

Abstract
Aims
The aims of the present study were to assess the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of BMS‐962212, a first‐in‐class factor XIa inhibitor, in Japanese and non‐Japanese healthy subjects.
Methods
This was a randomized, placebo‐controlled, double‐blind, sequential, ascending‐dose study of 2‐h (part A) and 5‐day (part B) intravenous (IV) infusions of BMS‐962212. Part A used four doses (1.5, 4, 10 and 25 mg h−1) of BMS‐962212 or placebo in a 6:2 ratio per dose. Part B used four doses (1, 3, 9 and 20 mg h−1) enrolling Japanese (n = 4 active, n = 1 placebo) and non‐Japanese (n = 4 active, n = 1 placebo) subjects per dose. The PK, PD, safety and tolerability were assessed throughout the study.
Results
BMS‐962212 was well tolerated; there were no signs of bleeding, and adverse events were mild. In parts A and B, BMS‐962212 demonstrated dose proportionality. The mean half‐life in parts A and B ranged from 2.04 to 4.94 h and 6.22 to 8.65 h, respectively. Exposure‐dependent changes were observed in the PD parameters, activated partial thromboplastin time (aPTT) and factor XI clotting activity (FXI:C). The maximum mean aPTT and FXI:C change from baseline at 20 mg h−1 in part B was 92% and 90%, respectively. No difference was observed in weight‐corrected steady‐state concentrations, aPTT or FXI:C between Japanese and non‐Japanese subjects (P > 0.05).
Conclusion
BMS‐962212 has tolerability, PK and PD properties suitable for investigational use as an acute antithrombotic agent in Japanese or non‐Japanese subjects.
Keywords: anticoagulation, factor XIa inhibition, Japanese, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject
Factor XI (FXI) is a component of the intrinsic pathway of the coagulation cascade and has been postulated to play an important role in thrombosis but only a minor role in haemostasis.
Clinical and experimental data suggest that FXIa inhibition may provide a novel mechanism for systemic anticoagulation, without increasing the risk of clinically significant bleeding.
In a phase II study using an antisense oligonucleotide, FXI inhibition was demonstrated to have superior efficacy to enoxaparin in patients undergoing total knee arthroplasty, without increasing bleeding risk; however, it took several weeks to achieve therapeutic anticoagulation levels in this study.
What this Study Adds
The present study is the first described clinical experience with an active site‐directed factor XIa small molecule inhibitor (BMS‐962212).
BMS‐962212 was well tolerated, with fast onset of pharmacodynamic (PD) responses and rapid elimination. Exposure‐dependent increases in activated partial thromboplastin time, and exposure‐dependent decreases in FXI clotting activity were observed.
No significant differences in pharmacokinetics or PD were observed in Japanese subjects compared with non‐Japanese subjects.
Introduction
The clinical benefits of anticoagulant therapy are well established for the prevention of thrombotic events in patients with existing cardiovascular disease. In the last decade, anticoagulant therapy has expanded also to include prevention of deep‐vein thrombosis and pulmonary embolism following knee or hip surgery, resulting in significant reductions in postsurgery mortality and morbidity. The growing role of anticoagulant therapy in the treatment and prevention of thrombosis is continually balanced by the potential complications and risks associated with excess bleeding. Although improvements in the therapeutic index of anticoagulant therapy have been made with the advent of small‐molecule inhibitors of factor Xa or thrombin relative to vitamin K antagonists, dose‐dependent bleeding continues to be observed 1 and therefore improving the risk‐to‐benefit ratio remains a central goal for antithrombotic drug discovery.
http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2360 is a component of the intrinsic pathway of the coagulation cascade and has been postulated to play an important role in thrombosis but only a minor role in haemostasis 2. Epidemiological data collected over the past 15 years indicate that FXI plays a greater part in thrombosis than in haemostasis. Epidemiology studies suggest that FXI deficiency may be associated with a lower risk of cardiovascular events and venous thromboembolism 3, 4, 5, whereas elevated FXI levels have been associated with an increased risk of thrombosis 6.
The attractiveness of FXI as a therapeutic target for anticoagulation is further supported by the lack of significant bleeding associated with FXI modulation. Spontaneous bleeding in humans with a deficiency in FXI (haemophilia C) is generally rare, and when observed it is limited to tissues with high fibrinolytic activity (e.g. oral cavity, nose and urinary tract) after episodes of direct injury or trauma 7. In fact, an increased tendency for bleeding has only been documented postsurgery in patients with very severe FXI deficiency (≤15–20 IU dl−1) 8; hence, despite the low risk of associated bleeding, rapid reversibility of FXI inhibition is still likely to be needed for situations of major trauma and emergency surgeries.
In preclinical models, inhibitors of activated FXI (FXIa) reduce thrombus formation without affecting haemostasis 2. The lack of a major bleeding diathesis in humans with severe FXI deficiency and the preclinical demonstration of robust antithrombotic efficacy with a direct FXIa inhibitor without significant bleeding liability provide a compelling rationale to investigate this mechanism for antithrombotic treatment in various therapeutic indications requiring anticoagulation. According to the scientific evidence accumulated so far, the inhibition of FXI may therefore provide a novel mechanism for systemic anticoagulation without increasing the risk of clinically significant bleeding in a variety of conditions predisposing to a high risk of thrombotic events 6, 9, 10, 11.
BMS‐962212 is a small‐molecule, selective FXIa inhibitor 12. In rabbit models, BMS‐962212 shows antithrombotic efficacy at doses which preserve haemostasis. Allometric scaling from pharmacokinetic (PK) studies in several preclinical species predicted a moderate clearance, with rapid elimination from the central compartment following the end of infusion. Based on PK and pharmacodynamic (PD) results from these preclinical models, an exposure dose range for the first‐in‐human study was selected. Preclinical studies suggest that the disposition of BMS‐962212 is primarily an active process that is mediated by transporters, organic anion transporting polypeptide (OATP) 1B1 and OATP1B3. The major route of elimination of BMS‐962212 is by both biliary and urinary excretion, and in vivo and in vitro data suggest that both multidrug resistance‐associated protein 2 and breast cancer resistance protein may play a role in the biliary excretion of BMS‐962212.
BMS‐962212 is envisioned as a safe and effective acute care antithrombotic agent for use in the hospital setting. A parenteral agent with a relatively short PK and PD half‐life is expected to achieve higher efficacy, faster onset and better control of antithrombotic effects compared with oral agents. To the best of our knowledge, this is the first publication describing the safety, PK and PD of a reversible small‐molecule FXIa inhibitor in humans.
The primary objective of the present study was to assess the safety and tolerability of BMS‐962212 when administered as intravenous (IV) infusions of 2 h or 5 days in healthy subjects. Secondary objectives included assessment of PK and dose proportionality of escalating doses of BMS‐962212; assessment of PD effects (known to be associated with anticoagulation) of escalating doses of BMS‐962212 in healthy subjects, as measured by activated partial thromboplastin time (aPTT) and FXI clotting activity (FXI:C); and assessment of PK and PD effects of 5‐day IV infusions of escalating doses of BMS‐962212 in Japanese subjects compared with non‐Japanese subjects.
Methods
The study was conducted in accordance with Good Clinical Practice (GCP) guidelines, as defined by the International Conference on Harmonization (ICH) and in accordance with the ethical principles underlying European Union Directive 2001/20/EC and the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The study was registered with ClinicalTrials.gov (NCT03197779). The protocols were approved by the Independent Ethics Committee and the Institutional Review Board (IRB) of IntegReview Ethical Review Board (Austin, TX, USA) and Aspire IRB (Santee, CA, USA) and the Food and Drug Administration. The approval number of the State Food and Drug Administration was 0910–0014. Prior to the beginning of the study, all subjects provided written informed consent. The study was conducted at three sites: West Coast Clinical Trials (WCCT) Global, LLC Cypress, CA, USA; California Clinical Trials Medical Group, Glendale, CA, USA; and Parexel International Baltimore EPCU, Baltimore, MD, USA.
Study design
This was a two‐part, randomized, double‐blind, placebo‐controlled, sequential ascending‐dose IV infusion study of BMS‐962212 in healthy subjects. The study evaluated BMS‐962212 following administration as a 2 h IV infusion (part A) and a 5‐day continuous IV infusion (part B). For parts A and B, dose panels were enrolled sequentially. Part A included four dose panels that evaluated continuous, fixed‐rate IV infusions of BMS‐962212 for 2 h at doses of 1.5, 4, 10 and 25 mg h−1 (panels 1, 2, 3 and 4). Following a transient increase in serum creatinine levels in some subjects in panel 1 (1.5 mg h−1), the protocol was amended to re‐evaluate this dose in a larger panel 1A prior to proceeding with dose escalation. In panel 1A, a 1.5 mg h−1 dose of BMS‐962212 was administered to 10 subjects (10 active and 10 placebo) concomitantly with iohexol [where iohexol plasma clearance was used to measure glomerular filtration rate (GFR), to investigate if BMS‐962212 had an impact on renal function]. Panel 1A data are available in the Supporting Information. Panels 1, 2, 3 and 4 each included eight subjects, who were randomized in a 3:1 ratio to receive BMS‐962212 or placebo. Subjects were admitted to the clinical site on day −2, dosed on day 1 and discharged on day 3. Part B commenced after PK, PD and safety data had been reviewed for the first four panels in part A (panels 1, 1A, 2 and 3) and had been deemed safe and sufficient to project appropriate exposures following 5‐day infusions. Part B included four dose panels that evaluated continuous, fixed‐rate IV infusions of BMS‐962212 for 5 days at doses of 1, 3, 9 and 20 mg h−1 (panels 5, 6, 7 and 8). Each panel in part B comprised 10 subjects, including five non‐Japanese subjects and five subjects of first‐degree Japanese origin. Within each dose panel, subjects were randomized in a 4:1 ratio (stratified by Japanese ancestry) to receive BMS‐962212 or placebo. Subjects were admitted to the clinical facility on day −2, dosed on days 1–6 and monitored for an additional 48 h following termination of the infusion before discharge on day 8.
Subjects
Men and women of non‐childbearing potential, with an age range of 18–45 years and a body mass index (BMI) of 18–32 kg m−2, who were healthy, as determined by medical history, physical examination, electrocardiogram (ECG) and clinical laboratory evaluations, were eligible to participate in the study. Inclusion criteria were updated following an amendment to the protocol after panel 1. Of note, subjects in panel 1 had a wider age range, of 18–55 years, compared with the remaining panels. Part B included a subset of subjects of Japanese origin, defined as residents of the United States who were first‐generation Japanese (born in Japan and not living outside of Japan for >10 years) and eating a standard Japanese diet. Non‐Japanese subjects in part B could include up to one Asian subject (including from India) per dose panel. A more detailed summary of the inclusion criteria, excluding those used for panel 1, is presented in Table S1.
Safety assessments
Safety was evaluated based on a medical review of adverse events (AEs) and the results of clinical laboratory tests, vital signs, ECGs, telemetry and physical examination. Based on observations of interstitial nephritis at supraclinical dose levels in nonclinical studies, the clinical laboratory evaluations in the present study comprised an extended renal safety assessment, including serum blood urea nitrogen (BUN), serum creatinine and several biomarkers of renal injury, including urinary neutrophil gelatinase‐associated lipocalin (NGAL), urinary monocyte chemoattractant protein 1 (MCP‐1) and serum cystatin C. AE severity was classified according to Medical Dictionary for Regulatory Activities criteria. The estimated GFR (eGFR) was calculated based on the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation using serum creatinine.
Sample collection
Sample collection time points, as well as handling and storage of the samples, are described in more detail in the Supporting Information.
Coagulation assays
Coagulation assays to investigate aPTT, FXI:C and prothrombin time/international normalized ratio (PT/INR) were performed.
Bioanalytical assay for BMS‐962212
Plasma samples were measured for BMS‐962212 by a validated liquid chromatography–tandem mass spectrometry method using a liquid–liquid extraction method. The analytical range was established at 0.1–100 ng ml−1 and a linear x−2 weighted fitting was used for the standard curve. The overall accuracy was demonstrated for the quality control samples with relative standard deviation as a percent (%Dev) ≤3.7%. Between‐run and within‐run precision were ≤4.2% and 7.0%, respectively. For the purposes of analysis, when BMS‐962212 plasma concentrations were lower than the lower limit of quantitation (LLQ; 0.1 ng ml−1), the value was set to one‐half the LLQ or 0.05 ng ml−1.
PK parameters
The PK parameters of BMS‐962212 were calculated from the plasma concentration–time data using noncompartmental methods in Phoenix WINNONLIN v7.0 (Certara, Princeton, NJ, USA). The area under the concentration–time curve (AUC) was calculated using the linear‐up, log‐down method within Phoenix. The terminal half‐life (t1/2) was estimated as ln2/λz, in which the slope of the terminal phase of the plasma concentration–time profile (λz) was determined by the least‐squares method (log linear regression of at least three data points) with a weighting factor of 1. Steady state was defined as the time to reach five times the t1/2 for each individual. In the 2‐h IV infusion part of the study (part A), the PK parameters assessed included: AUC for the sampling period (AUC0–t), where t was 24 h; AUC extrapolated to infinity (AUCinf); maximum plasma concentration (Cmax); time to Cmax (Tmax); t1/2; clearance (CL); and volume of distribution during the terminal phase (Vz). The PK parameters determined in the 5‐day IV infusion part of the study (part B) included: AUC0–t, where t was 144 h; Cmax; concentration at steady state (Css); Tmax; t1/2; CL; and volume of distribution at steady state (Vss). Weight‐corrected Css was calculated in part B by multiplying the Css of each individual by their weight.
Statistical analyses
Statistical analyses and reported PK and PD data were performed on all subjects who completed the study. The PK parameters AUCinf, AUC0–t, Cmax, t1/2, CL and Vz in Part A, and Cmax, t1/2, AUC0–t, AUCinf and Css in part B were analysed assuming normally distributed data. Summary PK parameters were described as the geometric mean [percentage coefficient of variation (%CV)]. To investigate whether PK parameters of Cmax, Css (part B only) and AUCinf increased dose proportionally, an analysis of variance was performed on the log‐transformed values in part A and part B. Differences between Japanese and non‐Japanese subjects in each dose panel for the PK parameters of Css and t1/2, and PD parameters of aPTT and FXI:C were analysed using an independent samples t‐test. Summary PD parameters were described as the mean and standard error of the mean, and as the percentage change from baseline.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 13, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 14.
Results
Demographics
All subjects completed part A of the study (n = 24 active, n = 8 placebo). Forty subjects completed part B (n = 32 active, n = 8 placebo). Two subjects, both on active treatment, did not complete the study and were subsequently replaced. These two subjects withdrew consent for reasons unrelated to treatment, and both were in part B (1 mg h−1 and 9 mg h−1 dose panels, respectively). All subjects in the study were male, and the age range in part A was (19–50 years) and in part B (20–45 years). Table 1 outlines the demographics and disposition of all subjects enrolled in the study.
Table 1.
Demographics and disposition of subjects
| Part A (2‐h IV infusion) | PLA (N = 8) | 1.5 mg h −1 (N = 6) | 4 mg h −1 (N = 6) | 10 mg h −1 (N = 6) | 25 mg h −1 (N = 6) | Total (N = 32) |
|---|---|---|---|---|---|---|
| Age, years | ||||||
| Mean (SD) | 36.1 (8.46) | 30.7 (8.48) | 32.2 (4.02) | 27.3 (6.25) | 29.0 (2.83) | 31.2 (7.17) |
| Gender, n (%) | ||||||
| Male | 8 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 32 (100) |
| Female | 0 | 0 | 0 | 0 | 0 | 0 |
| Weight, kg | ||||||
| Mean (SD) | 79.7 (8.55) | 77.2 (10.7) | 83.3 (13.3) | 72.6 (17.5) | 80.7 (19.5) | 78.8 (13.7) |
| Race, n (%) | ||||||
| White | 4 (50.0) | 1 (16.7) | 1 (16.7) | 3 (50.0) | 1 (16.7) | 10 (32.2) |
| Black/African American | 3 (37.5) | 3 (50.0) | 2 (33.3) | 3 (50.0) | 4 (66.7) | 15 (46.9) |
| Native Hawaiian or other Pacific Islander | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (3.1) |
| Other | 1 (12.5) | 2 (33.3) | 3 (50.0) | 0 | 0 | 6 (18.8) |
| Part B (5‐day IV infusion) | PLA (N = 8) | 1 mg h−1 (N = 9) | 3 mg h−1 (N = 8) | 9 mg h−1 (N = 9) | 20 mg h−1 (N = 8) | Total (N = 42) |
|---|---|---|---|---|---|---|
| Age, years | ||||||
| Mean (SD) | 32.4 (4.00) | 35.0 (8.26) | 33.5 (7.13) | 31.9 (4.51) | 31.4 (8.42) | 32.9 (6.52) |
| Gender, n (%) | ||||||
| Male | 8 (100) | 9 (100) | 8 (100) | 9 (100) | 8 (100) | 42 (100) |
| Female | 0 | 0 | 0 | 0 | 0 | 0 |
| Weight, kg | ||||||
| Mean (SD) | 80.4 (11.4) | 81.9 (13.7) | 83.7 (14.4) | 71.8 (10.2) | 73.2 (13.2) | 78.2 (12.9) |
| Race, n (%) | ||||||
| White | 3 (37.5) | 2 (22.2) | 2 (25.0) | 3 (33.3) | 3 (37.5) | 13 (31.0) |
| Black/African American | 1 (12.5) | 2 (22.2) | 2 (25.0) | 0 | 0 | 5 (11.9) |
| American Indian or Alaska Native | 0 | 0 | 0 | 1 (11.1) | 0 | 1 (2.4) |
| Japanese | 4 (50.0) | 5 (55.5) | 4 (50.0) | 4 (44.4) | 4 (50.0) | 21 (50.0) |
| Other | 0 | 0 | 0 | 1 (11.1) | 1 (12.5) | 2 (4.8) |
IV, intravenous; PLA, placebo; SD, standard deviation
Safety
No deaths or serious AEs were reported, and no subject discontinued treatment owing to an AE. All AEs reported were assessed by the investigator as mild in intensity, with the exception of one moderate AE of infusion site erythema, reported for a subject receiving a 5‐day continuous IV infusion of BMS‐962212 at a dose of 9 mg h−1 (part B, non‐Japanese cohort). A total of 16 AEs were reported in 12 of 68 subjects assigned to BMS‐962212 (17.6% of subjects), and four AEs were reported in three of 26 subjects receiving placebo (11.5%). In part A, six AEs were reported in four of the 34 subjects assigned to BMS‐962212 groups (11.7% of subjects) and 3 AEs were reported in two of the 18 subjects assigned to placebo (11.1% of subjects). No correlation between AE frequency and dose of BMS‐962212 was observed, with all of the reported AEs in the 1.5 mg h−1 and 4 mg h−1, and none in the 10 mg h−1 and 25 mg h−1 dose panels (Table S2A). In part B, 10 AEs were reported in eight of the 34 subjects assigned to BMS‐962212 (23.5% of subjects) and one AE was reported in one of the eight subjects assigned to placebo (12.5% of the subjects). No clear trend was seen between AE frequency and BMS‐962212 dose (two, one, six and one AE, respectively, in the 1, 3, 9 and 20 mg h−1 dose panels). The most frequent AEs were similarly distributed between active and placebo groups. In the BMS‐962212 groups, the most frequent AEs were: infusion site pain or irritation (5.9%), nausea (2.9%), headache (2.9%), upper respiratory tract infection (2.9%), flatulence (1.4%) and ecchymosis (1.4%). In the placebo groups, the most frequent AEs were: infusion site pain or irritation (7.7%), headache (3.8%) and upper respiratory tract infection (3.8%) (Table S2B). The frequency of AEs was 17.6% in Japanese and 29.4% in non‐Japanese subjects. There were no distinct AEs related to either the Japanese or non‐Japanese group. Of all the AEs in the BMS‐962212 groups, only one AE, of headache, was deemed by the principal investigator to be related to the study treatment.
In the first dose panel of part A (panel 1), a transient increase in serum creatinine levels was observed in five of the six subjects who received a 2‐h IV infusion of BMS‐962212 at a dose of 1.5 mg h−1 (see Figure S1A). Serum creatinine levels peaked at 12 h after initiation of the BMS‐962212 infusion, with a mean increase of 0.28 mg dl−1 (median increase 0.35 mg dl−1, range −0.1 mg dl−1 to 0.5 mg dl−1), and had returned to baseline levels by the 24‐h time point. A consequent mean percentage decrease in eGFR (based on serum creatinine) of −22.58% was observed at 12 h postdose. By comparison, the two placebo subjects in panel 1 showed no obvious change in serum creatinine levels.
Hence, to exclude the effects of BMS‐962212 on renal function, panel 1A was added to the study, in which GFR was measured directly using iohexol plasma clearance at baseline (day −2) and after a 2‐h IV infusion of 1.5 mg h−1 BMS‐962212 or placebo (day 1). No clinically relevant differences in iohexol plasma clearance were observed between day −2 and day 1, or between the BMS‐962212‐treated subjects and the placebo subjects. Furthermore, serum creatinine levels in the BMS‐962212‐treated subjects did not show any clinically meaningful increase relative to the individual time‐matched baseline levels on day −2 (see Figure S1B) or compared with the placebo subjects. The PK, aPTT and FXI:C of panel 1A are provided in Figure S2A,B,C, respectively.
In the subsequent dose panels in part A (4, 10 and 25 mg h−1) or part B (when administered as a 5‐day continuous IV infusion at 1, 3, 9 and 20 mg h−1), there were no clinically meaningful trends in any renal safety parameter. Specifically, there were no increases in mean levels of serum creatinine, cystatin‐C, BUN, urinary NGAL or urinary MCP‐1, and no decreases in mean eGFR. No subject had a serum creatinine or BUN value that met predefined criteria for a marked abnormality. Figure S3 depicts the individual serum creatinine levels at the highest dose panel in part B (20 mg h−1), demonstrating no change over time. Therefore, the totality of the renal safety data for part A and part B suggests that BMS‐962212 did not affect renal function.
PK
The summary statistics for BMS‐962212 PK parameters for parts A and B are shown in Table 2. BMS‐962212 peak concentrations were observed within 1–2 h after administration. The descending phase of the plasma concentration–time curves appeared to be biphasic, characterized by a relatively rapid initial phase followed by a slower terminal phase (Figure 1A,B). The terminal t1/2 was shorter in part A, ranging from 2.04 h to 4.94 h, compared with that observed in part B, where t1/2 ranged from 6.22 h to 8.65 h. The CL was consistent between panels within part A (ranging from 16.59 l h−1 to 21.47 l h−1) and part B (ranging from 16.78 l h−1 to 19.45 l h−1). All subjects had a <10% extrapolation for AUCinf values.
Table 2.
Pharmacokinetic parameters in 2‐h IV Infusion and 5‐day IV Infusion of subjects
| Part A (2‐h IV Infusion) | |||||||
|---|---|---|---|---|---|---|---|
| Cmax (ng ml−1) | Tmax (h) | AUC0–t (h.ng ml−1) | AUCinf (h.ng ml−1) | t1/2 (h) | CL (l h−1) | Vz (l) | |
| Panel 1 (1.5 mg h −1 ) (n = 6) | 81.90 (12.4) | 1.5 (1.5–2.0) | 121.90 (13.0) | 180.16 (11.1) | 2.04 (16.2) | 16.59 (12.3) | 72.59 (16.2) |
| Panel 2 (4 mg h −1 ) (n = 6) | 181.89 (16.9) | 2.0 (2.0–2.0) | 233.43 (13.3) | 376.56 (15.9) | 3.95 (30.1) | 21.47 (15.9) | 118.32 (46.0) |
| Panel 3 (10 mg h −1 ) (n = 6) | 504.81 (7.3) | 2.0 (1.0–2.0) | 664.15 (7.31) | 1046.43 (9.0) | 4.68 (21.7) | 19.18 (9.4) | 126.47 (26.8) |
| Panel 4 (25 mg h −1 ) (n = 6) | 1116.28 (42.1) | 2.0 (1.0–2.0) | 1736.70 (22.2) | 2725.14 (23.0) | 4.94 (28.6) | 18.74 (23.2) | 128.79 (35.9) |
| Part B (5‐day IV infusion) | |||||||
|---|---|---|---|---|---|---|---|
| Cmax (ng ml−1) | Tmax (h) | AUC0–t (h.μg ml−1) | AUCinf (h.μg ml−1) | t1/2 (h) | CL (l h−1) | Vz (l) | |
| Panel 5 (1 mg h −1 ) (n = 8) | 58.26 (24.8) | 24 (24–96) | 6.30 (23.1) | 6.38 (23.2) | 6.25 (28.1) | 18.81(25.3) | 188.09 (39.6) |
| Panel 6 (3 mg h −1 ) (n = 8) | 201.58 (21.8) | 60 (4–120) | 21.21 (18.8) | 21.45 (18.9) | 6.22 (14.78) | 16.78 (17.3) | 164.54 (27.3) |
| Panel 7 (9 mg h −1 ) (n = 8) | 520.32 (19.6) | 36 (12–96) | 54.77 (19.4) | 55.54 (19.5) | 8.65 (20.9) | 19.45 (18.6) | 216.06 (29.7) |
| Panel 8 (20 mg h −1 ) (n = 8) | 1170.54 (25.2) | 60 (12–120) | 124.19 (24.6) | 126.02 (24.8) | 7.65 (12.2) | 19.04 (25.9) | 218.90 (28.1) |
Cmax, AUC0‐t, AUCinf, t1/2, CL and Vz are described as geometric mean (CV%); Tmax is described as median (range).
AUC0–t, area under the concentration–time curve for the sampling period; AUCinf, area under the concentration–time curve extrapolated to infinity; CL, clearance; Cmax, maximum plasma concentration; CV%, percentage coefficient of variation; IV, intravenous; t1/2, terminal half‐life; Tmax, time to Cmax; Vz, volume of distribution during the terminal phase
Figure 1.
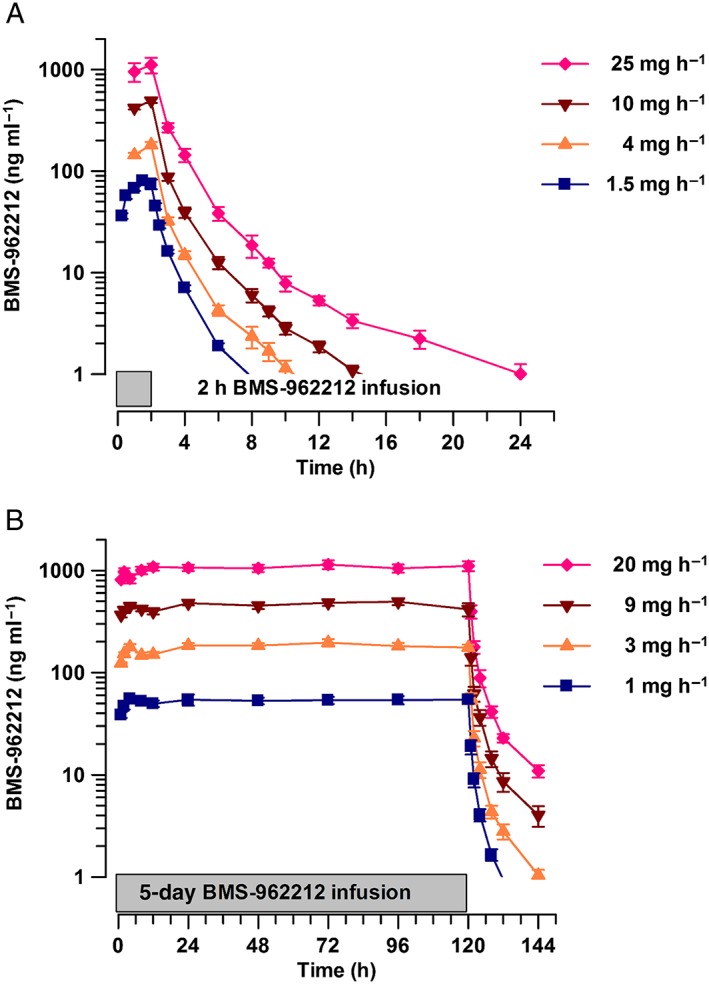
(A) Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), at the administration of doses of at 1.5, 4, 10 and 25 mg h−1 in part A. (B) Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), at the administration of doses of 1, 3, 9 and 20 mg h−1 in part B
Time course of aPTT and PT
BMS‐962212 demonstrated dose‐dependent increases in aPTT in part A and part B (Figure 2A,B). In part A, the maximal effects on aPTT were observed during the second hour of the infusion (hour 1 to hour 2) in all dose groups. The mean increase at the end of the IV infusion (2 h) compared with baseline was 1.4%, 35%, 51%, 81% and 94% in the placebo, 1.5, 4, 10 and 25 mg h−1 dose groups, respectively (Table 3). In part B, the maximal effect on aPTT was, again, achieved by approximately 2 h after initiation of the IV infusion on day 1 and was sustained throughout the 5‐day dosing period, with aPTT beginning to decrease within approximately 1 h after completion of the infusion (hour 121) and returning to baseline 4–12 h (hour 124 to hour 132) after completion of the infusion. The changes in aPTT in part B are described in the Japanese vs. non‐Japanese PK and PD section below. PT/INR did not change in either part A or part B (data not shown).
Figure 2.
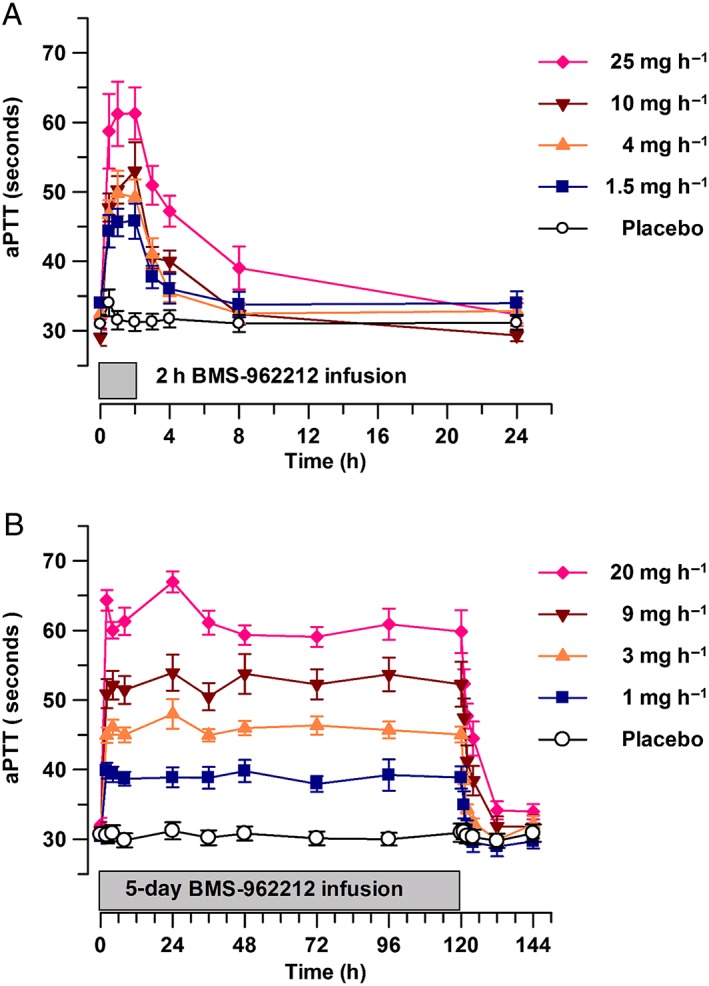
(A) Time course of activated partial thromboplastin time (aPTT) (mean ± standard error of the mean), stratified by BMS‐962212 dose panel in part A: placebo, 1.5, 4, 10 and 25 mg h−1. (B) Time course of aPTT (mean ± standard error of the mean), stratified by BMS‐962212 dose panel in part B: placebo, 1, 3, 9 and 20 mg h−1
Table 3.
Summary statistics for activated partial thromboplastin time (aPTT), stratified by BMS‐962212 dose panel, immediately prior to the end of infusion in part A and part B for non‐Japanese and Japanese subjects
| Part A (2‐h IV infusion) | |||
|---|---|---|---|
| Dose (mg h−1) | N | Mean aPTT (± SEM) (seconds) | % Change from baseline |
| Placebo | 8 | 31.3 ± 1.3 | 1.4 |
| 1.5 | 6 | 45.8 ± 2.5 | 35 |
| 4 | 6 | 49.2 ± 2.6 | 51 |
| 10 | 6 | 53.0 ± 4.2 | 81 |
| 25 | 6 | 61.3 ± 3.8 | 94 |
| Part B (5‐day IV infusion) (non‐Japanese subjects) | |||
| Placebo | 4 | 28.6 ± 1.0 | 1.0 |
| 1 | 4 | 42.4 ± 1.8 | 31 |
| 3 | 4 | 44.0 ± 1.9 | 49 |
| 9 | 4 | 52.6 ± 6.8 | 76 |
| 20 | 4 | 57.2 ± 4.0 | 83 |
| Part B (5‐day IV infusion) (Japanese subjects) | |||
| Placebo | 4 | 36.3 ± 3.6 | 12 |
| 1 | 4 | 37.7 ± 2.0 | 31 |
| 3 | 4 | 46.1 ± 1.2 | 47 |
| 9 | 4 | 52.0 ± 1.3 | 63 |
| 20 | 4 | 63.0 ± 4.9 | 92 |
IV, intravenous; SEM, standard error of the mean
Time course of FXI:C activity
BMS‐962212 decreased FXI:C in a dose‐dependent manner. In part A, the maximal effect on FXI:C activity was observed during the last hour of the infusion (hour 1 to hour 2) in all dose groups (Figures 3A). The mean decreases at the end of the IV infusion compared with baseline were −2.3%, −20%, −46%, −73% and −83% in the placebo, 1.5, 4, 10 and 25 mg h−1 dose groups, respectively (Table 4). In part B, FXI:C was reduced to a near‐maximal level within 2 h after initiation of the IV infusion on day 1 and was sustained throughout the 5‐day dosing period (Figure 3B). The FXI:C began to increase immediately and was observed at the first time‐point collection of 1 h after completion of the infusion (hour 121), and returned to near baseline by 4–12 h (hour 124 to hour 132) after completion of the infusion. The changes in FXI:C in part B are described in the Japanese vs. non‐Japanese PK and PD section below.
Figure 3.
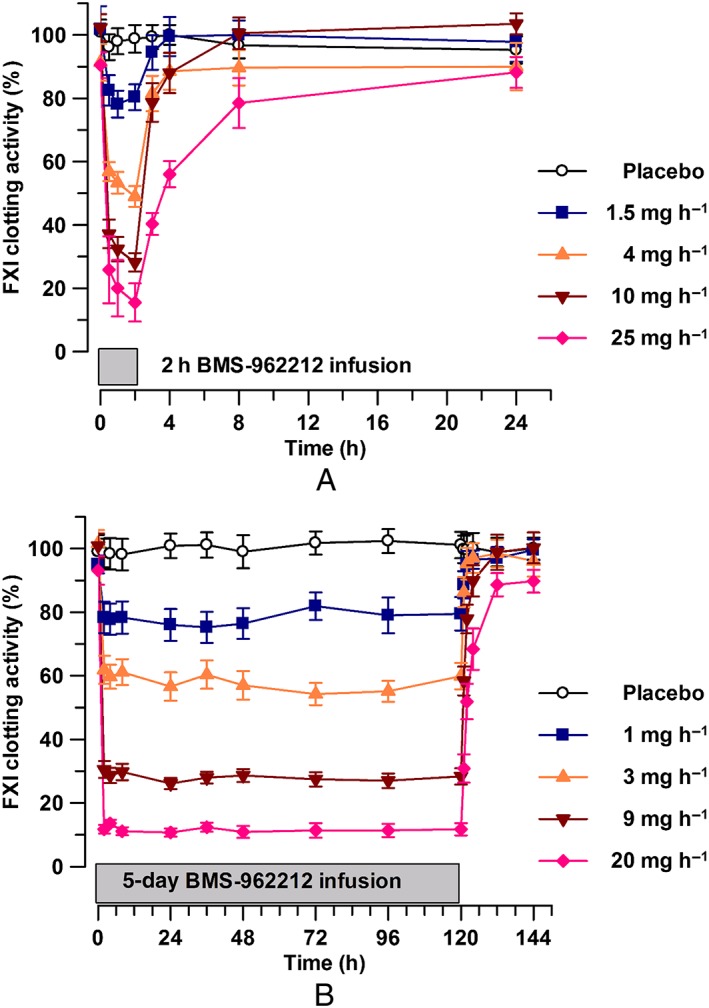
(A) Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by BMS‐962212 dose panel in part A: placebo, 1.5, 4, 10 and 25 mg h−1. (B) Time course of FXI clotting activity (mean ± standard error of the mean), stratified by BMS‐962212 dose panel in part B: placebo, 1, 3, 9 and 20 mg h−1
Table 4.
Summary statistics for factor XI clotting activity (FXI:C), stratified by BMS‐962212 dose panel, immediately prior to the end of infusion in part A and part B for non‐Japanese and Japanese subjects
| Dose (mg h−1) | N | Mean Observed FXI:C (± SEM) (%) | % Change from baseline |
|---|---|---|---|
| Part A (2‐h IV infusion) | |||
| Placebo | 8 | 99 ± 4.4 | −2.3 |
| 1.5 | 6 | 80.3 ± 4.1 | −20 |
| 4 | 6 | 49.0 ± 2.8 | −46 |
| 10 | 6 | 28.2 ± 2.9 | −73 |
| 25 | 6 | 15.5 ± 6.0 | −83 |
| Part B (5‐day IV infusion) (non‐Japanese subjects) | |||
| Placebo | 4 | 102 ± 3.6 | 0 |
| 1 | 4 | 70.3 ± 6.3 | −25 |
| 3 | 4 | 59.8 ± 5.3 | −40 |
| 9 | 4 | 26 ± 3.0 | −74 |
| 20 | 4 | 14 ± 3.5 | −85 |
| Part B (5‐day IV infusion) (Japanese subjects) | |||
| Placebo | 4 | 100.2 ± 8.2 | +1 |
| 1 | 4 | 78.8 ± 2.8 | −16 |
| 3 | 4 | 60.0 ± 7.2 | −42 |
| 9 | 4 | 30.8 ± 4.1 | −70 |
| 20 | 4 | 9.5 ± 1.2 | −90 |
IV, intravenous; SEM, standard error of the mean
Japanese vs. non‐Japanese PK and PD
Table 5 lists the BMS‐962212 Css, weight‐corrected Css and t1/2 in part B when stratified by Japanese and non‐Japanese ethnicity in each dose panel. Japanese subjects had numerically higher, albeit not significantly different, values for Css in the dose panels 1, 3 and 20 mg h−1 (P > 0.05). When corrected for weight, Css in dose panels 1 mg h−1 and 9 mg h−1 demonstrated a higher exposure in non‐Japanese subjects compared with Japanese subjects (P > 0.05). There was no difference in the t1/2 range in Japanese subjects (between 6.42 h and 8.28 h) vs. non‐Japanese subjects (between 6.05 h and 8.32 h) over all dose groups for part B (P > 0.05). The concentration–time, aPTT and FXI:C curves, stratified by Japanese and non‐Japanese ethnicity, at a dose of 20 mg h−1 in part B are shown in Figure 4A–C. In Japanese and non‐Japanese subjects, no difference was observed in the aPTT percentage change from baseline, with increases of 12–92% and 31–83%, respectively, across the dosing range for part B (P > 0.05) (Table 3). The percentage change from baseline in FXI:C between Japanese and non‐Japanese subjects across the dosing range for part B showed a similar reduction, 16–90% and 25–85%, respectively (P > 0.05) (Table 4). Dose proportionality was observed for Cmax and AUC in part A and Cmax, AUC and Css in part B (Table S3). When stratified by Japanese and non‐Japanese ethnicity in part B, dose proportionality was demonstrated for Cmax, AUC and Css. The concentration–time, aPTT and FXI:C curves, stratified by Japanese and non‐Japanese ethnicity, for dose panels placebo (aPTT and FXI:C only) and 1, 3 and 9 mg h−1 are depicted in Figure S4A–K.
Table 5.
Steady‐state concentration {geometric mean [percentage coefficient of variation (CV%)]} and half‐life [mean (CV%)], stratified by Japanese and Non‐Japanese ethnicity in part B
| Steady‐state concentration (ng ml −1 ) | Weight‐corrected steady‐state concentration (kg μg.ml −1 ) | t 1/2 (h) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Dose panel | Non‐Japanese ethnicity (n = 4) | Japanese ethnicity (n = 4) | P‐value | Non‐Japanese ethnicity (n = 4) | Japanese ethnicity (n = 4) | P‐value | Non‐Japanese ethnicity (n = 4) | Japanese ethnicity (n = 4) | P‐value |
| Panel 5 (1 mg h −1 ) (n = 8) | 49.88 (27.3) | 55.81 (20.5) | >0.05 | 4.34 (15.5) | 4.21 (21.6) | >0.05 | 5.30 (17) | 7.32 (33) | >0.05 |
| Panel 6 (3 mg h −1 ) (n = 8) | 157.76 (12.9) | 188.03 (17.9) | >0.05 | 14.10 (22.7) | 14.33 (32.9) | >0.05 | 6.05 (8) | 6.42 (21) | >0.05 |
| Panel 7 (9 mg h −1 ) (n = 8) | 458.34 (18.9) | 439.27 (18.9) | >0.05 | 34.47 (23.5) | 29.60 (18.9) | >0.05 | 8.32 (31) | 8.28 (21) | >0.05 |
| Panel 8 (20 mg h −1 ) (n = 8) | 887.21 (23.3) | 1161.68 (17.7) | >0.05 | 70.57 (32.4) | 76.07 (30.1) | >0.05 | 7.23 (18) | 8.28 (4) | >0.05 |
Figure 4.
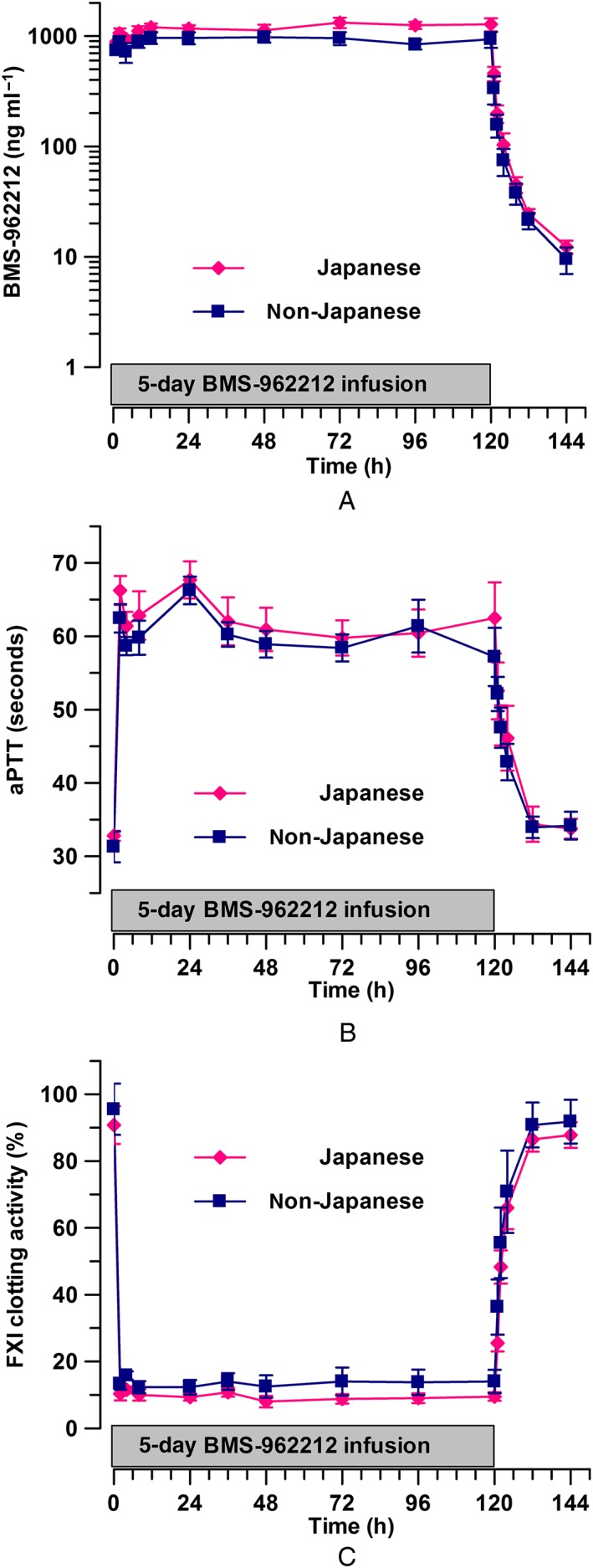
(A) Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, after administration of a dose of 20 mg h−1. (B) Time course of activated partial thromboplastin time (aPTT) (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, in part B at 20 mg h−1. and (C) Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, in Part B at 20 mg h−1
Discussion
The present study investigated the safety, tolerability, PK and PD of BMS‐962212, a first‐in‐class small‐molecule reversible factor XIa inhibitor. As a parenteral agent with a relatively short elimination t 1/2, BMS‐962212 would be expected to achieve higher efficacy, faster onset of action and better control of antithrombotic effects compared with oral agents. The ability to achieve anticoagulation rapidly through parenteral administration, followed by rapid restoration of normal coagulation is a highly desirable quality in the acute treatment arena. This profile, coupled with the anticipated improved bleeding risk profile of FXIa inhibitors, allows for investigation in several clinical indications. To date, there have been no publications reporting on the safety, PK and PD of a reversible small‐molecule factor XIa inhibitor in humans.
The present first‐in‐human study demonstrated that BMS‐962212 was well tolerated in healthy subjects when given as a 2‐h IV infusion over a 16‐fold dose range, and a 5 day IV infusion over a 20‐fold dose range. The majority of AEs observed were of mild intensity and resolved without intervention. AE frequency did not show a trend to increase with increasing doses of BMS‐962212, and only one out of the 16 AEs were deemed by the study investigators to be related to treatment. The frequency and type of AEs were similar in the placebo and active groups, with irritation at the infusion site being the most frequent (7.7% in the placebo vs. 5.9% in the active treatment groups). No dose‐limiting AEs were observed, and consequently the maximum tolerated dose for BMS‐962212 was not established.
The PK profile showed a rapid increase in exposure, as evidenced by near‐steady‐state BMS‐962212 plasma concentrations being achieved approximately 2 h after the start of continuous infusion. Furthermore, the biexponential elimination of the drug resulted in BMS‐962212 plasma concentrations declining by an order of magnitude in approximately 4 h during the initial elimination phase, followed by a slower terminal phase. The t1/2 of BMS‐962212 was approximately 2–5 h following a 2‐h IV infusion and 6–9 h after a 5‐day IV infusion. The disparity between the t1/2 observed in part A and that in part B is likely to have been due to the differences in the distribution profile of the drug after a 2‐h IV infusion (Vz ranging from 72 l to 129 l) compared with a 5‐day IV infusion (Vss ranging from 164 l to 218 l). BMS‐962212 exhibited dose‐proportional increases in Css across the 20‐fold dose range during the 5‐day IV infusion. Exposure parameters demonstrated moderate to low variability, with %CVs typically below 30% in healthy subjects, indicating that BMS‐962212 may be administered at a fixed dose to achieve a target concentration without dose adjustment. This distribution and elimination profile supports the investigation of BMS‐962212 in subjects with a need for acute anticoagulation where a rapid onset and offset of action may be most appropriate.
Furthermore, in Japanese subjects, a 5‐day IV infusion was shown to be well tolerated, with no significant differences in PK or PD parameters compared with non‐Japanese subjects. In the Japanese subjects, there was a trend towards higher concentrations in each of the dose panels, although a wide overlap in exposures was observed at each of the dose panels. When corrected by weight, the differences in Css no longer followed a trend. Similarly, no trend was observed in the t1/2 between Japanese and non‐Japanese subjects.
The PD investigated in the present study included aPTT, FXI:C and PT/INR. Consistent with FXIa inhibition, exposure‐dependent increases in aPTT and exposure‐dependent decreases in FXI:C were observed across dose panels, with no effect on PT/INR. The aPTT is used for therapeutic heparin monitoring in several indications requiring anticoagulation and presents a relevant PD biomarker to investigate the anticoagulant activity of a factor XIa inhibitor 15, 16. For BMS‐962212, the relationship between aPTT or FXI:C targets and the prevention of thromboembolic events needs to be established in larger clinical studies.
In the Leiden Thrombophilia Study, patients with FXI levels in the top 10% were nearly twice as likely to develop venous thromboembolism as the rest of the study population. Similarly, human subjects genetically deficient in FXI appear less prone to developing venous thrombosis 17. Severe FXI deficiency appears to confer protection from ischaemic stroke, and higher FXI levels were associated with an increased risk of ischaemic stroke in a study by the Atherosclerosis Risk in Communities study 5 and a study by Yang et al. 6. In young women enrolled in the Risk of Arterial Thrombosis In Relation to Oral Contraceptives study, levels of FXI:C and FXI antigen correlated with arterial thrombosis and stroke risk 18.
Currently, only one approach to inhibiting FXI specifically has been reported in the literature, using an antisense oligonucleotide (ASO) that demonstrated efficacy superior to enoxaparin in total knee arthroplasty 19. This ASO blocks FXI synthesis, and as the circulating t1/2 for FXI is 60–80 h, it can take several weeks to achieve a targeted level of plasma FXI reduction. The ASO also has a slow offset of action, and FXI levels can remain below normal for several weeks following discontinuation of ASO administration. Reducing FXI levels to ~20% of normal by the ASO proved more effective than enoxaparin in preventing postoperative venous thromboembolism, and appeared to be safe in regard to the risk of bleeding, providing a proof of concept in humans for FXI inhibition 19.
The small number of subjects used to investigate differences between Japanese and non‐Japanese individuals in the 5‐day IV infusion part of the study could be considered a limitation of the study; however, there are significant advantages associated with this innovative study design. Typically, investigation of PK in Japanese subjects is conducted in a standalone study, which can result in considerable interstudy variability, including batch–batch variability in biomarker assays, potential differences in comparison across dose panels, and a lag time associated with global development of the programme. Furthermore, through the use of population PK and PK/PD, the potential for differences based on covariate analysis can be rapidly assessed and investigated for the need to adjust the dose moving into phase II.
In summary, BMS‐962212 was well tolerated when given as an IV infusion over 2 h and 5 days in Japanese and non‐Japanese subjects. The PK of BMS‐962212 demonstrated dose proportionality over the dose range studied, and demonstrated a fast onset of action and a relatively rapid elimination. The PD of BMS‐962212 showed exposure‐dependent increases and exposure‐dependent decreases in aPTT and FXI:C, respectively. These results present a promising framework to develop BMS‐962212 in indications requiring acute care anticoagulation.
Competing Interests
All authors are current or former employees of Bristol‐Myers Squibb at the time of publication of this article.
The authors would like to thank and acknowledge the contribution of the following people to the manuscript: Lisa Christopher, for conducting transporter assays for characterization of the metabolic profile; Elizabeth Dierks, for investigating preclinical pharmacokinetic studies; and Silvi Chacko, for performing metabolite reaction phenotyping and characterization.
This study was wholly funded by Bristol Myers Squibb.
Supporting information
Data S1 Supplementary methods
Table S1 Inclusion criteria of study for panels in part A and part B, with the exception of panel 1 in part A
Table S2A Overview of number (percentage) of subjects reporting adverse events in part A
Table S2B Overview of number (percentage) of subjects reporting adverse events in part B
Table S3 Analysis of variance result for log‐transformed maximum plasma concentration (Cmax), area under the concentration–time curve (AUC) and concentration at steady state (Css) across dose panels in part A and part B, stratified by Japanese and non‐Japanese ethnicity
Figure S1A Serum creatinine (mg dl−1) over time, following a 2‐h intravenous infusion of BMS‐962212 at 1.5 mg h−1 in part A, panel 1 for individual subjects. Dashed lines represent upper and lower limits of normal
Figure S1B Serum creatinine (mg dl−1) over time, following intravenous administration of iohexol (day −2 and day 1) and a 2‐h intravenous infusion (day 1) of BMS‐962212 at 1.5 mg h−1 in part A, panel 1A for individual subjects. Dashed lines represent upper and lower limits of normal
Figure S2A Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean) at 1.5 mg h−1 in part A, panel 1A
Figure S2B Time course of activated partial thromboplastin time (aPTT) (mean ± standard error of the mean), stratified by dose panel in part A, panel 1A for placebo and 1.5 mg h−1
Figure S2C Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by dose panel in Part A, panel 1A for placebo and 1.5 mg h−1
Figure S3 Serum creatinine (mg dl−1) over time following a 5‐day IV infusion of BMS‐962212 at 20 mg h−1 in part B for individual subjects. Dashed lines represent upper and lower limits of normal
Figure S4A Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 1.5 mg h−1 dose
Figure S4B Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 3 mg h−1 dose
Figure S4C Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 9 mg h−1 dose
Figure S4D Time course of activated partial thromboplastin time (aPTT; mean ± SEM), stratified by Japanese and non‐Japanese ethnicity, for the placebo
Figure S4E Time course of activated partial thromboplastin time (aPTT; mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 1 mg h−1 dose
Figure S4F Time course of activated partial thromboplastin time (aPTT; mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 3 mg h−1 dose
Figure S4G Time course of activated partial thromboplastin time (aPTT; mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 9 mg h−1 dose
Figure S4H Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the placebo
Figure S4I Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 1 mg h−1 dose
Figure S4J Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 3 mg h−1 dose
Figure S4K Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 9 mg h−1 dose
Perera, V. , Luettgen, J. M. , Wang, Z. , Frost, C. E. , Yones, C. , Russo, C. , Lee, J. , Zhao, Y. , LaCreta, F. P. , Ma, X. , Knabb, R. M. , Seiffert, D. , DeSouza, M. , Mugnier, P. , Cirincione, B. , Ueno, T. , and Frost, R. J. A. (2018) First‐in‐human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS‐962212, a direct, reversible, small molecule factor XIa inhibitor in non‐Japanese and Japanese healthy subjects. Br J Clin Pharmacol, 84: 876–887. doi: 10.1111/bcp.13520.
References
- 1. Ezekowitz MD, Reilly PA, Nehmiz G, Simmers TA, Nagarakanti R, Parcham‐Azad K, et al Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100 (9): 1419–1426. [DOI] [PubMed] [Google Scholar]
- 2. Löwenberg EC, Meijers JCM, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost 2010; 8: 2349–2357. [DOI] [PubMed] [Google Scholar]
- 3. Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, et al Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood 2017; 129: 1210–1215. [DOI] [PubMed] [Google Scholar]
- 4. Salomon O, Steinberg DM, Koren‐Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood 2008; 111: 4113–4117. [DOI] [PubMed] [Google Scholar]
- 5. Suri MFK, Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Novel hemostatic factor levels and risk of ischemic stroke: the Atherosclerosis Risk In Communities (ARIC) study. Cerebrovasc Dis 2010; 29: 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol 2006; 126: 411–415. [DOI] [PubMed] [Google Scholar]
- 7. Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost 2009; 7: 84–87. [DOI] [PubMed] [Google Scholar]
- 8. Gomez K, Bolton‐Maggs P. Factor XI deficiency. Haemophilia 2008; 14: 1183–1189. [DOI] [PubMed] [Google Scholar]
- 9. Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol 2016; 36: 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al‐Horani RA, Desai UR. Factor XIa inhibitors: a review of the patent literature. Expert Opin Ther Pat 2016; 26: 323–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. David T, Kim Y, Ely LK, Rondon I, Gao H, O'Brien P, et al Factor XIa‐specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci Transl Med 2016; 8: 353ra112. [DOI] [PubMed] [Google Scholar]
- 12. Pinto DJP, Orwat MJ, Smith LM, Quan ML, Lam PYS, Rossi KA, et al Discovery of a parenteral small molecule coagulation factor XIa inhibitor clinical candidate (BMS‐962212). J Med Chem 2017; 60: 9703–9723. [DOI] [PubMed] [Google Scholar]
- 13. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smythe MA, Koerber JM, Westley SJ, Nowak SN, Begle RL, Balasubramaniam M, et al Use of the activated partial thromboplastin time for heparin monitoring. Am J Clin Pathol 2001; 115: 148–155. [DOI] [PubMed] [Google Scholar]
- 16. Ignjatovic V. Activated partial thromboplastin time In: Haemostasis. Methods in Molecular Biology (Methods and Protocols), ed Monagle P. Totowa, NJ: Humana Press, 2013; 111–120. [DOI] [PubMed] [Google Scholar]
- 17. Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med 2000; 342: 696–701. [DOI] [PubMed] [Google Scholar]
- 18. Siegerink B, Govers‐Riemslag JWP, Rosendaal FR, Cate HT, Algra A. Intrinsic coagulation activation and the risk of arterial thrombosis in young women: results from the Risk of Arterial Thrombosis in Relation to Oral Contraceptives (RATIO) case‐control study. Circulation 2010; 122: 1854–1861. [DOI] [PubMed] [Google Scholar]
- 19. Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, et al Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 2015; 372: 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supplementary methods
Table S1 Inclusion criteria of study for panels in part A and part B, with the exception of panel 1 in part A
Table S2A Overview of number (percentage) of subjects reporting adverse events in part A
Table S2B Overview of number (percentage) of subjects reporting adverse events in part B
Table S3 Analysis of variance result for log‐transformed maximum plasma concentration (Cmax), area under the concentration–time curve (AUC) and concentration at steady state (Css) across dose panels in part A and part B, stratified by Japanese and non‐Japanese ethnicity
Figure S1A Serum creatinine (mg dl−1) over time, following a 2‐h intravenous infusion of BMS‐962212 at 1.5 mg h−1 in part A, panel 1 for individual subjects. Dashed lines represent upper and lower limits of normal
Figure S1B Serum creatinine (mg dl−1) over time, following intravenous administration of iohexol (day −2 and day 1) and a 2‐h intravenous infusion (day 1) of BMS‐962212 at 1.5 mg h−1 in part A, panel 1A for individual subjects. Dashed lines represent upper and lower limits of normal
Figure S2A Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean) at 1.5 mg h−1 in part A, panel 1A
Figure S2B Time course of activated partial thromboplastin time (aPTT) (mean ± standard error of the mean), stratified by dose panel in part A, panel 1A for placebo and 1.5 mg h−1
Figure S2C Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by dose panel in Part A, panel 1A for placebo and 1.5 mg h−1
Figure S3 Serum creatinine (mg dl−1) over time following a 5‐day IV infusion of BMS‐962212 at 20 mg h−1 in part B for individual subjects. Dashed lines represent upper and lower limits of normal
Figure S4A Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 1.5 mg h−1 dose
Figure S4B Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 3 mg h−1 dose
Figure S4C Time course of BMS‐962212 plasma concentration (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 9 mg h−1 dose
Figure S4D Time course of activated partial thromboplastin time (aPTT; mean ± SEM), stratified by Japanese and non‐Japanese ethnicity, for the placebo
Figure S4E Time course of activated partial thromboplastin time (aPTT; mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 1 mg h−1 dose
Figure S4F Time course of activated partial thromboplastin time (aPTT; mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 3 mg h−1 dose
Figure S4G Time course of activated partial thromboplastin time (aPTT; mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 9 mg h−1 dose
Figure S4H Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the placebo
Figure S4I Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 1 mg h−1 dose
Figure S4J Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 3 mg h−1 dose
Figure S4K Time course of factor XI (FXI) clotting activity (mean ± standard error of the mean), stratified by Japanese and non‐Japanese ethnicity, for the 9 mg h−1 dose