

Abstract
Regulation of the Neisseria gonorrhoeae pilE gene is ill-defined. In this study, post-transcriptional effects on expression were assessed. In silico analysis predicts the formation of three putative stable stem–loop structures with favourable free energies within the 5′ untranslated region of the pilE message. Using quantitative reverse transcriptase PCR analyses, we show that each loop structure forms, with introduced destabilizing stem–loop mutations diminishing loop stability. Utilizing a series of pilE translational fusions, deletion of either loop 1 or loop 2 caused a significant reduction of pilE mRNA resulting in reduced expression of the reporter gene. Consequently, the formation of the loops apparently protects the pilE transcript from degradation. Putative loop 3 contains the pilE ribosomal binding site. Consequently, its formation may influence translation. Analysis of a small RNA transcriptome revealed an antisense RNA being produced upstream of the pilE promoter that is predicted to hybridize across the 5′ untranslated region loops. Insertional mutants were created where the antisense RNA is not transcribed. In these mutants, pilE transcript levels are greatly diminished, with any residual message apparently not being translated. Complementation of these insertion mutants in trans with the antisense RNA gene facilitates pilE translation yielding a pilus + phenotype. Overall, this study demonstrates a complex relationship between loop-dependent transcript protection and antisense RNA in modulating pilE expression levels.
Keywords: 5 UTR, stem–loops, transcription, RNA stability, antisense RNA
Introduction
Neisseria gonorrhoeae causes the sexually transmitted disease gonorrhea where the expression of pili on the cell surface has been shown to be crucial for infectivity (Kellogg et al., 1968; Swanson, 1973; Swanson et al., 1987). The pilus organelle consists of several proteins, with PilE polypeptide (encoded by the pilE gene) being the major component. Despite the importance of pili to the disease process, very little is known with regard to the regulation of expression of PilE polypeptide. The pilE promoter structure is complicated with three fully functional sense promoters being present (designated P1, P2 and P3), yet only the P1 promoter (sigma70 dependent) is used in the gonococcus (Fyfe et al., 1995; Carrick et al., 1997). In addition, two pilE antisense promoters have been identified: one located within a midgene region and the second one located at the 3′ end of the gene (Masters et al., 2016). Despite considerable effort having been expended on trying to identify regulatory proteins, only one transcriptional cofactor has been found in the form of the small DNA-binding protein, integration host factor (IHF). When IHF binds upstream of the pilE promoter, IHF binding facilitates the interaction of two specialized AT-rich promoter sequences (UP elements) with RNA polymerase that increases transcription approximately 10-fold (Hill et al., 1997; Fyfe & Davies, 1998).
mRNA turnover is believed to initiate in regions that are relatively free of bound ribosomes as their constant occupation on transcripts could enhance mRNA stabilization (Bechhofer & Dubnau, 1987). Consequently, 5′ and 3′ untranslated regions (UTRs) are considered to be good candidates as initial cleavage sites for RNases, with the formation of RNA secondary structures within these regions possibly influencing stability and/or translational efficiency (Régnier & Arraiano, 2000; Marzi et al., 2008). Loop structures are predicted in the Escherichia coli rpsT P1 mRNA and analysis has shown that the presence of a hairpin at the 5′ end of the rpsT P1 transcript hinders both the pyrophosphohydrolase activity of RppH and the single-stranded-dependent cleavage of RNase E, thus prolonging mRNA half-life (Deana et al., 2008). Consequently, the presence of any pilE mRNA secondary structural features may also moderate transcript turnover.
The presence of naturally occurring small antisense RNAs (sRNAs) has been widely reported (Georg & Hess, 2011). However, the regulatory functions of many of these RNAs are still yet to be determined (Waters & Storz, 2009). Most of the known sRNAs do not encode protein, with their pervasive transcription being initiated from intergenic or intragenic promoters (Wade & Grainger, 2014). A recent analysis of a gonococcal small RNA transcriptome revealed many such sRNAs (Wachter & Hill, 2015). Such trans-encoded sRNAs appear to act primarily by regulating mRNA translation/degradation via complementary binding in an antisense manner that often requires the aid of Hfq protein, which is an RNA chaperone. The absence of Hfq protein causes pleiotropic effects that occasionally involve bacterial pathogenicity (Hoe et al., 2013). Hfq also serves as a major post-transcriptional regulator of numerous stress-responsive genes (Sittka et al., 2007). In a gonococcal hfq mutant, the absence of Hfq protein has been shown to decrease pilE transcript levels, as well as to influence several pilus-associated phenotypes (Dietrich et al., 2009). In a Neisseria meningitidis hfq mutant, PilE polypeptide is absent (Pannekoek et al., 2009). Consequently, these observations suggest that a small RNA may be involved in moderating pilE expression.
In this study, post-transcriptional effects on pilE expression were investigated and a stabilizing role for stem–loop structures in the 5′ UTR of the pilE message was indicated. Furthermore, as one loop structure is predicted to occlude the pilE ribosomal binding site, evidence is presented whereby an sRNA is required for translation of the pilE message. Overall, the data indicate that pilE expression is regulated at the post-transcriptional level adding a further layer of complexity to the regulation of this important virulence determinant.
Methods
Strains and growth conditions.
N. gonorrhoeae strain MS11 (Rocky Mountain Laboratories, Hamilton, MT, USA) was used in this study. Gonococci were passaged daily on a gonococcal typing medium (Swanson, 1982) at 37 °C in a 5 % CO2 atmosphere. When grown in the presence of antibiotics, the final concentrations were as follows: chloramphenicol, 10 µg ml−1; kanamycin, 80 µg ml−1; erythromycin, 5 µg ml−1.
E. coli cells were grown using LB medium at 37 °C. When E. coli carried recombinants, the medium was supplemented with antibiotics at the following concentrations: carbenicillin, 100 µg ml−1; erythromycin, 200 µg ml−1; chloramphenicol, 20 µg ml−1; tetracycline, 15 µg ml−1; and kanamycin, 40 µg ml−1.
Construction of translational fusions.
pilE translational fusions were constructed by fusing the pilE 5′ UTR (which included DNA comprising the first 19 codons) in-frame to a reporter gene, either beta-galactosidase (lacZ) or chloramphenicol acetyl transferase (cat), that lacked their cognate ribosomal binding site (rbs). This procedure entailed amplifying pilE with the appropriate set of primers (Table 1) and initially cloning the fragment into the pCRII (Invitrogen) vector. For construction of the WTpilE : lacZ clone, the appropriate fragment was cut by EcoRI and BamHI enzymes and ligated in the vector pRS414 that carries a truncated lacZ gene preceded by a strong transcriptional terminator, resulting pRS414-WTpilE : lacZ(pTL2) vector. To create the loop 1 :: lacZ and the loop 2 :: lacZ deletion fusions, sequential PCR was employed that amplified the flanking regions to each loop followed by ligation of the fragments. The fragment containing the loop deletion was then cloned into vector pRS414 at the unique sites EcoRI and BamHI to produce pRS414-L1del : lacZ (pTL6) and pRS414-L2del : lacZ (pTL8) constructs.
Table 1. Primers used to make translational fusions.
| Primer | Sequence | Application |
|---|---|---|
| Fusion 1-HindIII | 5′-CCCAAGCTTCCGAAGCCATCCTTTTGGCCGAA-3′ | 5′ UTR-PCR |
| Fusion 2 | 5′-TCAGCCGTTGCCGGGTATTG-3′ | 5′ UTR-PCR |
| Fusion 3-BamHI | 5′-ATAGCGATCAGAATCATCAGCTCGATAAGGG-3′ | 5′ UTR-PCR |
| L1-L-KpnI | 5′-AAAAAGGTACCGACAACTGCGTATTATAAAGCAAG ATTCGTGCC-3′ | Loop 1 deletion |
| L1-R-KpnI | 5′- AAAAAGGTACCATGATGCCGATGGCGTAAGCC-3′ | Loop 1 deletion |
| EcoRV–pRS | 5′-CAGCAGGATATCCTGCACCATCGT-3′ | Loop 1 deletion |
| L2-L-KpnI | 5′-AAAAAAGGTACCATGCAATGTATTTAACCATCGGTTTTTTGTTGCG-3′ | Loop 2 deletion |
| L2-R-KpnI | 5′-AAAAAGGTACCTCCCCTTTCAATTAGGAGTAATTTTATGAATACCCTTCA-3′ | Loop 2 deletion |
| Fusion 4 | 5′-TCGATTTCTTTGCCGTCTTTGG-3′ | Loop 2 deletion |
| cat3-BamHI | 5′-AAGGATCCTGGAGAAAAAAATCACTGGATATA-3′ | cat gene PCR |
| MM13R | 5′-GTAAAACGACGGCCAGTGAATTGTA-3′ | cat gene PCR |
| Fusion 5 | 5′-AAACGGGAAGTAGGCTCCCATGAT-3′ | pilE :: lacZ fusions PCR |
A similar approach was used to make pilE : cat translational fusions. The promoter-less cat gene was obtained from pCR2.1-cat:DUS (S. A. Hill, unpublished). Various pilE fragments containing intact or the 5′ UTR loop deletions were amplified using the appropriate primers (Table 1), followed by cloning into the pCRII vector; a double SacI and Sau3AI digest released the fragment that was then inserted in the pCRII-cat vector. This protocol allowed us to generate pCRII-WTpilE : cat (pTL14), pCRII-L1del : cat (pTL15) and pCRII-L2del : cat (pTL16) constructs.
In order to introduce the pilE :: cat translational fusions into the gonococcal chromosome, the various pilE : cat fusions were cloned into a pBluescript-opaE:erm vector that carries the opaE gene with an ermC gene inserted in the unique SalI site. The fusions were then PCR amplified and inserted in pBluescript-opaE:erm at the unique XbaI site to create pBluescript-opaE :: erm : pilE :: cat constructs. These DNAs were then used to cross the fusions into the gonococcal opaE locus with transformants being selected through erythromycin resistance.
To investigate the effect of pilE antisense transcription on regulation of pilE across the 5′ loops, a kanamycin gene insertion was introduced into the pilE gene such that transcription of antisense RNAs across the loop structures would be disrupted. The pilE :: kan construct was generated by blunt-end ligation of the kan gene into the pilE BssHII site. The appropriate orientation of the kan insert was confirmed by PCR to ensure interruption of antisense transcription. The pUC8-pilE : kan plasmid DNA was then used to transform the N. gonorrhoeae MS11 carrying a pilE :: cat translational fusion to kanamycin resistance to create a GC pilE :: kan opaE :: erm : pilE :: cat strain.
Site-directed mutagenesis was performed as previously described (Wachter et al., 2015; Masters et al., 2016).
Construction and complementation of the asRNA7 ermC insertion knockout mutants.
Regions upstream of the pilE 5′ UTR in N. gonorrhoeae MS11 were disrupted with ermC insertions that were made by sequential ligation of PCR-generated DNA fragments and an ermC gene cassette. DNA transformation was then used to make the gonococcal mutants. The resulting constructs (∆asRNA7 : ermC1-6) were then tested for transcription of full-length pilE message through Northern blot and endpoint reverse transcriptase PCR analysis. To determine the translational efficiency of these insertion mutants, a cat gene was translationally fused to the 3′ end of pilE and assessed the chloramphenicol resistance and competency.
To construct a ∆asRNA7 : ermC complement, the genomic region encoding a small RNA upstream of the pilE 5′ UTR corresponding to ∆asRNA7 : ermC5 and ∆asRNA7 : ermC6 was amplified with primers 10605 (5′-CCGTATGTTAACGCGTAAATTCAAAAATC-3′) and 09449 (5′-GCAACAAAAAACCGATGGTTAAATACATTGC-3′) and ligated to a kan gene cassette containing a gonococcal DNA uptake sequence (DUS) in a TA cloning vector. For subsequent transformation into N. gonorrhoeae, the kan:DUS : asRNA7 construct was ligated into the opaE gene within a pBlueScript cloning vector. The pBlueScript-opaE : kan : DUS : asRNA7 construct was then crossed into the opaE locus of N. gonorrhoeae strain MS11. Chromosomal DNA from the ∆asRNA7 : ermC6 mutational constructs was then used to transform the opaE : kan : DUS : asRNA7 cells, generating mutants ∆asRNA7 : ermC6 : opaE : asRNA7 : kan1, ∆asRNA7 : ermC6 : opaE : asRNA7 : kan2 and ∆asRNA7 : ermC6 : opaE : asRNA7 : kan3. Translational efficiencies of WT, ∆asRNA7 : ermC5 and ∆asRNA7 : ermC6 were determined through transformation efficiency and phenotypic assays.
RNA analysis.
The conditions employed for RNA extraction, quantitative real-time PCR (qRT-PCR) analysis and primer extension analysis were as described previously (Wachter et al., 2015; Masters et al., 2016). The primer pairs used for loop analysis and translational fusion analysis are found in Table 2; primer pairs for the assessment of the gonococcal insertion mutants have been previously described (Wachter et al., 2015).
Table 2. Oligonucleotide primers used in qRT-PCR analysis involving 5′ UTR loops.
| Primer | Nucleotide sequence | Application |
|---|---|---|
| Loop 1 | 5′-TTGTCGCAACAAAAAACCGATGGTTAAA- 3′ | Loop 1-forward |
| Loop 2 | 5′-AAAAAAGGTACCATGATGCCGATGGCGTAAGCC-3′ | Loop 2-forward |
| Loop 3 | 5′-CCCTTTCAATTAGGAGTAATTTTATGAATACCCTTC-3′ | Loop 3-forward |
| RC3 | 5′-GTCGGCATTTTGGCGGCAGTCG-3′ | Outside the loops-forward |
| RC4 | 5′-GATAGCGATCAGAATCATCAGCTCGATAAGGG-3′ | Loop 1-reverse |
| tsp5 | 5′-CCGTGTAGTCTTGGTAGGCGGGAA-3′ | Loop 2-reverse |
| 09173 | 5′-TTGACCTTCGGCCAAAAGGATGGCTTCGGAAAC-3′ | Loop 3-reverse |
| tsp4 | 5′-CGCCGGCAGAAGTGTTGTTTTC-3′ | Outside the loops-reverse |
| lacZ fusion 1 | 5′-GTCGGCATTTTGGCGGCAGTCG-3′ | |
| lacZ fusion 2 | 5′-GCTTCTGGTGCCGGAAACCAGGC-3′ | |
| cat fusion 1 | 5′-CACTGGATATACCACCGTTGATATATCCCAATCGCATCGTAAAGAACA-3′ | |
| cat fusion 2 | 5′-GTGAACACTATCCCATATCACCAGCTCACCGTCTTTCATTGC-3′ |
Loop formation assay.
The assay was utilized to determine whether the 5′ UTR stem–loops are formed under non-denaturing in vitro conditions using qRT-PCR analysis. E. coli carrying the pilE-containing vector pVD203 was grown to the exponential phase (OD600 reaching 0.5) at which point pilE mRNAs were extracted. pilE-specific, reverse-transcribed cDNAs were then subjected to qRT-PCR reactions utilizing the forward primers that were designed such that each primer resided within each predicted loop sequence. The reverse primers were designed such that they recognized nucleic acid sequences outside of the loop structures and that the amplified products that were produced were of a similar size, 160 bp. If the stem–loop structures are formed due to base pairing following RNA purification under native conditions, then PCRs using the primers within each loop should produce little or no products as base-paired loop sequences are not available for hybridization with the primers. In control experiments, the addition of betaine (a denaturing agent) in the qRT-PCR mix was employed to disrupt loop formation during the qRT-PCR experiments.
Computer modeling analysis.
Mfold (Zuker, 2003) and RNAstructure (Bellaousov et al., 2013) web servers were used for analysis of RNA secondary structures within the 5′ UTR of pilE.
Results
Protection of the pilE transcript through loop formation
When the IHF binding site, which is located immediately upstream of the pilE promoter, was deleted, transcription was diminished approximately 10-fold (Hill et al., 1997; Fyfe & Davies, 1998). However, stable residual full-length pilE message is evident when total RNA is assessed by both Northern blotting and primer extension analysis (Hill et al., 1997). However, this residual message does not appear to be translated (Fig. S1, available in the online Supplementary Material). Consequently, we explored the possibility that pilE mRNA structural elements may have contributed to the above-mentioned observations. In silico analysis of the 5′ UTR of the pilE message using the Mfold (Zuker, 2003) and RNA structure (Bellaousov et al., 2013) applications revealed the potential for three thermodynamically stable stem–loop structures to form (Fig. 1a). Consequently, transcript levels, and/or protein expression, may be a subject to post-transcriptional regulation by such cis-embedded elements, especially as one of the predicted loops (designated loop 3 or L3) contains the pilE rbs and the AUG start codon. When primer extension analysis was performed using pilE mRNA, several secondary premature 5′ endpoints were observed in addition to the prominent signal for the true transcriptional start site (Fig. 1b; arrows). These secondary 5′ endpoints mapped to AU-rich regions in the predicted loop 2 (Fig. 1a; arrows). The pilE gene of N. gonorrhoeae was reported to possess three promoter sequences (designated P1, P2 and P3), with the sigma70-type P1 being the only active pilE promoter in GC (Fyfe et al., 1995). None of the 5′ endpoints maps to the tsp of these alternative promoters. However, recent analysis of transcription within the pil system has revealed the existence of alternative promoter usage using non-cognate promoter elements (Wachter et al., 2015). Whether alternative promoter usage accounts for the secondary 5′ endpoints has not been explored in the current manuscript. Consequently, the secondary 5′ endpoints mapped within the loop 2 of pilE mRNA may be products of either endonucleolytic cleavage or non-cognate promoter usage.
Fig. 1.

RNA secondary structural analysis. (a) pilE mRNA was analysed using the Mfold algorithm that revealed three potential secondary structural features with highly favourable predicted free energy values within the 5′ UTR of pilE. The loops are designated loops 1, 2 and 3. The rbs is indicated in loop 3. (b) Primer extension analysis of pilE mRNA. The arrows in both panels (b) highlight potential alternative 5′ mRNA endpoints. (c) Schematic diagram indicating the location of the alternative 5′ mRNA endpoints.
qRT-PCR was used to determine whether these putative pilE-specific RNA loop structures form. In these assays, primer pairs were designed such that the forward primer was located within each putative loop structure, with the reverse primer being located outside of the predicted loop structures. Primer design was such that the amplified products were similar in size (160 bp). Consequently, if base pairing occurred within the predicted loop structures, then it was hypothesized that the loop structures would be less accessible to the forward primer during the amplification process (Fig. 2a). pilE mRNA was prepared following transcription of the pilE gene carried on plasmid pVD203 (Bergström et al., 1986); the qRT-PCR data presented in Fig. 2b indicate the formation of all three loop structures in the pilE 5′ UTR mRNA.
Fig. 2.

Loop formation in pilE mRNA. (a) Schematic representation indicating the relative location of each forward primer. (b) qRT-PCR analysis indicating the relative amount of cDNA using the above-mentioned primers. When the loop primers were used in the qRT-PCR analysis, less-amplified products were observed when compared to the product from the primer located outside of the loop (RC3). The error bars reflect ±sd; n=3. L1 primer vs RC3, P<0.001; L2 primer vs RC3, P<0.001; L3 primer vs RC3, P<0.001.
The formation of the pilE 5′ UTR stem–loops was further investigated through site-directed mutagenesis where the predicted loop sequences were changed such that RNA secondary structure would be disrupted. Mutagenic primers were designed for each loop that would impede complementary base pairing (Fig. 3a), with individual loop stability again being assessed by the qRT-PCR assay. When the loops were mutated, the forward primer was able to gain access to the RNA yielding a product (Fig. 3b), indicating that the previous negative observations were due to pilE 5′ UTR loop formation. The data are presented in a log10 scale of pilE expression compared to expression of the internal control amp gene carried on the plasmid. The higher the log value indicates more-amplified product and relates to the loops not forming. A similar qRT-PCR approach was also utilized to examine loop stability when the putative loops were individually deleted (Fig. 3c). Again, the presence of an amplified product indicates loop destabilization. For example, when loop 2 or 3 was deleted, the RNA became accessible to the loop 1 primer; when loops 1 and 3 were deleted, the RNA became accessible to the loop 2 primer; and when loop 1 was deleted, the RNA became accessible to the loop 3 primer. However, in contrast to these observations, when loop 2 was deleted, loop 3 is still able to form.
Fig. 3.

Stability of the putative loop structures. (a) Nucleotide sequence of the 5′ UTR of pilE (in uppercase letters) and relative position of the primers used in qRT-PCRs; each putative loop sequences is boxed. The loop sequences (boldface uppercase letters) were mutated to alternative sequences (boldface lowercase letters below the sequence) that were predicted to disrupt loop formation. (b) qRT-PCR analysis using constructs containing nucleotide substitutions. (c) qRT-PCR analysis of loop deletion mutants. In (b) and (c), expression levels of pilE transcripts were calculated by subtracting expression of the internal control beta-lactamase gene carried on the plasmid. The error bars reflect ±sd; n=4. In (b), P values of the mutated constructs are compared to the non-mutated pilE construct: Loop 1 primer : L1mut, P<0.001; L2mut, P<0.001; L3mut, P<0.001 : Loop 2 primer : L1mut, P<0.001; L2mut, P<0.001; L3mut, P<0.001; Loop 3 primer: L1mut, P<0.001; L2mut, P<0.001; L3mut, P<0.001. In (c), P values of the mutated constructs are compared to the non-mutated pilE construct: Loop 1 primer: L1del, P<0.001; L2del, P=0.004; L3del, P=0.001; Loop 2 primer: L1del, P<0.001; L2del, P<0.001; L3del, P=0.001; Loop 3 primer: L1del, P<0.001; L2del, P<0.001; L3del, P<0.001.
Overall, these combined experiments indicated that (i) the stem–loops form through complementary base pairing within the pilE 5′ UTR (Fig. 2b), (ii) mutating an individual loop through site-directed mutagenesis also affects the formation/stability of the other loops (Fig. 3b) and (iii) individually deleting any of the loops also disrupts loop stability except for the one exception noted above (Fig. 3c).
The pilE 5′ UTR provides a protective role in maintaining pilE transcript levels
To determine what effect loop formation plays in pilE expression, we focused on the loop deletions and constructed a series of translational fusions, where loop 1 and loop 2 were individually deleted. The deleted pilE 5′ UTR segments were then fused in-frame to one of two truncated reporter genes, beta-galactosidase (lacZ), or, chloramphenicol acetyl transferase (cat), with each reporter gene lacking its own rbs. The deleted constructs were then compared to an equivalent WT construct; relative expression was measured by growth on solid medium, by qRT-PCR analysis and by biochemical analysis. qRT-PCRs were performed using RNAs that were isolated from the different constructs under the same conditions, thus reflecting their relative protective role. Since loop 3 contains the rbs that is needed for translation of the reporter gene, a loop 3 deletion mutant could only be used to determine relative RNA stability. The data presented in Fig. 4a show the growth of each beta-galactosidase fusion strain on solid medium containing the lacZ indicator X-Gal. The data presented in Fig. 4b assess the relative RNA levels by qRT-PCR analysis from the various pilE :: lacZ fusions, with Fig. 4c showing the corresponding biochemical analysis. From these combined experiments, we conclude that when loop 1, loop 2 or loop 3 is individually deleted, there is a significant reduction in the amount of RNA compared to WT (for all three deletion constructs, P<0.001; n=3), which is also reflected at the protein level for loop 1 and loop 2 mutants (P<0.001; n=15). Moreover, because very little relative RNA is observed with the loop 2 deletion fusion (Fig. 4b) (even though loop 3 still forms; Fig. 3c), this implies that loop 1 is likely to be the critical loop structure for protection of the pilE message. Qualitatively similar results were also obtained from comparable pilE :: cat translational fusions where the individual loops were deleted, as well as in lacZ translational fusions where the formation of the loop structures was impaired through site-directed mutagenesis. Consequently, these data imply a protective role for the pilE 5′ UTR stem–loops.
Fig. 4.

pilE translational fusion analysis. The effects of the 5′ UTR loops on the expression of pilE ::lacZ translational fusions in E. coli. (a) Growth of the different pilE::lacZ translational fusion strains (WT, loop 1 deletion and loop 2 deletion) on solid medium containing X-Gal. (b) qRT-PCR analysis of the various deletion strains compared to WT that is set at unity. Error bars reflect ±sd; n=3; L1del, P<0.001; L2del, P<0.001; and L3del, P<0.001. The expression levels were compared to a 16S RNA control. (c) Biochemical analysis of beta-galactosidase activity. The error bars reflect ±sd; n=15; when compared to WT, L1del (P<0.001) and L2del (P<0.001).
Analysis of pilE :: cat translational fusions in gonococci
To determine whether the pilE 5′ UTR loops play a similar role in the gonococcus, pilE::cat translational fusions were placed ectopically on the gonococcal chromosome within the opaE locus. As each fusion construct contained the pilE leader peptide encoding sequence, only mRNA analysis could be performed as any protein product would be secreted from the cell. The data presented in Fig. 5a show the expression levels of the fused cat RNAs produced by the WT and loop deletion constructs. Consistent with the E. coli data, individual loop deletions caused a significant decrease in expression of the reporter gene at the RNA level when compared to WT (P<0.001 for both deletion fusions; n=4).
Fig. 5.

pilE :: cat translational fusions in gonoccocci. (a) qRT-PCR analysis of the various deletion strains compared to WT that was set at unity. Expression levels were compared to a 16S RNA control. The error bars reflect ±sd; n=6; when compared to WT, L1del.cat (P=0.042) and L2del.cat (P<0.001). (b) Schematic representation of the two gonococcal constructs. It shows where the insertion mutations are located on the chromosome at the pilE and opaE loci. The insertion of the kanamycin gene prevents antisense transcription across the loop regions of the pilE from the intragenic promoters. (c) qRT-PCR analysis of the pilE::cat expression in these constructs. The opaE :: pilE :: cat is normalized to unity. The error bars reflect ±sd; n=4.
In the above-mentioned experiment, a WT copy of the pilE gene was also present within the cells (genotype pilE+ opaE :: pilE :: cat; Fig. 5b). Consequently, pilE antisense RNA that originates from the mid-gene intragenic promoter is also being produced within these cells. Consequently, this antisense RNA may bind across the 5′ UTR fusion loops (Masters et al., 2016). When the resident pilE gene was mutated through a kanamycin gene insertion that blocks pilE antisense RNA production across the loops, a twofold to threefold increase in cat RNA level was observed (Fig. 5c; P<0.001; n=4). Consequently, this observation suggests that expression of pilE cis-antisense RNA may affect stem–loop formation by making the pilE transcript more susceptible to degradation.
sRNA predicted to bind to the 5′ UTR of the pilE transcript
In many systems, trans regulatory elements are often found adjacent to the gene in question. Therefore, to test this possibility that a regulatory element resides upstream of the pilE promoter, gonococcal strains were constructed such that non-homologous gene inserts encoding erythromycin resistance were placed at regular intervals (six insertional mutants were constructed) upstream of the IHF binding site; a chloramphenicol acetyl transferase (cat) gene lacking its cognate rbs was also fused in-frame downstream of the pilE gene (Fig. 6a). RNA was then isolated from each mutant and pilE mRNA production was assessed by Northern blotting. From the blots presented in Fig. 6b, pilE transcription is apparent in all strains except for the two insertion mutants that are closest to the IHF binding site (oligonucleotide 245; insertions 5 and 6; 63 bp and 18 bp upstream of the IHF binding site; respectively). When an ermC gene cassette was placed 89 bp upstream of the IHF binding site (insert 4), pilE mRNA was observed. The production of pilS-derived sRNA (oligonucleotide 246) did not appear to be affected (Wachter et al., 2015). However, with the use of the more sensitive endpoint PCR amplifying reverse-transcribed cDNAs (real-time PCR assay), pilE message was still apparent in mutants 5 and 6. This result suggests that the pilE RNA is still being transcribed in the insertion mutants 5 and 6, albeit less efficiently when compared to transcription from the other strains, yielding less pilE transcript in the mutants 5 and 6. The difference between the observations with real-time PCR analysis and the Northern blot analysis is likely due to the sensitivity of the two assays. However, the translational efficiency of cells containing these inserts was greatly reduced as these cells displayed neither chloramphenicol resistance nor competence as the cells were non-piliated (Fig. 6c). Competence is measured by the ability of the bacteria to take up exogenous Neisseria-specific DNA via DNA transformation and is tightly linked to the piliation status of the organism as mutations causing loss of pilus expression lead to transformation incompetence; in this study, a negative competence score reflected a transformation frequency of <1×10–8 transformants per millilitre per microgram of DNA in contrast to a positive competence score of at least 1×10–3 transformants per millilitre per microgram of DNA (Koomey et al., 1991; Tønjum & Koomey, 1997). Therefore, given the above-mentioned observations, we further explored whether a regulatory element resided between inserts 4 and 6.
Fig. 6.
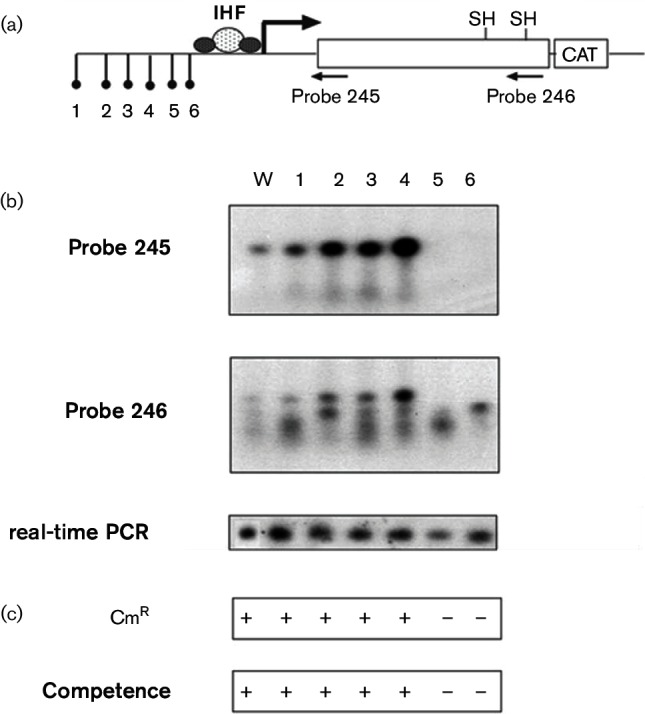
Effect of upstream insertions on pilE transcription and translation. (a) Schematic representation of the pilE gene showing the relative positions of the ermC insertions and cat translational fusion. Distance of insertions from the pilE IHF binding site: 1, 562 bp; 2, 277 bp; 3, 162 bp; 4, 89 bp; 5, 63 bp; 6, 18 bp. (b) Transcriptional analysis of pilE with Northern blot utilizing a pilE-specific and pilS-specific probes (probes 245 and 246, respectively; Wachter et al., 2015) and endpoint PCR using pilE reversed-transcribed cDNA templates (real-time PCR) of WT and the ermC insertion cells. (c) Translational analysis as determined by resistance of the gonococcal mutants to chloramphenicol (10 µg m–1) and competence for DNA transformation (a positive score reflects a transformation efficiency of approximately 1×10–3 transformants per millilitre per microgram DNA; a negative score reflects a transformation efficiency of <1×10–8 transformants per millilitre per microgram DNA).
The previously described N. gonorrhoeae small RNA transcriptome (Wachter & Hill, 2015) was assessed for potential sRNA molecules within this region and a single sRNA (designated asRNA7) was found. An oligonucleotide probe was designed to recognize this antisense sRNA species, and a strong signal was observed at approximately 100 bp on a Northern blot (Fig. 7a). In silico hybridization analysis was then performed between the putative asRNA7 and the 5′ UTR of the pilE transcript, with complementary binding being predicted with a favourable free energy (△G=−28.3 kcal mol−1) (Fig. 7b). Therefore, if asRNA7 binds to the 5′ UTR of the pilE transcript, such binding could potentially denature the secondary loop structures and expose the ribosomal binding site, thus allowing for translation. Complementation of insertion mutants 5 and 6 with the asRNA7 gene placed within the opaE locus (opaE :kan:DUS:asRNA7) caused the cells to become piliated, regain competence and express WT levels of pilE mRNA (Fig. 8). Consequently, asRNA7 apparently stabilizes the pilE transcript in readiness for translation.
Fig. 7.
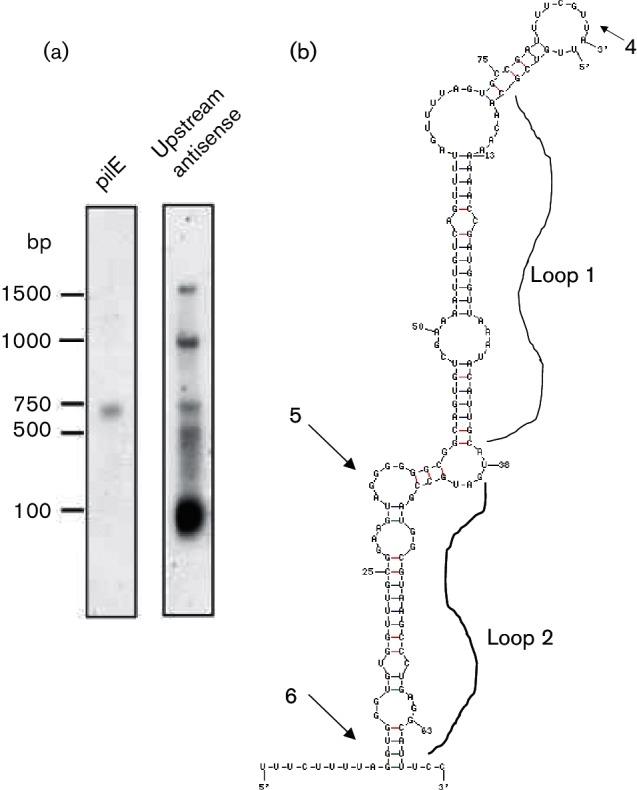
Analysis of asRNA7. (a) Northern blot analysis of total RNA isolated from N. gonorrhoeae. The left panel is probed with a pilE-specific probe 245 (Wachter et al., 2015); the right panel is probed with an oligonucleotide designed to bind to asRNA7. (b) Predicted interactions of asRNA7 and pilE 5′ UTR mRNA. asRNA7 has the potential to bind the 5′ UTR of pilE and expose the ribosomal binding site. The sites of the ermC insertions 4, 5 and 6 are indicated by arrows on the figure. This predicted interaction would be energetically favourable, with a ∆G=−28.7 kcal mol−1.
Fig. 8.

qRT-PCR analysis of asRNA7 constructs. qRT-PCR analysis of WT, asRNA7 insertion mutant 6 (∆asRNA7 : ermC6) and asRNA7 complements 1, 2 and 3 (∆asRNA7 : ermC6:opaE : asRNA7 : kan1, 2 and 3) utilizing primer pairs specific for recA and the 5′ end of pilE (Masters et al., 2016). The relative log difference as compared to an external RNA3 control. The error bars reflect ±sd; n=6; P<0.001.
Discussion
The impetus for the current study was the in silico identification of three putative stem–loop structures in the 5′ UTR of the pilE transcript. The loops were shown to form in pilE mRNA (Fig. 2) and disruption of these sequences, either by site-directed mutagenesis or by individually removing the loop sequences, destabilized loop stability causing the mRNA to be more susceptible to degradation (Figs 3, 4 and 5). Consequently, the 5′ UTR loops appear to be able to protect pilE mRNA from degradation. Similar observations have been made where loop structures within the 5′ UTR of the ompA, rne and cspE mRNA in E. coli (Arnold et al., 1998; Uppal et al., 2008; Schuck et al., 2009), as well as within the 5′ UTR of the ermC mRNA in Bacillus subtilis (Bechhofer & Dubnau, 1987), protect the mRNA, as in each case the presence of a loop prolonged mRNA half-lives. How the pilE loop structures protect the RNA is currently under investigation.
Loop structures in a 5′ UTR can determine the fate of transcripts not only by controlling stability but also by influencing translational efficiency (Régnier & Arraiano, 2000; Marzi et al., 2008). The presence of RNA secondary structures in the pilE 5′ UTR region may explain why residual pilE mRNA remained when the IHF binding site located upstream of the pilE promoter was deleted (Hill et al., 1997). However, what was not evident in that study was why this residual mRNA was not translated into PilE polypeptide (Fig. S1). A possible explanation for the lack of translation is that in the construction of the pilE IHF deletion mutants, not only was the IHF binding site deleted but also other upstream DNA was removed, causing the asRNA7 gene to be absent as well (Hill et al., 1997). Consequently, without asRNA7, the loop structures would remain, with the pilE ribosome binding site still being occluded, thus preventing translation. Likewise, with the insertion mutants 5 and 6, where asRNA7 antisense RNA is also absent, pilE mRNA in each of these mutants is unstable with any residual pilE transcripts also apparently not being translated (Figs 6 and 8). Interestingly, the insertions at the positions 2, 3 and 4 appear to stimulate pilE transcription (Fig. 6b, probe 245). Since these locations are within the vicinity of the pilE-specific, G4-associated asRNA and its promoter region (Cahoon & Seifert, 2013), it could be that these insertions negate any cis-mediated effects caused by G4 asRNA transcription that, in turn, enhances production of the asRNA7 from its promoter resulting in an increase in stabilization of the pilE primary transcript in these mutants. When the asRNA7 gene complements insertion mutants 5 and 6, pilE mRNA is again observed and is translated yielding a pilus + phenotype. Therefore, as the asRNA7 antisense RNA is predicted to bind to the pilE 5′ UTR across loops 1 and 2, it would appear that asRNA7 serves as a small RNA that facilitates mRNA protection, and, after binding, loop 3 presumably opens allowing access to the previously occluded ribosome binding site. Consequently, the pilE transcript could now be translated into PilE polypeptide. A slightly similar scenario has recently been presented regarding an operon involved in Type IV DNA secretion in the gonococcus. In this study, an RNA switch mechanism that involves two putative stem–loop structures contained within the 5′ UTR of the secretion operon has been proposed, one of which occludes the rbs within a putative loop structure; this occluded rbs is then released under certain conditions thus allowing for translation (Ramsey et al., 2015).
In a previous study, it was demonstrated that there exists an inverse relationship between the level of pilE sense RNA levels and antisense RNA production across the pilE gene (Masters et al., 2016). Consequently, a titration model was proposed whereby the presence of pilE antisense transcription helped determine the amount of pilE sense transcript levels. In the analysis of the cat translational fusions in the gonococcus (Fig. 5), elimination of pilE-specific antisense RNA derived from the midgene antisense promoter allowed twofold to threefold more message to be observed, suggesting that pilE antisense transcription may either impede loop formation in the 5′ UTR or alternatively compete with asRNA7 for binding to loops 1 and 2. Therefore, it would seem that there needs to be an orchestrated coordination of antisense RNA production across the pilE locus (both within the pilE gene and upstream with transcription of the asRNA7 gene) in order to obtain optimal transcript levels and to maintain appropriate PilE polypeptide levels. Whether this is achieved by differential promoter strengths, coordinated IHF binding or varying supercoiling fluxes across the pilE locus is currently unknown. Regardless, what has become apparent in this study is that, for a gene where no apparent regulatory protein has been identified, a complex regulatory circuit exists to maintain transcript levels operating in conjunction with a sophisticated translational scheme in order to optimize production of this important virulence determinant in the gonococcus.
Acknowledgement
This study was supported by National Institutes of Health grant 1R15 AI072720-01A1 to S. A. H.
Supplementary Data
Abbreviations:
- DUS
DNA uptake sequence
- IHF
integration host factor
- qRT-PCR
quantitative real-time PCR
- rbs
ribosomal binding site
- sRNA
small antisense RNA
- UTR
untranslated region
Footnotes
Edited by: P. W. O'Toole
Edited by: D. Grainger
One supplementary figure is available with the online Supplementary Material.
References
- Arnold T. E., Yu J., Belasco J. G.(1998). mRNA stabilization by the ompA 5′ untranslated region: two protective elements hinder distinct pathways for mRNA degradation. RNA 4319–330. [PMC free article] [PubMed] [Google Scholar]
- Bechhofer D. H., Dubnau D.(1987). Induced mRNA stability in Bacillus subtilis. Proc Natl Acad Sci U S A 84498–502. 10.1073/pnas.84.2.498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellaousov S., Reuter J. S., Seetin M. G., Mathews D. H.(2013). RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 41W471–W474. 10.1093/nar/gkt290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström S., Robbins K., Koomey J. M., Swanson J.(1986). Piliation control mechanisms in Neisseria gonorrhoeae. Proc Natl Acad Sci U S A 833890–3894. 10.1073/pnas.83.11.3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon L. A., Seifert H. S.(2013). Transcription of a cis-acting, noncoding, small RNA is required for pilin antigenic variation in Neisseria gonorrhoeae. PLoS Pathog 9e1003074. 10.1371/journal.ppat.1003074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrick C. S., Fyfe J. A., Davies J. K.(1997). The normally silent sigma54 promoters upstream of the pilE genes of both Neisseria gonorrhoeae and Neisseria meningitidis are functional when transferred to Pseudomonas aeruginosa. Gene 19889–97. 10.1016/S0378-1119(97)00297-7 [DOI] [PubMed] [Google Scholar]
- Deana A., Celesnik H., Belasco J. G.(2008). The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature 451355–359. 10.1038/nature06475 [DOI] [PubMed] [Google Scholar]
- Dietrich M., Munke R., Gottschald M., Ziska E., Boettcher J. P., Mollenkopf H., Friedrich A.(2009). The effect of hfq on global gene expression and virulence in Neisseria gonorrhoeae. FEBS J 2765507–5520. 10.1111/j.1742-4658.2009.07234.x [DOI] [PubMed] [Google Scholar]
- Fyfe J. A., Carrick C. S., Davies J. K.(1995). The pilE gene of Neisseria gonorrhoeae MS11 is transcribed from a sigma 70 promoter during growth in vitro. J Bacteriol 1773781–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe J. A., Davies J. K.(1998). An AT-rich tract containing an integration host factor-binding domain and two UP-like elements enhances transcription from the pilEp1 promoter of Neisseria gonorrhoeae. J Bacteriol 1802152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georg J., Hess W. R.(2011). cis-Antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev 75286–300. 10.1128/MMBR.00032-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S. A., Samuels D. S., Carlson J. H., Wilson J., Hogan D., Lubke L., Belland R. J.(1997). Integration host factor is a transcriptional cofactor of pilE in Neisseria gonorrhoeae. Mol Microbiol 23649–656. 10.1046/j.1365-2958.1997.2321612.x [DOI] [PubMed] [Google Scholar]
- Hoe C. H., Raabe C. A., Rozhdestvensky T. S., Tang T. H.(2013). Bacterial sRNAs: regulation in stress. Int J Med Microbiol 303217–229. 10.1016/j.ijmm.2013.04.002 [DOI] [PubMed] [Google Scholar]
- Kellogg D. S., Cohen I. R., Norins L. C., Schroeter A. L., Reising G.(1968). Neisseria gonorrhoeae II. Colonial variation and pathogenicity during 35 months in vitro. J Bacteriol 96596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koomey M., Bergstrom S., Blake M., Swanson J.(1991). Pilin expression and processing in pilus mutants of Neisseria gonorrhoeae: critical role of Gly-1 in assembly. Mol Microbiol 5279–287. 10.1111/j.1365-2958.1991.tb02108.x [DOI] [PubMed] [Google Scholar]
- Marzi S., Fechter P., Chevalier C., Romby P., Geissmann T.(2008). RNA switches regulate initiation of translation in bacteria. Biol Chem 389585–598. 10.1515/BC.2008.055 [DOI] [PubMed] [Google Scholar]
- Masters T. L., Wachter S., Wachter J., Hill S. A.(2016). H-NS suppresses pilE intragenic transcription and antigenic variation in Neisseria gonorrhoeae. Microbiology 162177–190. 10.1099/mic.0.000199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannekoek Y., Huis in 't Veld R., Hopman C. T., Langerak A. A., Speijer D., van der Ende A.(2009). Molecular characterization and identification of proteins regulated by Hfq in Neisseria meningitidis. FEMS Microbiol Lett 294216–224. 10.1111/j.1574-6968.2009.01568.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey M. E., Bender T., Klimowicz A. K., Hackett K. T., Yamamoto A., Jolicoeur A., Callaghan M. M., Wassarman K. M., van der Does C., Dillard J. P.(2015). Targeted mutagenesis of intergenic regions in the Neisseria gonorrhoeae gonococcal genetic island reveals multiple regulatory mechanisms controlling type IV secretion. Mol Microbiol 971168–1185. 10.1111/mmi.13094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Régnier P., Arraiano C. M.(2000). Degradation of mRNA in bacteria: emergence of ubiquitous features. BioEssays 22235–244. [DOI] [PubMed] [Google Scholar]
- Schuck A., Diwa A., Belasco J. G.(2009). RNase E autoregulates its synthesis in Escherichia coli by binding directly to a stem-loop in the rne 5′ untranslated region. Mol Microbiol 72470–478. 10.1111/j.1365-2958.2009.06662.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittka A., Pfeiffer V., Tedin K., Vogel J.(2007). The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol Microbiol 63193–217. 10.1111/j.1365-2958.2006.05489.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J.(1973). Studies on gonococcus infection. IV. Pili: their role in attachment of gonococci to tissue culture cells. J Exp Med 127571–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J.(1982). Colony opacity and protein II compositions of gonococci. Infect Immun 37359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J., Robbins K., Barrera O., Corwin D., Boslego J., Ciak J., Blake M., Koomey J. M.(1987). Gonococcal pilin variants in experimental gonorrhea. J Exp Med 1651344–1357. 10.1084/jem.165.5.1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tønjum T., Koomey M.(1997). The pilus colonization factor of pathogenic neisserial species: organelle biogenesis and structure/function relationships – a review. Gene 192155–163. 10.1016/S0378-1119(97)00018-8 [DOI] [PubMed] [Google Scholar]
- Uppal S., Akkipeddi V. S., Jawali N.(2008). Posttranscriptional regulation of cspE in Escherichia coli: involvement of the short 5′-untranslated region. FEMS Microbiol Lett 27983–91. 10.1111/j.1574-6968.2007.01009.x [DOI] [PubMed] [Google Scholar]
- Wachter J., Hill S. A.(2015). Small transcriptome analysis indicates that the enzyme RppH influences both the quality and quantity of sRNAs in Neisseria gonorrhoeae. FEMS Microbiol Lett 3621–7. 10.1093/femsle/fnu059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter J., Masters T. L., Wachter S., Mason J., Hill S. A.(2015). pilS loci in Neisseria gonorrhoeae are transcriptionally active. Microbiology 1611124–1135. 10.1099/mic.0.000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade J. T., Grainger D. C.(2014). Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol 12647–653. 10.1038/nrmicro3316 [DOI] [PubMed] [Google Scholar]
- Waters L. S., Storz G.(2009). Regulatory RNAs in bacteria. Cell 136615–628. 10.1016/j.cell.2009.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M.(2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 313406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.