

Abstract
Aims
Although substantial progress has been made in understanding of ontogeny of drug metabolism, there is still a gap of knowledge in developmental pharmacogenetics in neonates. We hypothesized that both age and pharmacogenetics might explain the developmental pattern of CYP2C19. We conducted a population pharmacokinetic–pharmacogenetic study to quantify the developmental pharmacogenetics of CYP2C19 in neonates and young infants using omeprazole as a probe drug.
Methods
Pharmacokinetic samples were collected from 51 Caucasian neonates and young infants, who were receiving omeprazole treatment. Population pharmacokinetic–pharmacogenetic analysis of omeprazole and its metabolites was performed using NONMEM.
Results
Data fitted a one‐compartment parent and metabolite model with first‐order absorption and elimination. CYP2C19 and CYP3A4 are predominantly involved in the metabolism of omeprazole despite their relatively low activities compared to adults. The clearance of omeprazole converted to 5‐hydroxy‐omeprazole (CLOMZ‐M1) increases with postnatal age. In CYP2C19 poor and intermediate metabolizers, model‐predicted CLOMZ‐M1 are 12.5% (5–95% percentile: 3–14.9%) and 44.9% (5–95% percentile: 29.9–72.6%) of the value in extensive/ultrarapid metabolizer, respectively. Model‐predicted absorption rate constant of omeprazole is 6.93 (5–95% percentile: 3.01–14.61) times higher in ABCB1 homozygous mutant patients, 1.86 (5–95% percentile: 0.86–3.47) times higher in ABCB1 heterozygous patients than that in ABCB1 homozygous wild‐type patients.
Conclusions
Developmental pharmacogenetics of CYP2C19 was quantitatively described in neonates and young infants using omeprazole as a probe drug. Our findings emphasize the importance of semiphysiological developmental pharmacokinetic modelling approach when evaluating developmental pharmacogenetics of drugs with multiple routes of biotransformation.
Keywords: ABCB1, CYP2C19, neonates, ontogeny, pharmacogenetics, pharmacokinetics
What is Already Known about this Subject
Developmental changes in drug metabolizing enzyme expression and activity during childhood make a major contribution to the overall pharmacokinetic difference between adults and children.
Ontogeny of CYP2C19 has been demonstrated in vitro, however, the quantitative data are still missing in vivo and large interindividual variability remains unexplained.
What this Study Adds
A population pharmacokinetic–pharmacogenetic model was successfully developed to quantify the developmental patterns of CYP2C19 in neonates and young infants using omeprazole as a probe drug.
Both CYP2C19 genotype and age contribute to the developmental pharmacokinetics of omeprazole and its metabolites.
A semiphysiological developmental pharmacokinetic modelling approach should be encouraged to evaluate developmental pharmacogenetics of drugs with multiple routes of biotransformation.
Introduction
Developmental changes in http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=242 (CYP) enzyme expression and activity during childhood make a major contribution to the developmental pharmacokinetics of extensively metabolized drugs 1, 2. Distinct patterns of isoform‐specific ontogeny have been demonstrated antenatally and postnatally, and contribute substantially to the observed differences in therapeutic efficacy and safety in children 1, 3. Although significant progress has been achieved in understanding of ontogeny of drug metabolism, there is still a gap in knowledge of developmental pharmacogenetics in neonates.
http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=262#1328 plays an important role in the oxidative biotransformation of several important groups of drugs, including anticancer, antidepressants, antihypertensive drugs and inhibitor proton pump inhibitors 4, 5. Ontogeny of CYP2C19 has been demonstrated in vitro. Koukouritaki et al. 6 reported that CYP2C19 catalytic activity was at 12–15% of mature value and similar throughout the fetal period. Its expression did not change at birth, increased over the first 5 months after birth and achieved adult level at age 10 years. In vivo data also demonstrated a relatively lower activity of CYP2C19 in neonates as compared to older infants, children, and adolescents, as reflected by total clearance of pantoprazole 7, 8.
We hypothesized that both age and pharmacogenetics might explain the developmental pattern of CYP2C19 in neonates. Therefore, we conducted a population pharmacokinetic–pharmacogenetic study to quantify the developmental pharmacogenetics of CYP2C19 in neonates and young infants using omeprazole as a probe drug.
Omeprazole was selected as a probe drug based on the following reasons:
It is frequently used to treat neonatal gastroesophageal reflux, a common disease in neonates 9, 10.
CYP2C19 is the predominant enzyme involved in the biotransformation of omeprazole. Following oral administration, omeprazole is rapidly absorbed in the gastrointestinal tract and highly bound to plasma proteins (>95%, mainly albumin). It is completely metabolized into inactive metabolites in the liver, and further excreted in faeces (mainly by biliary excretion) and in urine 11. Both CYP2C19 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1337 play important roles in the metabolism of omeprazole. However, the affinity for CYP3A4 is approximately 10 times less than for CYP2C19 12.
The significant impact of genetic polymorphism of CYP2C19 on omeprazole disposition has been reported in adults. The exposure to omeprazole is about 6–8‐fold higher in poor metabolizers (PM) compared with that of extensive metabolizers (EM) 10, 13, 14.
Methods
Clinical trial
The current investigation is an independent part of an omeprazole dose‐finding study in Caucasian infants with gastroesophageal reflux. This study was designed in accordance with legal requirements and the Declaration of Helsinki. The study protocol was approved by the Ethics Committee (Comité de Protection des Personnes, Hôpital Saint Louis, Paris, France) and was registered in the ClinicalTrials.gov registry (No. NCT01657578). Informed consent forms were obtained from patients' parents or guardians.
Omeprazole was administered once daily in the morning. According to the Bayesian approach, five doses of omeprazole were initially selected (1–3 mg kg–1 day–1). Pharmacokinetic samples were obtained between 0.5 and 4, and 4 and 12 h after administration of the first dose.
Analytical method of omeprazole and its metabolites
The analytical method of omeprazole and its metabolites was adapted from Rezk et al. 15. Briefly, plasma concentrations of omeprazole, 5‐hydroxy‐omeprazole and omeprazole sulfone were determined by high‐performance liquid chromatography with ultraviolet detection. The calibration curve ranged from 10 to 1000 ng ml–1 for omeprazole, and 25 to 1000 ng ml–1 for 5‐hydroxy‐omeprazole and omeprazole sulfone. The interday and intraday coefficients of variation (CVs) of controls were 3.2% and 2.8% for omeprazole, 13.9% and 3.0% for 5‐hydroxy‐omeprazole, and 9.0% and 3.4% for omeprazole sulfone, respectively. The lower limits of quantification (LOQ, associated interday and intraday CVs) were 10 ng ml–1 (8.1% and 5.3%) for omeprazole, 25 ng ml–1 (10.5% and 5.1%) for 5‐hydroxy‐omeprazole, and 25 ng ml–1 (6.0% and 4.0%) for omeprazole sulfone.
Genotyping
DNA was extracted using a QIAmp DNA Blood Mini Kit (Qiagen, Chatsworth, CA, USA) from whole blood sample. A Nanodrop spectrophotometer (Labtech, Palaiseau, France) was used to quantify DNA concentration. TaqMan allelic discrimination technique with 3′‐minor groove binding quencher probes (ABI Prism 7900; Applied Biosystems, Foster City, CA, USA) was used to perform CYP2C19 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=152#768 genotyping.
For CYP2C19 polymorphisms, primers and probes were obtained from Applied Biosystems (CYP2C19*2, rs4244285; CYP2C19*17, rs12248560). For ABCB1 polymorphisms (C3435T, rs28365046; G2677T/A, rs2032582; C1236T, rs1045642), the design of primers and probes and determination of technical conditions were carried out in our laboratory, as previously reported 16.
Population pharmacokinetic–pharmacogenetic modelling of omeprazole and its metabolites
Nonlinear mixed effects modelling program (NONMEM V 7.2; Icon Development Solutions, USA) was used for pharmacokinetic analysis. The estimation of pharmacokinetic parameters and their variabilities were carried out using a first‐order conditional estimation method with interaction option.
An exponential model was selected to estimate interindividual variability of the pharmacokinetic parameter and could be expressed as follows:
where ηi represent the individual pharmacokinetic parameter value of the ith subject; θmean and θi are the typical value of the parameter in the population and the variability between subjects (assumed to follow a normal distribution), respectively.
Covariate analysis used a forward and backward selection method. The stepwise covariate modelling 17 and likelihood ratio test were used to evaluate the potential impact of each variable on pharmacokinetic parameter. During the forward selection step, a covariate was included into the model if a significant (P < 0.05, χ2 distribution with one degree of freedom) decrease (reduction >3.84) in the objective function value (OFV) from the base model was achieved. Then, all the significant covariates were added simultaneously into the full model. At the backward selection step, the contribution of each significant covariate was removed one by one from the full model and was retained in the final model if a significant increase in the OFV was obtained.
Both graphical and statistical criteria were used for model validation. Goodness‐of‐fit plots, including observed (DV) vs. population (PRED) and individual prediction (IPRED); conditional weighted residuals (CWRES) vs. time and PRED were generalized based on model parameters for diagnostic purposes 18. The stability and performance of the final model was also evaluated by a nonparametric bootstrap with re‐sampling and replacement 500 times using PsN (v2.30). Then, the estimated parameters from the bootstrap procedure were compared with those estimated from the original data set. The final model was also assessed by normalized prediction distribution errors (NPDE) 19. One thousand simulation were performed using the final model parameters. NPDE results were shown graphically by the NPDE R package (v1.2), including QQ‐plot and histogram of the NPDE. The NPDE is assumed to follow normal distribution.
The predictive performance of the final model was further evaluated in external validation with an independent group of neonates and young infants (n = 8, number of samples = 12). The individual concentration of each patient was predicted by Bayesian estimation method (‘MAXEVAL = 0’ in the ESTIMATION step) using NONMEM. Prediction error (PE) was calculated using the following equation:
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 20, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 21, 22.
Results
Among fifty‐four patients who participated to the clinical study, fifty‐one with available pharmacokinetic data and DNA samples were included in the present analysis. The mean (standard deviation) gestational age at birth was 31.3 (3.9; range 24–41) weeks and the mean (standard deviation) weight was 2108 (609; range 780–3800) g. A summary of patients' characteristics is presented in Table 1.
Table 1.
Baseline characteristics and frequencies of CYP2C19 and ABCB1 genotypes in 51 neonates
| n | % | Mean (SD) | Median (range) | |
|---|---|---|---|---|
| Patients | 51 | |||
| Sex | 24M/27F | |||
| Gestational age at birth (weeks) | 31 (3.9) | 31 (24–41) | ||
| Current weight (g) | 2108 (609) | 2130 (780–3800) | ||
| PNA (days) | 41 (21) | 38 (7–87) | ||
| Omeprazole dose (mg/d) | 3.5 (1.6) | 3.6 (0.8–7.3) | ||
| Omeprazole dose (mg/kg/d) | 1.7 (0.6) | 2.0 (1.0–2.5) | ||
| CYP2C19 | ||||
| Poor metabolizer (PM) | ||||
| *2/*2 | 2 | 3.9 | ||
| Intermediate metabolizer (IM) | ||||
| *1/*2 | 7 | 13.7 | ||
| *2/*17 | 4 | 7.8 | ||
| Extensive metabolizer (EM) | ||||
| *1/*1 | 21 | 41.2 | ||
| Ultrarapid metabolizer (UM) | ||||
| *1/*17 | 14 | 27.5 | ||
| *17/*17 | 3 | 5.9 | ||
| ABCB1 | ||||
| C1236T | ||||
| C/C | 27 | 52.9 | ||
| C/T | 21 | 41.2 | ||
| T/T | 3 | 5.9 | ||
| G2677T/A | ||||
| G/G | 34 | 66.7 | ||
| G/T | 15 | 29.4 | ||
| G/A | ‐ | ‐ | ||
| T/T | 1 | 2.0 | ||
| T/A | 1 | 2.0 | ||
| C3435T | ||||
| C/C | 25 | 49.0 | ||
| C/T | 22 | 43.1 | ||
| T/T | 4 | 7.8 |
For population pharmacokinetic modelling, 73 plasma omeprazole concentrations from the 51 patients were available. The median concentrations were 217 [range from <LOQ (n = 6) to 2658] ng ml–1 for omeprazole, 111 [range from <LOQ (n = 7) to 842] ng ml–1 for 5‐hydroxy‐omeprazole and 92 [range from <LOQ (n = 22) to 764] ng ml–1 omeprazole sulfone, respectively. Half of the LOQ value was used to handle concentrations below the LOQ in pharmacokinetics modelling. Data fitted a one‐compartment parent and metabolite model with first‐order absorption and elimination (Figure 1). The model was parameterized in terms of absorption rate constant (Ka), apparent volumes of distribution of omeprazole, 5‐hydroxy‐omeprazole and omeprazole sulfone (V1/F, V2/F and V3/F), apparent clearances of omeprazole, 5‐hydroxy‐omeprazole and omeprazole sulfone (CLp/F, CLM1/F and CLM2/F) and clearances of omeprazole converted to 5‐hydroxy‐omeprazole and omeprazole sulfone (CLOMZ‐M1 and CLOMZ‐M2). Since 5‐hydroxy‐omeprazole and omeprazole sulfone were not administered alone and the true fractions of omeprazole converted to metabolites are unknown, CLOMZ‐M and V/F of metabolites are unidentifiable. Hence, the apparent volumes of distribution of the metabolites (V2/F and V3/F) were fixed to 1 litre 23. Consequently, estimated CLOMZ‐M was interpreted as the ratio of the clearance of omeprazole converted to metabolite to the distribution volume of metabolite. The estimation of CLOMZ/F by the final model gave a negligible value (<0.0001 l h–1). Interindividual variability was included in the model using an exponential relationship and was then estimated for Ka, V1/F, CLOMZ‐M1, CLOMZ‐M2, CLM1/F and CLM2/F. Residual variability was best included by an addictive relationship.
Figure 1.
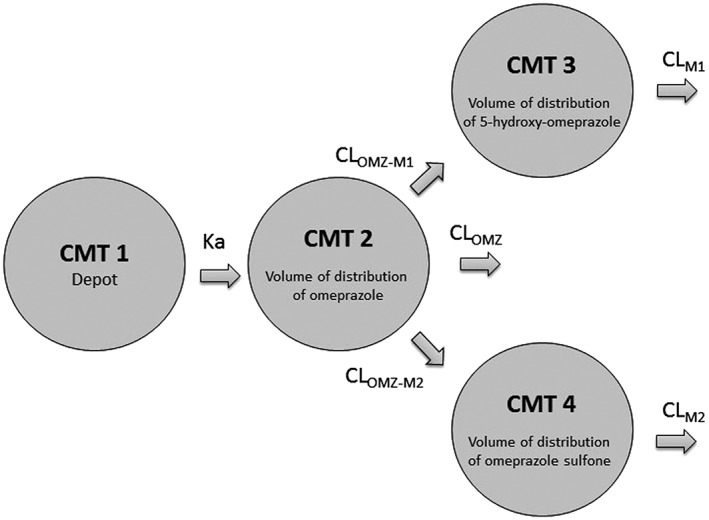
Pharmacokinetic model of omeprazole and its metabolites, where Ka is the absorption rate constant, CLOMZ‐M1 is the clearance of omeprazole converted to 5‐hydroxy‐omeprazole; CLOMZ‐M2 is the clearance of omeprazole converted to omeprazole sulfone; CLOMZ is the clearance of omeprazole; CLM1 is the clearance of 5‐hydroxy‐omeprazole; CLM2 is the clearance of omeprazole sulfone
The systematic covariate analysis identified ABCB1 C3435T genotype as the most important covariate influencing Ka, leading to a significant drop in the OFV (6.98 points, P < 0.01). For CLOMZ‐M1, CYP2C19 genotype was the most important covariate, causing a significant drop in the OFV of 18.25 points (P < 0.01). A further significant decrease in the OFV (6.32 points, P < 0.05) was achieved by including postnatal age on CLOMZ‐M1. The significant impact of these identified covariates was confirmed by the backward exclusion process.
Model diagnostics showed acceptable goodness‐of‐fit for the final model of omeprazole and metabolites. As shown in Figure 2A, predictions are unbiased. In the diagnostic plot of CWRES vs. time, no trend was demonstrated (Figure 2B). Moreover, the mean pharmacokinetic parameters obtained from the bootstrap were in agreement with respective values estimated from the final model, indicating that the final model was stable and the estimated parameters were accurate (Table 2). NPDE results are presented in Figure 2C,D. The mean and variance of NPDE were 0.001 and 1.310 for omeprazole, 0.119 and 0.899 for 5‐hydroxy‐omeprazole, and 0.149 and 0.860 for omeprazole sulfone indicating a good fit of the model to the individual data.
Figure 2.
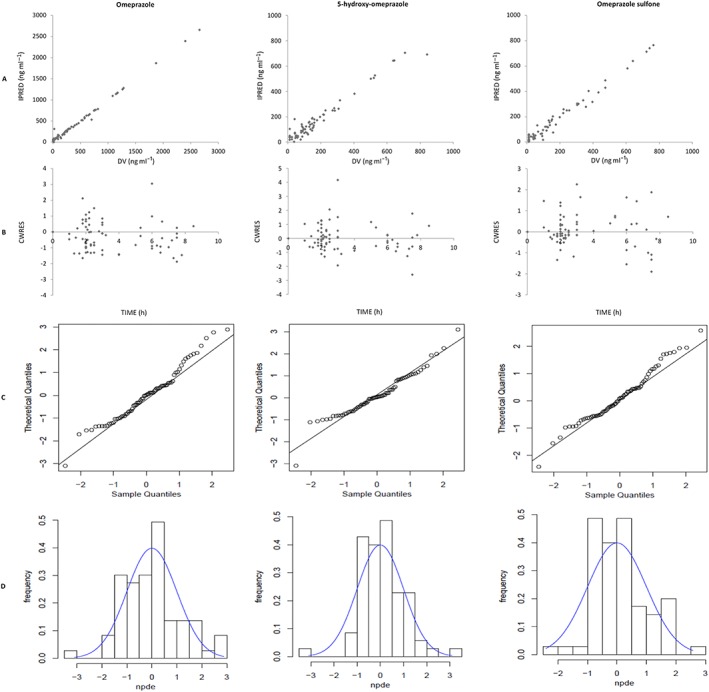
Model evaluation for omeprazole, 5‐hydroxy‐omeprazole and omeprazole sulfone: (A) Individual predicted (IPRED) vs. observed concentrations (DV); (B) conditional weighted residuals (CWRES) vs. time; (C) QQ‐plot of the distribution of the normalized prediction distribution errors (NPDE) vs. the theoretical N (0,1) distribution; (D) Histogram of the distribution of the NPDE, overlaid with the density of the standard Gaussian distribution
Table 2.
Population pharmacokinetic parameters of omeprazole and its metabolites and bootstrap validation
| Parameters | Final model | Bootstrap n = 500 | ||
|---|---|---|---|---|
| Final estimate | RSE (%) | Median | 5th–95th percentile | |
| Ka (h –1 ) | ||||
| Ka = θ1× F ABCB1 | 0.0497 | 29.4 | 0.0509 | 0.0294–0.0870 |
| F ABCB1 WT | 1 FIX | |||
| F ABCB1 HET | 1.86 | 44.4 | 1.78 | 0.86–3.47 |
| F ABCB1 MUT | 6.93 | 44.6 | 6.79 | 3.01–14.61 |
| V 1 /F (l) | 0.513 | 25.2 | 0.508 | 0.295–0.921 |
| V 2 /F (l) | 1 FIX | |||
| V 3 /F (l) | 1 FIX | |||
| CL OMZ‐M1 (l h –1 ) | ||||
| CL OMZ‐M1 = θ2× F PNA × F CYP2C19 | ||||
| θ2 | 0.658 | 21.4 | 0.676 | 0.474–0.943 |
| F CYP2C19 EM/UM | 1 FIX | |||
| F CYP2C19 IM | 0.449 | 25.6 | 0.463 | 0.299–0.726 |
| F CYP2C19 PM | 0.125 | 44.5 | 0.111 | 0.030–0.149 |
| F PNA = (PNA/38) θ3 | 0.472 | 29.2 | 0.482 | 0.185–0.795 |
| CL OMZ‐M2 (l h –1 ) | 0.140 | 8.4 | 0.142 | 0.119–0.165 |
| CL M1 /F (l h –1 ) | 0.846 | 18.4 | 0.863 | 0.614–1.167 |
| CL M2 /F (l h –1 ) | 0.130 | 27.3 | 0.135 | 0.062–0.206 |
| Interindividual variability (%) | ||||
| Ka | 130.0 | 29.4 | 124.9 | 94.1–150.1 |
| V 1 /F | 99.0 | 37.0 | 97.3 | 67.1–130.4 |
| CL OMZ‐M1 | 52.6 | 25.3 | 50.8 | 39.9–59.0 |
| CL OMZ‐M2 | 35.9 | 47.3 | 34.9 | 19.3–48.7 |
| CL M1 /F | 55.9 | 30.8 | 54.1 | 38.1–66.6 |
| CL M2 /F | 68.8 | 103.4 | 74.4 | 35.8–114.6 |
| Residual additive (ng ml –1 ) | 55.6 | 50.2 | 50.4 | 31.6–71.0 |
Ka: absorption rate constant (h–1); V1/F: apparent volume of distribution of omeprazole; V2/F: apparent volume of distribution of 5‐hydroxy‐omeprazole; V3/F: apparent volume of distribution of omeprazole sulfone; CLOMZ‐M1: clearance of omeprazole converted to 5‐hydroxy‐omeprazole; CLOMZ‐M2: clearance of omeprazole converted to omeprazole sulfone; CLM1/F: apparent clearance of 5‐hydroxy‐omeprazole; CLM2/F: apparent clearance of omeprazole sulfone; FABCB1: scaling factors applied for ABCB1 homozygous wild‐type (WT); heterozygous (HET) and homozygous mutant (MUT); FCYP2C19: scaling factors applied for CYP2C19 extensive metabolizer (EM), ultrarapid metabolizer (UM); intermediate metabolizer (IM) and poor metabolizer (PM); PNA: postnatal age in days
The predictive performance of the developed model was further checked in the external validation with an independent group of eight neonates and young infants. The mean gestational age and weight were 32 (range 28.0–36.0) weeks and 2178 (range 780–2750) g, respectively. A total of 12 concentrations were available and ranged from <LOQ (n = 1) to 2021 ng ml–1 for omeprazole, 41–735 ng ml–1 for 5‐hydroxy‐omeprazole and <LOQ (n = 1) to 665 ng ml–1 omeprazole sulfone. The median PEs were 4.6% (5th–95th percentile range: –4.5 to 24.6%) for omeprazole, 2.0% (5th–95th percentile: –5.4 to 20.9%) for 5‐hydroxy‐omeprazole, and –2.6% (5th–95th percentile: –15.3 to 55.1%) for omeprazole sulfone, indicating a good and consistent predictive performance of the developed model on new patients.
The final population pharmacokinetic parameters are given in Table 2. CLOMZ‐M1 increases with postnatal age. In CYP2C19 poor and intermediate metabolizers, model‐predicted CLOMZ‐M1 are 12.5% and 44.9% of the value in an extensive/ultrarapid metabolizer, respectively. Figure 3 shows CYP2C19 genotype‐based ontogeny of CLOMZ‐M1. Model‐predicted Ka is 6.93 times higher in ABCB1 homozygous mutant patients, 1.86 times higher in ABCB1 heterozygous patients than that in ABCB1 homozygous wild‐type patients.
Figure 3.
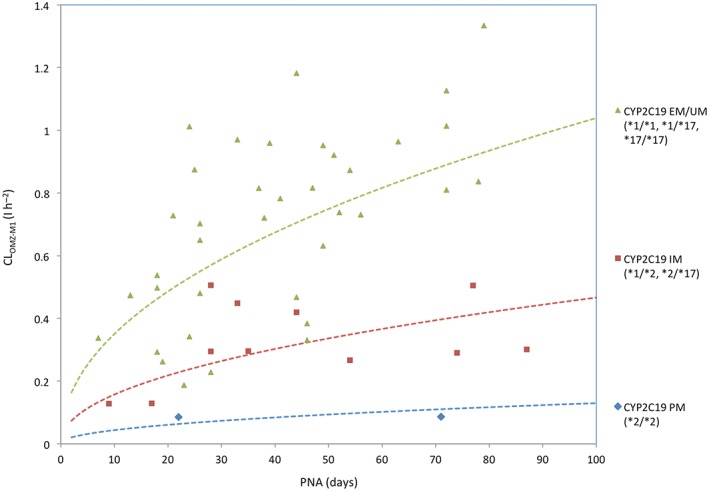
CYP2C19 genotype‐based ontogeny of clearance of omeprazole converted to 5‐hydroxy‐omeprazole. CLOMZ‐M1: Clearance of omeprazole converted to 5‐hydroxy‐omeprazole; EM: extensive metabolizer; UM: ultrarapid metabolizer; IM: intermediate metabolizer; PM: poor metabolizer; PNA: postnatal age. Dashed lines represent the typical ontogeny patterns of clearance of omeprazole converted to 5‐hydroxy‐omeprazole according to CYP2C19 genotype
Discussion
The developmental pharmacogenetics of CYP2C19 was studied for the first time in neonates and young infants using omeprazole as a probe drug to quantify the impact of age and pharmacogenetics on the disposition of omeprazole and its metabolites. Our results showed that a one‐compartment parent and metabolites model with first‐order absorption and elimination was optimal for omeprazole population pharmacokinetic modelling.
The estimated value of clearance of omeprazole in unchanged form was nearly 0, indicating that, like in adults and children, omeprazole is extensively metabolized by the liver in neonates. The median (range) of weight‐normalized clearances of omeprazole converted to 5‐hydroxy‐omeprazole and omeprazole sulfone are 0.287 (0.036–1.711) and 0.142 (0.065–0.316) l h–1 kg–1, respectively. The median (range) of apparent weight‐normalized volume distribution of omeprazole is 0.24 (0.09–0.77) l kg–1, similar to that reported in adults 24.
Our results demonstrate that CLOMZ‐M1 increases with postnatal age. In CYP2C19 poor and intermediate metabolizers, model‐predicted CLOMZ‐M1 are 12.5% and 44.9% of the value in extensive/ultrarapid metabolizers, respectively. This observation is well correlated with in vitro and in vivo findings. As early as 8 weeks of gestation, CYP2C19 protein and catalytic activities could achieve 12–15% of mature values and did not change throughout the prenatal period until birth. Its expression increased over the first 5 postnatal months 6, suggesting that postnatal age is a determinant of CYP2C19 ontogeny. In addition, large variability in CYP2C19 expression was observed during the ontogeny process, as illustrated by approximately 21‐fold variability between 5 months and 10 years of age 6. Using developmental pharmacokinetic modelling approach, the complex effects of CYP2C19 pharmacogenetics and ontogeny were incorporated in omeprazole metabolism. This semiphysiological model was more powerful to evaluate the impact of developmental pharmacogenetics of CYP2C19 on omeprazole pharmacokinetics, as the metabolite pathways were quantitatively described in this model. The contribution of developmental pharmacogenetics of CYP2C19 might have potential clinical implication. Reduced drug clearance leads to increased drug exposure, which might increase the risk of adverse events. The clinical utility of pharmacogenetic‐based dosing regimen of omeprazole in neonates needs to be evaluated in future study. A genetic based ontogeny process is demonstrated in Figure 3. In vivo, the combined impact of CYP2C19 pharmacogenetic polymorphism and ontogeny on pantoprazole pharmacokinetics was also demonstrated in neonates. Ward et al. reported that pantoprazole apparent clearance was increased with postnatal age in neonates and infants aged 1–19 weeks and the two CYP2C19 PM patients had a substantially higher AUC than EM 25.
As CYP3A4 and P‐gp are also involved in the metabolism and absorption of omeprazole 26, their impacts were also evaluated in the present study. ABCB1 is expressed at the apical surface of mature enterocytes in the small intestine to prevent the absorption of omeprazole from the intestinal lumen by active extrusion 26. In the duodenum, mRNA and P‐gp protein were present in the enterocytes from age 1 month with a high interindividual variability and no influence of age was observed 27. To our knowledge, the impact of age on ABCB1 expression in intestinal tissue remains unknown in the neonatal period. There are limited data in other tissues and cell types, however, the results are contradictory. Pilarski et al. evaluated ABCB1 activity in T lymphocytes and found that its activity was lower in young children (<10 years) than in older patients with levels increasing in adults up to the age of 55 years 28. In contrast, Machado et al. demonstrated that the highest level of ABCB1 activity was achieved in cord blood. Its activity decreased rapidly after birth and remained in a slower way throughout life 29. This finding was partly supported by Giraud et al., who reported that ABCB1 activity in T and natural killer cell types achieved the peak value at birth, and decreased until 6 months after birth and stabilized between 6 months and 2 years 30. In vivo, Fanta et al. found that ABCB1 genotype had significant impacts on oral bioavailability of cyclosporine in paediatric renal transplant recipients older than 8 years, thereby, affected cyclosporine oral dose requirement, however, this association was not significant in young children 31. Our data illustrate that ABCB1 genotype has significant impact on the absorption rate constant of omeprazole, but not on the amount of absorption. Ka is significantly higher in ABCB1 homozygous mutant and heterozygous patients than that in ABCB1 homozygous wild‐type patients.
CYP3A is abundant in the liver and gut and contributes to the first‐pass metabolism of many orally administrated drugs. In vitro, CYP3A mRNA expression decreases and protein level increases with age, reflecting a post‐transcriptional regulatory mechanism 32. In vivo, midazolam oral bioavailability is significantly higher in preterm infants than in adults, suggesting developmentally low intestinal and hepatic CYP3A activities 33. Our results did not show significant impact of age on clearance of omeprazole converted to omeprazole sulfone, indicating that the activity of CYP3A4 did not change dramatically during the neonatal period. A pooled analysis with children and adults would be required to illustrate the developmental pattern of CYP3A4 across a wide age range.
Our study had some limitations. As the main objective of our study is to describe the developmental pattern of CYP2C19, the clinical outcomes were not assessed. Developmental pharmacokinetics and pharmacodynamics need to be evaluated in a following study. The number of CYP2C19 poor metabolizer patients was very limited in Caucasian population. This might influence the accurate quantification of developmental pattern for this special subgroup of patients. The ontogeny of CYP2C19 in other ethnic background children is urgently required. In addition, due to the limited number of patients, there was not sufficient variability to conclude the impact of developmental pharmacogenetics of CYP3A4 on omeprazole pharmacokinetics. Moreover, the unexplained variability is still large in our study, indicating the significant contribution of other covariate on omeprazole pharmacokinetics, such as albumin, renal function etc. These missing covariates should be further evaluated.
In conclusion, the developmental pharmacogenetics of CYP2C19 are quantitatively described in neonates and young infants using omeprazole as a probe drug. A population pharmacokinetic and pharmacogenetic model was developed and validated for omeprazole and its metabolites in neonates and young infants. CYP2C19 was predominantly involved in the metabolism of omeprazole despite the relatively low activity as compared to adults. The developmental clearance of omeprazole converted to 5‐hydroxy‐omeprazole was dependent on individual CYP2C19 genotype. ABCB1 polymorphism had a significant impact on the absorption rate constant of omeprazole. Our findings emphasize the importance of considering multiple routes of drug biotransformation and their relative quantitative importance when evaluating developmental pharmacogenetics of drugs.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). None of the authors have any other relationships or activities that could appear to have influenced the submitted work.
This work is supported by National Science and Technology Major Projects for “Major New Drugs Innovation and Development (2017ZX09304029–002), Young Taishan Scholars Program, Young Scholars Program of Shandong University, Global Research in Paediatrics Network of Excellence (GRIP, EU‐funded FP7 project, Grant Agreement number 261060) and a grant from the French Ministry of Health (AOM05123).
Contributors
W.Z. and E.J.‐A. designed the research. S.L. and V.B. participated to the pharmacokinetic study and contributed to the discussion and interpretation of the results. W.Z. wrote the manuscript, which was critically reviewed by all authors.
Zhao, W. , Leroux, S. , Biran, V. , and Jacqz‐Aigrain, E. (2018) Developmental pharmacogenetics of CYP2C19 in neonates and young infants: omeprazole as a probe drug. Br J Clin Pharmacol, 84: 997–1005. doi: 10.1111/bcp.13526.
Contributor Information
Wei Zhao, Email: zhao4wei2@hotmail.com.
Evelyne Jacqz‐Aigrain, Email: evelyne.jacqzaigrain@gmail.com.
References
- 1. Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Drug metabolism for the paediatrician. Pharmacol Ther 2008; 118: 250–267. [DOI] [PubMed] [Google Scholar]
- 2. de Wildt SN, Tibboel D, Leeder JS. Drug metabolism for the paediatrician. Arch Dis Child 2014; 99: 1137–1142. [DOI] [PubMed] [Google Scholar]
- 3. Kearns GL, Abdel‐Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology – drug disposition, action, and therapy in infants and children. N Engl J Med 2003; 349: 1157–1167. [DOI] [PubMed] [Google Scholar]
- 4. Goldstein JA. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Br J Clin Pharmacol 2001; 52: 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kearns GL, Leeder JS, Gaedigk A. Impact of the CYP2C19*17 allele on the pharmacokinetics of omeprazole and pantoprazole inchildren: evidence for a differential effect. Drug Metab Dispos 2010; 38: 894–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koukouritaki SB, Manro JR, Marsh SA, Stevens JC, Rettie AE, McCarver DG, et al Developmental expression of human hepatic CYP2C9 and CYP2C19. J Pharmacol Exp Ther 2004; 308: 965–974. [DOI] [PubMed] [Google Scholar]
- 7. Knebel W, Tammara B, Udata C, Comer G, Gastonguay MR, Meng X. Population pharmacokinetic modeling of pantoprazole in pediatric patients from birth to 16 years. J Clin Pharmacol 2011; 51: 333–345. [DOI] [PubMed] [Google Scholar]
- 8. Ward RM, Kearns GL. Proton pump inhibitors in pediatrics: mechanism of action, pharmacokinetics, pharmacogenetics, and pharmacodynamics. Paediatr Drugs 2013; 15: 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kearns GL, Andersson T, James LP, Gaedigk A, Kraynak RA, Abdel‐Rahman SM, et al Omeprazole disposition in children following single‐dose administration. J Clin Pharmacol 2003; 43: 840–848. [DOI] [PubMed] [Google Scholar]
- 10. Tafuri G, Trotta F, Leufkens HG, Martini N, Sagliocca L, Traversa G. Off‐label use of medicines in children: can available evidence avoid useless paediatric trials? The case of proton pump inhibitors for the treatment of gastroesophageal reflux disease. Eur J Clin Pharmacol 2009; 65: 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Litalien C, Théorêt Y, Faure C. Pharmacokinetics of proton pump inhibitors in children. Clin Pharmacokinet 2005; 44: 441–466. [DOI] [PubMed] [Google Scholar]
- 12. Andersson T. Pharmacokinetics, metabolism and interactions of acid pump inhibitors. Focus on omeprazole, lansoprazole and pantoprazole. Clin Pharmacokinet 1996; 31: 9–28. [DOI] [PubMed] [Google Scholar]
- 13. Sakai T, Aoyama N, Kita T, Nishiguchi K, Nishitora Y, Hohda T, et al CYP2C19 genotype and pharmacokinetics of three proton pump inhibitors in healthy subjects. Pharm Res 2001; 18: 721–727. [DOI] [PubMed] [Google Scholar]
- 14. Shirai N, Furuta T, Moriyama Y, Okochi H, Kobayashi K, Takashima M, et al Effects of CYP2C19 genotypic differences in the metabolism of omeprazole and rabeprazole on intragastric pH. Aliment Pharmacol Ther 2001; 15: 1929–1937. [DOI] [PubMed] [Google Scholar]
- 15. Rezk NL, Brown KC, Kashuba AD. A simple and sensitive bioanalytical assay for simultaneous determination of omeprazole and its three major metabolites in human blood plasma using RP‐HPLC after a simple liquid‐liquid extraction procedure. J Chromatogr B Analyt Technol Biomed Life Sci 2006; 844: 314–321. [DOI] [PubMed] [Google Scholar]
- 16. Zhao W, Elie V, Roussey G, Brochard K, Niaudet P, Leroy V, et al Population pharmacokinetics and pharmacogenetics of tacrolimus in de novo pediatric kidney transplant recipients. Clin Pharmacol Ther 2009; 86: 609–618. [DOI] [PubMed] [Google Scholar]
- 17. De Cock RF, Piana C, Krekels EH, Danhof M, Allegaert K, Knibbe CA. The role of population PK‐PD modelling in paediatric clinical research. Eur J Clin Pharmacol 2011; 67 (S1): 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res 2007; 24: 2187–2197. [DOI] [PubMed] [Google Scholar]
- 19. Brendel K, Comets E, Laffont C, Laveille C, Mentré F. Metrics for external model evaluation with an application to the population pharmacokinetics of gliclazide. Pharm Res 2006; 23: 2036–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, et al The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 2017; 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morris D, Podolski J, Kirsch A, Wiehle R, Fleckenstein L. Population pharmacokinetics of telapristone (CDB‐4124) and its active monodemethylated metabolite CDB‐4453, with a mixture model for total clearance. AAPS J 2011; 13: 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stedman CA, Barclay ML. Review article: comparison of the pharmacokinetics, acid suppression and efficacy of proton pump inhibitors. Aliment Pharmacol Ther 2000; 14: 963–978. [DOI] [PubMed] [Google Scholar]
- 25. Ward RM, Tammara B, Sullivan SE, Stewart DL, Rath N, Meng X, et al Single‐dose, multiple‐dose, and population pharmacokinetics of pantoprazole in neonates and preterm infants with a clinical diagnosis of gastroesophageal reflux disease (GERD). Eur J Clin Pharmacol 2010; 66: 555–561. [DOI] [PubMed] [Google Scholar]
- 26. Pauli‐Magnus C, Rekersbrink S, Klotz U, Fromm MF. Interaction of omeprazole, lansoprazole and pantoprazole with P‐glycoprotein. Naunyn Schmiedebergs Arch Pharmacol 2001; 364: 551–557. [DOI] [PubMed] [Google Scholar]
- 27. Fakhoury M, Litalien C, Medard Y, Cavé H, Ezzahir N, Peuchmaur M, et al Localization and mRNA expression of CYP3A and P‐glycoprotein in human duodenum as a function of age. Drug Metab Dispos 2005; 33: 1603–1607. [DOI] [PubMed] [Google Scholar]
- 28. Pilarski LM, Paine D, McElhaney JE, Cass CE, Belch AR. Multidrug transporter P‐glycoprotein 170 as a differentiation antigen on normal human lymphocytes and thymocytes: modulation with differentiation stage and during aging. Am J Hematol 1995; 49: 323–335. [DOI] [PubMed] [Google Scholar]
- 29. Machado CG, Calado RT, Garcia AB, Falcão RP. Age‐related changes of the multidrug resistance P‐glycoprotein function in normal human peripheral blood T lymphocytes. Braz J Med Biol Res 2003; 36: 1653–1657. [DOI] [PubMed] [Google Scholar]
- 30. Giraud C, Declèves X, Perrot JY, Manceau S, Pannier E, Firtion G, et al High levels of P‐glycoprotein activity in human lymphocytes in the first six months of life. Clin Pharmacol Ther 2009; 85: 289–295. [DOI] [PubMed] [Google Scholar]
- 31. Fanta S, Niemi M, Jönsson S, Karlsson MO, Holmberg C, Neuvonen PJ, et al Pharmacogenetics of cyclosporine in children suggests an age‐dependent influence of ABCB1 polymorphisms. Pharmacogenet Genomics 2008; 18: 77–90. [DOI] [PubMed] [Google Scholar]
- 32. Mooij MG, de Koning BA, Huijsman ML, de Wildt SN. Ontogeny of oral drug absorption processes in children. Expert Opin Drug Metab Toxicol 2012; 8: 1293–1303. [DOI] [PubMed] [Google Scholar]
- 33. de Wildt SN, Kearns GL, Hop WC, Murry DJ, Abdel‐Rahman SM, van den Anker JN. Pharmacokinetics and metabolism of oral midazolam in preterm infants. Br J Clin Pharmacol 2002; 53: 390–392. [DOI] [PMC free article] [PubMed] [Google Scholar]