) Western immunoblot using anti-FLAG antibodies. (c) Western immunoblot using anti-HA antibodies. For each strain, equal aliquots of total protein (first lane, crude) and proteins captured by anti-FLAG magnetic beads (second lane, bound) were separated using SDS-PAGE. Note that soluble RsoA accumulates poorly in the absence of SigO co-expression. (d) Bound protein band intensities from (b) and (c) were quantified, and the efficiency of RsoA co-sedimentation was reported as the ratio of RsoA to SigO in each treatment. This figure is a composite of several images from the same experiment.
All of the point mutants of RsoAFL tested (F67A, E69V, F70A, M74T) accumulated in cells as abundantly as the wt fusion protein (Fig. 6c, crude lanes), and approximately similar amounts of SigONterm-FLAG were sedimented by the magnetic beads in each test (Fig. 6b, bound lanes). In repeated experiments, the F67A mutant showed the most obvious and reproducible decrease in its ability to interact with bead-captured SigONterm-FLAG (Fig. 6c, d), along with the F70A mutant, relative to co-sedimentation of wt RsoA. Increased binding and wash stringencies in additional experiments (data not shown) failed to completely eliminate pull-down of some F67A mutant protein. Both the E69V and M74T mutant proteins also frequently showed a qualitatively reduced affinity for SigO relative to the wt protein (Fig. 6c, d). Qualitatively, the results of the pull-down assays closely parallel the results we obtained from the BACTH assays (Figs 2b and 4).
We next used the co-sedimentation assay to test the effects of N-terminal RsoA deletions on the ability of RsoA to interact with SigO, using deletion derivatives analogous to those originally tested using two different BACTH assays (Figs 3 and S2). A qualitative BACTH assay was performed with all RsoA deletion derivatives, demonstrating that the imposition of the HA epitope tag on the proteins did not alter the previously observed interaction patterns with SigO (Fig. S5). The Δ1–14, Δ1–35 and Δ1–55 amino acid derivatives all accumulated similar to wt fusion protein levels (see Fig. S6, lanes 1–4) and all deletion derivatives co-sedimented with SigONterm-FLAG (Fig. 7b, c), thus corroborating the results from our genetic analysis that RsoA region 2.3 (i.e. amino acids 56–79) is sufficient for interaction with SigO. We imparted the F67A substitution into the Δ1–55 deletion derivative to test whether the mutation would impair binding as it did when the full RsoA protein was tested. Note that the imposition of the F67A mutation (and also an F67S mutation, see Fig.S7) resulted in aberrant mobility during electrophoresis, perhaps reflecting a structural perturbation in the mutant protein. As with the full-length protein, this mutation substantially impaired co-sedimentation with SigONterm-FLAG, relative to the non-mutated Δ1–55 deletion protein (Fig. 7c), though as in Fig. 6, some F67A mutant protein co-sedimented with SigO even when the stringency of the binding and bead washing steps were increased.
Fig. 7.

Pull-down of RsoA-HA deletion mutants by SigONterm-FLAG. Proteins were co-expressed as CyaA T18 and T25 fusion proteins from separate plasmids in E. coli BL21. (a) Coomassie-stained loading control. (b) Western immunoblot using anti-FLAG antibodies. (c) Western immunoblot using anti-HA antibodies. For each strain, equal aliquots of total protein (first lane, crude) and proteins captured by anti-FLAG magnetic beads (second lane, bound) were separated using SDS-PAGE. ΔN, N-terminal deletions from RsoA (amino acids deleted in brackets). The full-length RsoA protein is 79 amino acids long. Note that soluble RsoA accumulates poorly in the absence of SigO co-expression. This figure is a composite of several images from the same experiment.
Our various data establish that the C-terminal region 2.3-like segment of RsoA is sufficient to account for the interaction between SigO and RsoA and that, not surprisingly, mutations impairing the SigO–RsoA interaction map uniquely to this region of RsoA.
Delineating a minimal RsoA interaction element
The protein–protein interaction element we have identified resides in the C-terminal 24 amino acids of RsoA, and the deletion of three amino acids from the C-terminal end of RsoA had no effect on protein interaction (e.g. Fig. 3). To gain an appreciation of the minimal amino acid requirements for RsoA region 2.3 interaction with SigO, we translationally fused a 21-amino acid region of the C-terminal end of RsoA, and subsets of this region, to CyaA via a poly-glycine linker and tested it for interaction using the BACTH assay (Table 1).
Table 1. Delineation of RsoA protein–protein interaction motif.
| Construct* | Sequence | β-Galactosidase Act. mean (sd)† | %‡ |
|---|---|---|---|
| (−) cont | – – | 36 (4) | 3 |
| 21 Aa (wt) | GGGGGGADMLLCQDVPGFWEFILYMVD | 1296 (62) | 100 |
| 21 Aa (F67A) | GGGGGGADMLLCQDVPGAWEFILYMVD | 46 (5) | 4 |
| 18 Aa | GGGGGG– – –LLCQDVPGFWEFILYMVD | 1091 (107) | 84 |
| 15 Aa | GGGGGG– – – – – –QDVPGFWEFILYMVD | 974 (69) | 75 |
| 12 Aa | GGGGGG– – – – – – – – – PGFWEFILYMVD | 114 (13) | 9 |
| 10 Aa | GGGGGG– – – – – – – – – – –FWEFILYMVD | 59 (6) | 5 |
| 9 Aa | GGGGGG– – – – – – – – – – – –WEFILYMVD | 66 (5) | 5 |
*Test peptides are fused to T25 CyaA fragment via polyglycine linker. T18 CyaA fragment is fused to full-length SigO and proteins are expressed in E. coli strain BTH101. Negative (−) control is pKT25 without insert.
†Mean and standard deviation based on Miller units (n=3).
‡Percentage activity of wt 21 amino acid fusion protein and SigO fusion protein interaction.
The 21 residue fusion protein interacted with full-length SigO to a degree similar to other region 2.3 constructs and an F67A substitution abolished interaction, as expected. An element consisting of only 15 amino acids still displayed considerable interaction capability. We did not further refine the segment required for interaction or delete any residues from the C-terminus beyond the terminal three amino acids, but the data indicate that the RsoA interaction element is shorter than 24 amino acids and is not larger than about 15 amino acid residues.
Effect of RsoA mutations on transcription in B. subtilis
It was previously shown (MacLellan et al., 2009a) that RsoA interacts with purified full-length SigO, as well as the N-terminal half of SigO, but not with other tested B. subtilis σ factors. Additionally, co-expression of SigO and RsoA is required to activate transcription from target promoters in B. subtilis in vitro and in vivo (MacLellan et al., 2008, 2009a). We wished to test the effects of mutations in RsoA on its co-activation of transcription, particularly those that impair the interaction between RsoA and SigO. We tested all of the region 2.3 RsoA site-directed alanine substitution mutations, and the four mutations identified from our random mutagenesis screen for their effects on co-activation of transcription in vivo from a target promoter (PoxdC) transcriptionally fused to lacZ. Almost all of the mutations in RsoA region 2.3 either partially or completely inhibit transcription activation by SigO–RsoA (Fig. 8). The most dramatic decreases in transcriptional activation coincide with RsoA mutations that resulted in impaired interaction with SigO in either the BACTH experiments or the co-sedimentation experiments (e.g. F67A, F67S, E69V and F70A). These data suggest that a productive interaction between RsoA region 2.3 and the amino-terminus of SigO may be required for transcription activation by the SigO–RsoA two-subunit σ factor. It is important to note, however, that these mutations have not been examined for misfolding effects and it is likely, based upon its effects on protein mobility in some contexts, that mutation of residue F67 does cause a misfolding effect with the interaction domain. This could result in transcription activation defects that are not specifically related to the loss of interaction between RsoA and SigO. Other residues not playing an obvious role in the interaction also reduce transcription when mutated (Fig. 8), presumably because they are important for other reasons yet to be defined.
Fig. 8.

Transcription activation from PoxdC-lacZ fusion by wt and region 2.3 mutant alleles of RsoA co-expressed with SigO in B. subtilis. SigO and RsoA expression induced by 2 % xylose from separate ectopic (amyE and lacA, respectively) locations. Quantitative assays conducted using β-galactosidase assays and activity reported as mean and sd (n=3). Lower panel: immunoblot detection of RsoA-HA expression. A non-specific band (ns) acts as a loading control. This image is a composite of two immunoblots conducted in the same experiment. Negative control is expression of SigO protein in the absence of RsoA expression.
Discussion
Co-expression of SigO and RsoA is required to activate transcription from a specific set of related promoters in B. subtilis (see Fig. S1) (MacLellan et al., 2008). The 23 kDa SigO protein appears to be a partially inactivated group IV σ factor possessing a well-defined region 4 and highly degenerate region 2. Earlier work established that SigO region 4 interacts with the flap region of the core β subunit, but we could not detect an interaction between either full-length or the N-terminus of SigO and the clamp helices of the β′ subunit (MacLellan et al., 2009a). Electromobility shift assays suggested that SigO alone could bind cRNAP and target the complex to cognate promoter DNA. However, activation of transcription in vivo and stabilization of open complex in vitro required RsoA.
The orgin of RsoA is uncertain, but we suspect that this 79-amino acid (10 kDa) protein arose as the result of a partial σ factor gene duplication event and that the rsoA gene codes for an amino terminal σ factor fragment carrying contiguous peptide segments highly similar to those found in σ70 regions 2.2 and 2.3. These same peptides also correspond to the most highly conserved amino acids amongst RsoA orthologues (see Figs 1 and S8 for an alignment of 44 non-redundant RsoA orthologues). Prior investigations revealed that RsoA interacts specifically with the N-terminal half of SigO (but not other B. subtilis σ factors) and the β′ RNAP subunit (MacLellan et al., 2009a). We declared this system a legitimate two-subunit σ factor since both proteins appeared related to σ70 family proteins on the basis of conserved amino acid sequence. Unlike typical transcriptional activators that sometimes directly contact region 4 of the σ factor and are situated towards the posterior end of a transcription complex, RsoA is likely situated proximal to the −10 promoter element, based on its interaction with the N-terminus of SigO and the core β′ clamp helices. In terms of its proximity to the anterior end of a transcription complex, RsoA is similar to other recently characterized transcription factors including S. coelicolor RbpA (Tabib-Salazar et al., 2013), Chlamydia trachomatis GrgA (Bao et al., 2012) and E. coli Crl (Banta et al., 2013; Monteil et al., 2010), though these latter proteins lack sequence similarity to σ factor. In addition to interacting with σs, Crl is situated close to the β′ clamp helices and may interact with the core subunit (Liu et al., 2016).
N-terminal and C-terminal peptide deletion analyses combined with alanine substitutions and a random mutagenesis approach demonstrate that the region 2.3-like segment of RsoA constitutes a protein–protein interaction motif that, in isolation, is sufficient to mediate interaction with SigO. Several point mutations in RsoA region 2.3 partially impair the interaction, but mutation of residue F67 (F67A and F67S) substantially abolishes interaction and the ability of the two-subunit system to activate transcription in cells. F67 is conserved in all identified RsoA orthologues and spatially aligns with phenylalanine residues conserved in all group 1 σ factors (e.g. F186 in B. subtilis SigA and F427 in E. coli σ70) and many alternative σ factors (e.g. F86, F72 and F73 in B. subtilis SigHWY, respectively, and F73 in E. coli σE) (see Fig. 1).
Earlier work focusing on the seven conserved aromatic amino acids in region 2.3 of SigA first revealed that this region is required for promoter melting/open complex formation (Juang & Helmann, 1994). Notably, an F186A mutant displayed anomalous DNA melting activity different from most other region 2.3 mutations. The expression of SigA F186A in cells could neither support growth nor cause toxicity when co-expressed with wt SigA, suggesting that this mutation caused structural perturbation in the protein and core binding defects (Rong & Helmann, 1994). Similar melting defects were observed with the analogous F427 mutant of E. coli σ70 (Panaghie et al., 2000). Structural studies of E. coli σ70 (Malhotra et al., 1996) revealed that F427 was buried within a local hydrophobic fold, probably explaining why mutation at the same site in B. subtilis SigA caused structural irregularity and subsequent loss of activity. Interestingly, mutation of F67 in RsoA (which co-aligns with SigA F186 and σ70 F427) also causes loss of activity (loss of binding activity with SigO and loss of transcription activity in vivo). Thus, not only RsoA F67 is spatially conserved with phenylalanine residues in σ70, SigA and many alternative σ factors but also its mutation has a similarly pronounced consequence. When the 24-amino acid region 2.3-like segment is fused to CyaA, mutation of F67 to either alanine or serine results in aberrant mobility during electrophoresis (Fig. S7), an observation that might indicate structural perturbation as a result of the mutation. In RsoA, the peptide P65-G66-F67-W68-E69-F70-I71 constitutes the most highly conserved segment in region 2.3 that, in an overall sense, is highly hydrophobic [of the 24 residues we broadly designate as region 2.3, 14 amino acids (61 %) are moderately to strongly hydrophobic].
RsoA region 2.3 is reminiscent of a so-called short Linear Motif (sLiM) owing to its demonstrated role in mediating interaction with SigO. sLiMs are small, solvent-accessible peptide segments, usually found at the carboxyl termini of proteins, that mediate protein–protein interactions by interacting with cognate elements in partner proteins (Davey et al., 2012; Neduva & Russell, 2005). They have been identified in a large number of viral, prokaryotic and eukaryotic proteins, particularly in cell-signalling proteins or proteins that are part of larger multi-subunit complexes. sLiMs have been called ‘evolutionarily plastic’ and ‘particularly evolvable’ interaction elements, since only a small number of residues in the peptide play a dominant role in mediating an interaction. Therefore, one or a few amino acid substitutions in a protein are enough to convert a peptide into a novel and functionally important interaction motif (Davey et al., 2012; Van Roey et al., 2014; Wagner & Lynch, 2008). We think it at least plausible that RsoA originated from a partial σ factor gene duplication event and that region 2.3, presumably a single-strand promoter DNA-binding element in the ancestral protein, acquired a new function as a sLiM-like protein–protein interaction element. We speculate that those residues in RsoA that appear conserved with those in canonical σ factors may be important for the SigO–RsoA interaction for structural reasons and may also have been co-opted to play a role in mediating the protein–protein interaction. Determining the specific roles of amino acids in the region 2.3-like element will require further investigation, although it seems likely that F67, at the very least, probably plays a structural role and that the maintenance of structure within the region is required for interaction with SigO. Given the small peptide nature of this interaction element, it is possible that some amino acids are important for both structure and interaction.
Surprisingly, the region 2.3-like element of RsoA also mediates the previously reported (MacLellan et al., 2009a) weak interaction of this protein with the RNAP β′ subunit. This result now explains why extensive mutation in region 2.2 of RsoA (note that it is amino acids within region 2.2 of canonical σ factors that mediate interaction with the β′ subunit) had little or no effect on the interaction (MacLellan et al., 2009a). Weakly mimicking, as it does, the much stronger interaction with the N-terminus of SigO, it is tempting to suspect that the RsoA–β′N interaction may be artifactual. On the other hand, Sengupta et al. (2015) demonstrated that RsoA does indeed independently interact with cRNAP and it is therefore possible that the region 2.3 segment of RsoA mediates this interaction with core polymerase.
The precise mechanistic roles of SigO and RsoA in a transcription complex are not yet clear. It may be that RsoA performs a bridging role by interacting with both the core β′ subunit and SigO, and it orients a divergent (but still functional) domain in SigO for appropriate interaction with promoter DNA. Alternatively, the region 2.3-like peptide of RsoA may indeed act as a canonical region 2.3 and directly bind non-template promoter DNA and stabilize open complex. Any model supporting the latter scenario, however, would need to account for the observation that RsoA region 2.3 also constitutes a protein–protein interaction element that mediates a weak interaction with the β′ subunit and a strong interaction with the N-terminus of SigO.
Acknowledgements
X. X. and M. C. D. contributed equally to this work. Initial studies related to this work were performed in the John D. Helmann laboratory, Cornell University (supported by National Institutes of Health grant GM047446). This work was supported by Natural Sciences and Engineering Research Council of Canada grant 386710-2010 to SRM.
Supplementary Data
Abbreviations:
- BACTH
bacterial two-hybrid
- cRNAP
core RNA polymerase
- ECF
extracytoplasmic function
- HA
haemagglutinin
Footnotes
Edited by: T. Msadek
Edited by: P. Zuber
Eight supplementary figures and two supplementary tables are available with the online Supplementary Material.
References
- Arthur T. M., Burgess R. R.(1998). Localization of a sigma70 binding site on the N terminus of the Escherichia coli RNA polymerase beta' subunit. J Biol Chem 27331381–31387. 10.1074/jbc.273.47.31381 [DOI] [PubMed] [Google Scholar]
- Banta A. B., Chumanov R. S., Yuan A. H., Lin H., Campbell E. A., Burgess R. R., Gourse R. L.(2013). Key features of σS required for specific recognition by Crl, a transcription factor promoting assembly of RNA polymerase holoenzyme. Proc Natl Acad Sci U S A 11015955–15960. 10.1073/pnas.1311642110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X., Nickels B. E., Fan H.(2012). Chlamydia trachomatis protein GrgA activates transcription by contacting the nonconserved region of σ66. Proc Natl Acad Sci U S A 10916870–16875. 10.1073/pnas.1207300109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti A., Bouveret E.(2012). The bacterial two-hybrid system based on adenylate cyclase reconstitution in Escherichia coli. Methods 58325–334. 10.1016/j.ymeth.2012.07.018 [DOI] [PubMed] [Google Scholar]
- Bhandari V., Ahmod N. Z., Shah H. N., Gupta R. S.(2013). Molecular signatures for Bacillus species: demarcation of the Bacillus subtilis and Bacillus cereus clades in molecular terms and proposal to limit the placement of new species into the genus Bacillus. Int J Syst Evol Microbiol 632712–2726. 10.1099/ijs.0.048488-0 [DOI] [PubMed] [Google Scholar]
- Burgess R. R., Travers A. A., Dunn J. J., Bautz E. K.(1969). Factor stimulating transcription by RNA polymerase. Nature 22143–46. 10.1038/221043a0 [DOI] [PubMed] [Google Scholar]
- Campagne S., Marsh M. E., Capitani G., Vorholt J. A., Allain F. H.(2014). Structural basis for −10 promoter element melting by environmentally induced sigma factors. Nat Struct Mol Biol 21269–276. 10.1038/nsmb.2777 [DOI] [PubMed] [Google Scholar]
- Cole C., Barber J. D., Barton G. J.(2008). The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36W197–W201. 10.1093/nar/gkn238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., Gibson T. J.(2012). Attributes of short linear motifs. Mol Biosyst 8268–281. 10.1039/C1MB05231D [DOI] [PubMed] [Google Scholar]
- deHaseth P. L., Helmann J. D.(1995). Open complex formation by Escherichia coli RNA polymerase: the mechanism of polymerase-induced strand separation of double helical DNA. Mol Microbiol 16817–824. 10.1111/j.1365-2958.1995.tb02309.x [DOI] [PubMed] [Google Scholar]
- Dove S. L., Hochschild A.(2004). A bacterial two-hybrid system based on transcription activation. Methods Mol Biol 261231–246. 10.1385/1-59259-762-9:231 [DOI] [PubMed] [Google Scholar]
- Feklistov A., Darst S. A.(2011). Structural basis for promoter −10 element recognition by the bacterial RNA polymerase σ subunit. Cell 1471257–1269. 10.1016/j.cell.2011.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geszvain K., Gruber T. M., Mooney R. A., Gross C. A., Landick R.(2004). A hydrophobic patch on the flap-tip helix of E.coli RNA polymerase mediates sigma(70) region 4 function. J Mol Biol 343569–587. 10.1016/j.jmb.2004.08.063 [DOI] [PubMed] [Google Scholar]
- Gribskov M., Burgess R. R.(1986). Sigma factors from E. coli, B. subtilis, phage SP01, and phage T4 are homologous proteins. Nucleic Acids Res 146745–6763. 10.1093/nar/14.16.6745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundlach J., Rath H., Herzberg C., Mäder U., Stülke J.(2016). Second messenger signaling in Bacillus subtilis: Accumulation of cyclic di-AMP inhibits biofilm formation. Front Microbiol 7804. 10.3389/fmicb.2016.00804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hachmann A. B., Angert E. R., Helmann J. D.(2009). Genetic analysis of factors affecting susceptibility of Bacillus subtilis to daptomycin. Antimicrob Agents Chemother 531598–1609. 10.1128/AAC.01329-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann J. D.(2002). The extracytoplasmic function (ECF) sigma factors. Adv Microb Physiol 4647–110. [DOI] [PubMed] [Google Scholar]
- Helmann J. D., Chamberlin M. J.(1988). Structure and function of bacterial sigma factors. Annu Rev Biochem 57839–872. 10.1146/annurev.bi.57.070188.004203 [DOI] [PubMed] [Google Scholar]
- Johnston E. B., Lewis P. J., Griffith R.(2009). The interaction of Bacillus subtilis sigmaA with RNA polymerase. Protein Sci 182287–2297. 10.1002/pro.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang Y. L., Helmann J. D.(1994). A promoter melting region in the primary sigma factor of Bacillus subtilis. Identification of functionally important aromatic amino acids. J Mol Biol 2351470–1488. 10.1006/jmbi.1994.1102 [DOI] [PubMed] [Google Scholar]
- Karimova G., Pidoux J., Ullmann A., Ladant D.(1998). A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 955752–5756. 10.1073/pnas.95.10.5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznedelov K., Minakhin L., Niedziela-Majka A., Dove S. L., Rogulja D., Nickels B. E., Hochschild A., Heyduk T., Severinov K.(2002). A role for interaction of the RNA polymerase flap domain with the sigma subunit in promoter recognition. Science 295855–857. 10.1126/science.1066303 [DOI] [PubMed] [Google Scholar]
- Liu B., Zuo Y., Steitz T. A.(2016). Structures of E. coli σS-transcription initiation complexes provide new insights into polymerase mechanism. Proc Natl Acad Sci U S A 1134051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonetto M., Gribskov M., Gross C. A.(1992). The sigma 70 family: sequence conservation and evolutionary relationships. J Bacteriol 1743843–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Yang X., Kandemir H., Mielczarek M., Johnston E. B., Griffith R., Kumar N., Lewis P. J.(2013). Inhibitors of bacterial transcription initiation complex formation. ACS Chem Biol 81972–1980. 10.1021/cb400231p [DOI] [PubMed] [Google Scholar]
- MacLellan S. R., Wecke T., Helmann J. D.(2008). A previously unidentified sigma factor and two accessory proteins regulate oxalate decarboxylase expression in Bacillus subtilis. Mol Microbiol 69954–967. 10.1111/j.1365-2958.2008.06331.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan S. R., Helmann J. D., Antelmann H.(2009a). The YvrI alternative σ factor Is essential for acid stress induction of oxalate decarboxylase in Bacillus subtilis. J Bacteriol 191931–939. 10.1128/JB.01435-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan S. R., Guariglia-Oropeza V., Gaballa A., Helmann J. D.(2009b). A two-subunit bacterial σ-factor activates transcription in Bacillus subtilis. Proc Natl Acad Sci 10621323–21328. 10.1073/pnas.0910006106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A., Severinova E., Darst S. A.(1996). Crystal structure of a sigma 70 subunit fragment from E. coli RNA polymerase. Cell 87127–136. 10.1016/S0092-8674(00)81329-X [DOI] [PubMed] [Google Scholar]
- Missiakas D., Raina S.(1998). The extracytoplasmic function sigma factors: role and regulation. Mol Microbiol 281059–1066. 10.1046/j.1365-2958.1998.00865.x [DOI] [PubMed] [Google Scholar]
- Monteil V., Kolb A., Mayer C., Hoos S., England P., Norel F.(2010). Crl binds to domain 2 of σ(S) and confers a competitive advantage on a natural rpoS mutant of Salmonella enterica serovar Typhi. J Bacteriol 1926401–6410. 10.1128/JB.00801-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K. S.(2013). X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J Biol Chem 2889126–9134. 10.1074/jbc.M112.430900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K. S.(2015). Structural biology of bacterial RNA polymerase. Biomolecules 5848–864. 10.3390/biom5020848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K. S., Darst S. A.(2003). Bacterial RNA polymerases: the wholo story. Curr Opin Struct Biol 1331–39. 10.1016/S0959-440X(02)00005-2 [DOI] [PubMed] [Google Scholar]
- Neduva V., Russell R. B.(2005). Linear motifs: evolutionary interaction switches. FEBS Lett 5793342–3345. 10.1016/j.febslet.2005.04.005 [DOI] [PubMed] [Google Scholar]
- Paget M. S. B., Helmann J. D.(2003). The sigma70 family of sigma factors. Genome Biol 4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G., Aiyar S. E., Bobb K. L., Hayward R. S., de Haseth P. L.(2000). Aromatic amino acids in region 2.3 of Escherichia coli sigma 70 participate collectively in the formation of an RNA polymerase-promoter open complex. J Mol Biol 2991217–1230. 10.1006/jmbi.2000.3808 [DOI] [PubMed] [Google Scholar]
- Rong J. C., Helmann J. D.(1994). Genetic and physiological studies of Bacillus subtilis sigma A mutants defective in promoter melting. J Bacteriol 1765218–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S., Prajapati R. K., Mukhopadhyay J.(2015). Promoter escape with bacterial two-component σ factor suggests retention of σ region two in the elongation complex. J Biol Chem 29028575–28583. 10.1074/jbc.M115.666008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7 539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabib-Salazar A., Liu B., Doughty P., Lewis R. A., Ghosh S., Parsy M. L., Simpson P. J., O'Dwyer K., Matthews S. J., Paget M. S.(2013). The actinobacterial transcription factor RbpA binds to the principal sigma subunit of RNA polymerase. Nucleic Acids Res 415679–5691. 10.1093/nar/gkt277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Takayanagi Y., Fujita N., Ishihama A., Takahashi H.(1993). Heterogeneity of the principal sigma factor in Escherichia coli: the rpoS gene product, sigma 38, is a second principal sigma factor of RNA polymerase in stationary-phase Escherichia coli. Proc Natl Acad Sci U S A 903511–3515. 10.1073/pnas.90.8.3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roey K., Uyar B., Weatheritt R. J., Dinkel H., Seiler M., Budd A., Gibson T. J., Davey N. E.(2014). Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev 1146733–6778. 10.1021/cr400585q [DOI] [PubMed] [Google Scholar]
- Wagner G. P., Lynch V. J.(2008). The gene regulatory logic of transcription factor evolution. Trends Ecol Evol 23377–385. 10.1016/j.tree.2008.03.006 [DOI] [PubMed] [Google Scholar]
- Zuo Y., Steitz T. A.(2015). Crystal structures of the E. coli transcription initiation complexes with a complete bubble. Mol Cell 58534–540. 10.1016/j.molcel.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.