

Abstract
The inhibitory activity of coronaridine congeners on human (h) α4β2 and α7 nicotinic acetylcholine receptors (AChRs) is determined by Ca2+ influx assays, whereas their effects on neurons in the ventral inferior (VI) aspect of the mouse medial habenula (MHb) are determined by patch-clamp recordings. The Ca2+ influx results clearly establish that coronaridine congeners inhibit hα3β4 AChRs with higher selectivity compared to hα4β2 and hα7 subtypes, and with the following potency sequence, for hα4β2: (±)-18-methoxycoronaridine [(±)-18-MC] > (+)-catharanthine > (±)-18-methylaminocoronaridine [(±)-18-MAC] ∼ (±)-18-hydroxycoronaridine [(±)-18-HC]; and for hα7: (+)-catharanthine > (±)-18-MC > (±)-18-HC > (±)-18-MAC. Interestingly, the inhibitory potency of (+)-catharanthine (27 ± 4 μM) and (±)-18-MC (28 ± 6 μM) on MHb (VI) neurons was lower than that observed on hα3β4 AChRs, suggesting that these compounds inhibit a variety of endogenous α3β4* AChRs. In addition, the interaction of bupropion with (−)-ibogaine sites on hα3β4 AChRs is tested by [3H]ibogaine competition binding experiments. The results indicate that bupropion binds to ibogaine sites at desensitized hα3β4 AChRs with 2-fold higher affinity than at resting receptors, suggesting that these compounds share the same binding sites. In conclusion, coronaridine congeners inhibit hα3β4 AChRs with higher selectivity compared to other AChRs, by interacting with the bupropion (luminal) site. Coronaridine congeners also inhibit α3β4*AChRs expressed in MHb (VI) neurons, supporting the notion that these receptors are important endogenous targets for their anti-addictive activities.
Keywords: Nicotinic acetylcholine receptor, Coronaridine congeners, 18-Methoxycoronaridine, (+)-Catharanthine, Medial habenula, Brain slices
1. Introduction
Pharmacologically, coronaridine congeners, including (−)-ibogaine and (±)-18-methoxycoronaridine [(±)-18-MC], behave as noncompetitive antagonists (NCAs) of several nicotinic acetylcholine receptors (AChRs) (Glick et al., 2002a; Pace et al., 2004; Arias et al., 2010a,b, 2011; Arias et al., 2015). From the therapeutic point of view, these compounds decrease drug self-administration in animals (Glick et al., 2000, 2002a,b; Maissoneuve and Glick, 2003) and interrupt drug dependence in humans (reviewed in Alper et al., 2008). In this regard, clinical trials are being conducted by the company Savant HWP to determine the potential therapeutic use and safety of 18-MC for nicotine and other drugs dependence.
An important distinction between the toxicity of (±)-18-MC and (−)-ibogaine is that the former compound has less side effects than the latter, which may produce hallucinogenic, cardiac (e.g., arrhythmia and bradycardia), and tremogenic effects, especially after prolonged use (Glick et al., 2000; Maissoneuve and Glick, 2003). The safer activity of (±)-18-MC compared to (−)-ibogaine has been ascribed to its higher receptor selectivity towards α3β4 AChRs. However, the only experiment suggesting such receptor selectivity is based on a qualitative comparison between voltage-clamp recordings showing that 20 μM (±)-18-MC inhibits only α3β4 AChRs, whereas 20 μM (−)-ibogaine inhibits both α3β4 and α4β2 subtypes (Glick et al., 2002a). To clarify this subject in a more thorough manner, the inhibitory potency (IC50) of several coronaridine congeners, including (±)-18-MC, (+)-catharanthine, (±)-18-methylaminocoronaridine [(±)-18-MAC], and (±)-18-hydroxycoronaridine [(±)-18-HC] (see molecular structures in Fig. 1), is determined on hα4β2 and hα7 AChRs and subsequently compared to that previously obtained on hα3β4 AChRs (Arias et al., 2015).
Fig. 1.
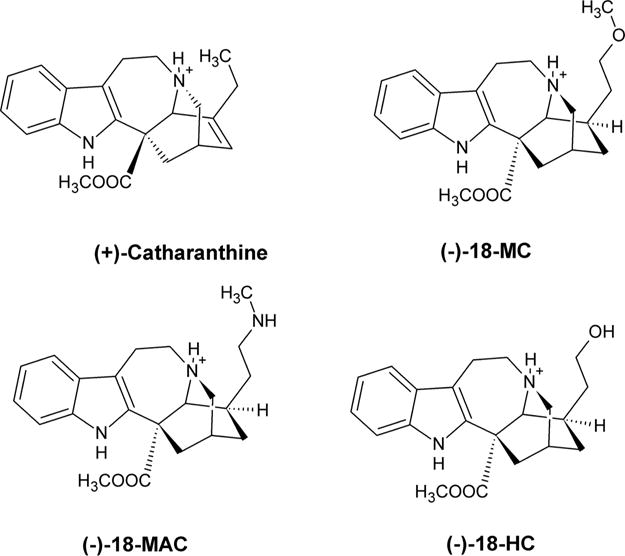
Molecular structure of coronaridine congeners in the protonated state, including (−)-18-MC [(−)-18-methoxycoronaridine], (−)-18-HC [(−)-18-hydroxycoronaridine], (−)-18-MAC [(−)-18-methylaminocoronaridine], and (+)-catharanthine [(+)-3,4-didehydrocoronaridine].
Previous studies support the hypothesis that the inhibition of α3β4* AChRs expressed in the habenulo-interpeduncular cholinergic pathway modulates the dopaminergic brain reward circuitry located in the mesocorticolimbic system, and that this is the main mechanism underlying the anti-addictive properties of coronaridine congeners (McCallum et al., 2012; Glick et al., 2002a,b, 2011; Maissoneuve and Glick, 2003; reviewed in Ortells and Arias, 2010). Experiments showing that the local administration of 18-MC in the medial habenula (MHb) diminishes nicotine self-administration (Glick et al., 2011) support the above mentioned hypothesis. The same as results showing that lower doses of the antidepressant bupropion and 18-MC maintain the beneficial activity with less side effects (Maissoneuve and Glick, 2003), probably due to the demonstrated bupropion-induced α3β4 AChR inhibition (reviewed in Arias et al., 2014). Nevertheless, no evidence of direct inhibition of habenular α3β4* AChRs by 18-MC, nor a structural interaction between bupropion and (−)-ibogaine sites at α3β4 AChRs, has been clearly demonstrated. In this regard, we sought to determine the activity of (±)-18-MC and (+)-catharanthine on MHb by brain slice electrophysiology recordings of ventral inferior (VI) MHb neurons, strongly expressing α3β4* AChRs (Quick et al., 1999; Shih et al., 2014, 2015), as well as to determine the pharmacological interaction of bupropion with (−)-ibogaine sites at hα3β4 AChRs in different conformational states by radioligand competition binding experiments (Arias et al., 2015).
A better understanding of the functional interaction and selectivity of coronaridine congeners for neuronal AChRs, especially α3β4* AChRs, expressed in heterologous cells and endogenous neurons is crucial to develop novel analogs for safer anti-addictive therapies.
2. Materials and methods
2.1. Materials
[3H]Ibogaine (23 Ci/mmol) and ibogaine hydrochloride were obtained through the National Institute on Drug Abuse (NIDA) (NIH, Baltimore, USA). [3H]Epibatidine (45.1 Ci/mmol) was purchased from PerkinElmer Life Sciences Products, Inc. (Boston, MA, USA). (±)-18-Methoxycoronaridine hydrochloride [(±)-18-MC] was purchased from Obiter Research, LLC (Champaign, IL, USA). (±)-18-Methylaminocoronaridine [(±)-18-MAC], (+)-catharanthine hydrochloride, and (±)-18-hydroxycoronaridine [(±)-18-HC] were a gift from Dr. Kuehne (University of Vermont, VT, USA). (±)-Epibatidine and QX-314 were obtained from Tocris Bioscience (Ellisville, MO, USA). Fluo-4 was obtained from Molecular Probes (Eugene, Oregon, USA). Euthasol (sodium pentobarbital, 100 mg/kg; sodium phenytoin, 12.82 mg/kg) was obtained from LeVet Pharma (Oudewater, Netherlands). Polyethylenimine, acetylcholine (ACh), (−)-nicotine hydrogen tartrate, bupropion hydrochloride, probenecid, and bovine serum albumin (BSA), were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Salts were of analytical grade.
2.2. Mice
An animal study protocol pertaining to this study (#IS00003604) was reviewed and approved by the Northwestern University Institutional Animal Care and Use Committee. Procedures also followed the guidelines for the care and use of animals provided by the National Institutes of Health (NIH) Office of Laboratory Animal Welfare. Mice were housed at 22 °C on a 12-h light/dark cycle with food and water ad libitum. Mice were weaned on postnatal day 21 and housed with same-sex littermates. Experiments were conducted on C57BL/6J mice obtained from Jackson Laboratories. All studies were restricted to male mice, age 8–24 weeks.
2.3. Ca2+ influx measurements in cells expressing hα4β2 or hα7 AChRs
Ca2+ influx measurements were performed on HEK293-hα4β2 and GH3-hα7 cells as previously described (Arias et al., 2010c, 2016). Briefly, 5 × 104 cells per well were seeded 72 h prior to the experiment on black 96-well plates (Costar, New York, USA) and incubated at 37 °C in a humidified atmosphere (5% CO2/95% air). Under these conditions, the majority of expressed hα4β2 AChRs have the (α4)3(β2)2 stoichiometry (see Arias et al., 2016, and references therein). 16–24 h before the experiment, the medium was changed to 1% BSA in HEPES-buffered salt solution (HBSS) (130 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 0.8 mM MgSO4, 0.9 mM NaH2PO4, 25 mM glucose, 20 mM Hepes, pH 7.4). On the day of the experiment, the medium was removed by flicking the plates and replaced with 100 μL HBSS/1% BSA containing 2 mM Fluo-4 and 2.5 mM probenecid. The cells were then incubated at 37 °C in a humidified atmosphere (5% CO2/95% air) for 1 h.
To determine the antagonistic activity of each coronaridine conger (Fig. 1), plates were flicked to remove excess of Fluo-4, washed twice with HBSS/1% BSA, refilled with 100 μL of HBSS containing different concentrations of the ligand under study, and finally pre-incubated for 5 min. Plates were finally placed in the cell plate stage of the fluorescent imaging plate reader (FLIRP: Molecular Devices, Sunnyvale, CA, USA), and (±)-epibatidine (0.1 μM for hα4β2 AChRs or 1.0 μM for hα7 AChRs) added from the agonist plate to the cell plate using the 96-tip pipettor simultaneously to fluorescence recordings for a total length of 3 min. A baseline consisting of 5 measurements of 0.4 s each was recorded. (±)-The laser excitation and emission wavelengths are 488 and 510 nm, at 1 W, and a CCD camera opening of 0.4 s.
2.4. Brain slice preparation
Brain slices were prepared as previously described (Shih et al., 2014, 2015). Mice were anesthetized with Euthasol (sodium pentobarbital, 100 mg/kg; sodium phenytoin, 12.82 mg/kg) before transcardiac perfusion with oxygenated (95% O2/5% CO2), 4 °C N-methyl-D-glucamine (NMDG)-based recovery solution that contains (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4·7H2O, and 0.5 CaCl2·2H2O; 300–310 mOsm; pH 7.3–7.4). Brains were immediately dissected after the perfusion and held in oxygenated, 4 °C recovery solution for one minute before cutting a brain block containing the MHb and sectioning the brain with a vibratome (VT1200S; Leica). Coronal slices (250 μm) were sectioned through the medial habenula and transferred to oxygenated, 33 °C recovery solution for 12 min. Slices were then kept in holding solution (containing in mM: 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 2 MgSO4·7H2O, and 2 CaCl2·2H2O; 300–310 mOsm; pH 7.3–7.4) for 60 min or more before recordings.
Brain slices were transferred to a recording chamber being continuously superfused at a rate of 1.5–2.0 mL/min with oxygenated 32 °C recording solution. The recording solution contained (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 12.5 glucose, 2 MgSO4·7H2O, and 2 CaCl2·2H2O; 300–310 mOsm; pH 7.3–7.4). Patch pipettes were pulled from borosilicate glass capillary tubes (1B150F-4; World Precision Instruments) using a programmable microelectrode puller (P-97; Sutter Instrument). Tip resistance ranged from 4.5 to 8.0 MΩ when filled with internal solution. The following internal solution was used (in mM): 135 potassium gluconate, 5 EGTA, 0.5 CaCl2, 2 MgCl2, 10 HEPES, 2 MgATP, and 0.1 GTP; pH adjusted to 7.25 with Tris base; osmolarity adjusted to 290 mOsm with sucrose. This internal solution also contained QX-314 (2 mM) for improved voltage control.
2.5. Patch clamp recording
Neurons within brain slices were visualized with infrared or visible differential interference contrast (DIC) optics. Neurons in the ventral inferior (VI) aspect of the MHb were targeted for recordings, as previously described (Shih et al., 2014, 2015). Electrophysiology experiments were conducted using a Scientifica SliceScope upright microscope. A computer running pCLAMP 10 software was used to acquire whole-cell recordings along with an Axopatch 200B amplifier and an A/D converter (Digidata 1440A). pClamp software and acquisition hardware were from Molecular Devices. Data were sampled at 10 kHz and low-pass filtered at 1 kHz. Immediately prior to gigaseal formation, the junction potential between the patch pipette and the superfusion medium was nulled. Series resistance was uncompensated.
To record physiological events following local application of drugs, a drug-filled pipette was moved to within 20–40 μm of the recorded neuron using a second micromanipulator. The drug (dissolved in recording solution) was dispensed onto the recorded neuron by using a Picopump (World Precision Instruments) at an ejection pressure of 12 psi for 250 ms. The ejection volume varied depending on the goal of the experiment. Atropine (1 μM) was present in the superfusion medium when using ACh application to prevent activation of muscarinic AChRs.
2.6. [3H]Ibogaine competition binding experiments using hα3β4 AChRs-containing membranes
To determine whether the antidepressant bupropion binds to the coronaridine binding sites at hα3β4 AChRs in different conformational states, [3H]ibogaine competition binding experiments were performed using hα3β4 AChR-containing membranes prepared from HEK293-hα3β4 cells as previously described (Arias et al., 2010b). In this regard, hα3β4 AChR membranes (1.0–1.5 mg/mL) were suspended in binding saline buffer (50 mM Tris–HCl, 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, pH 7.4) and pre-incubated with 16.6 nM [3H]ibogaine in the absence (receptors are mainly in the resting state) and presence of 1 μM (−)-nicotine (receptors are mainly in the desensitized state) for 30 min at room temperature (RT). The total volume was divided into aliquots, and increasing concentrations of the ligand under study were added to each tube and incubated for 2 h at RT. The nonspecific binding was determined in the presence of 100 μM (−)-ibogaine.
AChR-bound [3H]ibogaine was then separated from free ligand by a filtration assay using a 48-sample harvester system with GF/B Whatman filters (Brandel Inc., Gaithersburg, MD), previously soaked with 0.5% polyethylenimine for 30 min. The membrane-containing filters were transferred to scintillation vials with 3 mL of Bio-Safe II (Research Product International Corp, Mount Prospect, IL), and the radioactivity was determined using a Beckman 6500 scintillation counter (Beckman Coulter, Inc., Fullerton, CA).
2.7. Analysis methods
The concentration–response results from heterologous cells and MHb neurons, as well as from the radioligand competition binding experiments were curve-fitted by nonlinear least squares analysis using the Prism software (GraphPad Software, San Diego, CA), and the EC50, IC50, and nH values for the studied ligands calculated. The obtained IC50 values for bupropion were transformed into an inhibition constant (Ki) values using the Cheng–Prusoff relationship (Cheng and Prusoff, 1973):
| (1) |
where [[3H]ligand] is the initial concentration of [3H]ibogaine or [3H] epibatidine, and Kdligand is the dissociation constant for [3H]ibogaine at the hα3β4 AChR (0.46 μM; Arias et al., 2010c).
To compare the affinity of bupropion at hα3β4 AChRs in different conformational states, the Student t-test (two-tail) was used.
3. Results
3.1. Inhibitory potency and AChR selectivity for coronaridine congeners
The activation potency of (±)-epibatidine on hα4β2 and hα7 AChRs was first determined by assessing the fluorescence change in the respective hα4β2- (Fig. 2) and hα7-expressing cells (Fig. 3) after (±)-epibatidine stimulation. The respective EC50 values for (±)-epibatidine (12 ± 5 and 26 ± 4 nM) are in the same range as previous determinations (Arias et al., 2010c, 2016).
Fig. 2.
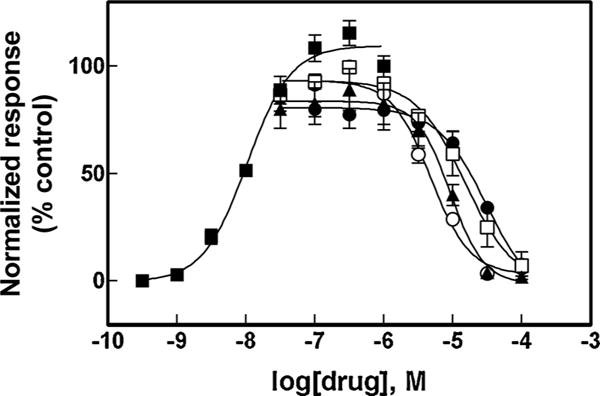
Effect of coronaridine congeners on (±)-epibatidine-induced Ca2+ influx in HEK293-hα4β2 cells. Increased concentrations of (±)-epibatidine (■) activated hα4β2 AChRs with potency EC50 = 12 ± 5 nM (n = 25). Subsequently, cells were pre-treated (5 min) with several concentrations of (+)-catharanthine (▲), (±)-18-MC (○), (±)-18-MAC (□), and (±)-18-HC (●), followed by addition of 0.1 μM (±)-epibatidine. Response was normalized to the maximal (±)-epibatidine response which was set as 100%. The plots are representative of four determinations, where the error bars are the S.D. The calculated IC50 and nH values are summarized in Table 1.
Fig. 3.
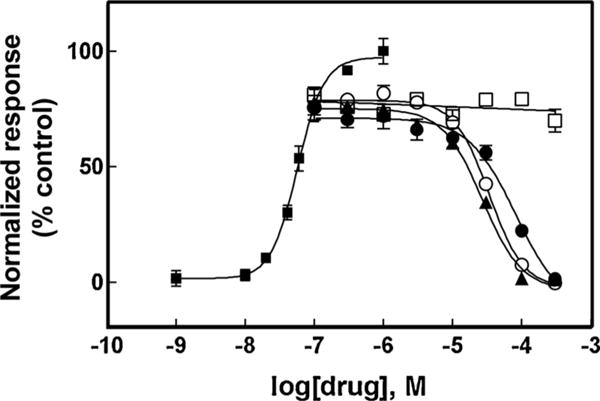
Effect of coronaridine congeners on (±)-epibatidine-induced Ca2+ influx in GH3-hα7 cells. Increased concentrations of (±)-epibatidine (■) activated hα7 AChRs with potency EC50 = 26 ± 4 nM (n = 25). Subsequently, cells were pre-treated (5 min) with several concentrations of (+)-catharanthine (▲), (±)-18-MC (○), (±)-18-MAC (□), and (±)-18-HC (●), followed by addition of 1.0 μM (±)-epibatidine. Response was normalized to the maximal (±)-epibatidine response which was set as 100%. The plots are representative of four determinations, where the error bars are the S.D. The calculated IC50 and nH values are summarized in Table 1.
The inhibitory activity of several coronaridine congeners (Fig. 1) was subsequently assessed by pre-incubating each compound with hα4β2- (Fig. 2) and hα7-expressing cells (Fig. 3) for 5 min before (±)-epibatidine stimulation (0.1 and 1.0 μM, respectively). Interestingly, each coronaridine congener inhibited (±)-epibatidine-induced hα4β2 and hα7 AChR activity with IC50 values that depend on the ligand structure and AChR subtype (Table 1). For example, the ligand potency for the hα4β2 AChR follows the rank order: (±)-18-MC > (+)-catharanthine > (±)-18-MAC ∼ (±)-18-HC, which is slightly different to that for hα7 AChRs: (+)-catharanthine > (±)-18-MC > (±)-18-HC > (±)-18-MAC (Table 1).
Table 1.
Inhibitory potency (IC50) of coronaridine congeners at hα4β2 and ha7 AChRs determined by Ca2+ influx assays.
| Congener | hα4β2
|
hα7
|
hα4β2/hα3β4c (x-fold) | hα7/hα3β4c (x-fold) | ||
|---|---|---|---|---|---|---|
| IC50, μMa | nHb | IC50, μMa | nHb | |||
| (±)-18-MC | 6.3 ± 1.3 | 1.85 ± 0.17 | 34.7 ± 5.0 | 2.21 ± 0.50 | 4.3 | 23.6 |
| (+)-Catharanthine | 12.6 ± 1.8 | 1.48 ± 0.25 | 21.8 ± 2.5 | 1.43 ± 0.20 | 18.5 | 32.0 |
| (±)-18-MAC | 20.7 ± 3.0 | 1.40 ± 0.14 | No effect | – | 7.9 | – |
| (±)-18-HC | 24.8 ± 1.2 | 1.27 ± 0.06 | 73.3 ± 10.8 | 1.64 ± 0.20 | 8.8 | 26.1 |
Hill coefficient.
The calculated ratios were obtained using the data on hα3β4 AChRs previously published (Arias et al., 2015).
Comparing the IC50 values obtained in the hα4β2 and hα7 AChRs with that obtained in hα3β4 AChRs (Arias et al., 2015), the following receptor selectivity sequence was obtained: hα3β4 > hα4β2 > hα7. Illustrating this selectivity, the values for (+)-catharanthine at hα3β4 AChRs are 18- and 32-fold higher than that for the respective hα4β2 and hα7 AChRs (Table 1).
The results showing that the nH values for the majority of the congeners [with exception of (±)-18-HC] are higher than unity (Table 1) indicate that the inhibitory process is mediated by a cooperative mechanism. A cooperative mechanism, in turn, suggests that there is potentially more than one binding site or several mechanisms of inhibition.
3.2. Inhibition of ACh-evoked currents from MHb (VI) neurons by coronaridine congeners
Patch-clamp recordings on MHb (VI) neurons showed that 100 μM ACh puffs activated endogenous AChRs (see control traces in Figs. 4A and 5A). MHb (VI) neurons were identified primarily by their close proximity (< 50–70 μm) to the 3rd ventricle within the ventral aspect of the MHb. They were secondarily distinguished from other nearby brain areas (e.g., thalamus and lateral habenula) via the presence of slow (1–8 Hz) tonic firing (Shih et al., 2014).
Fig. 4.
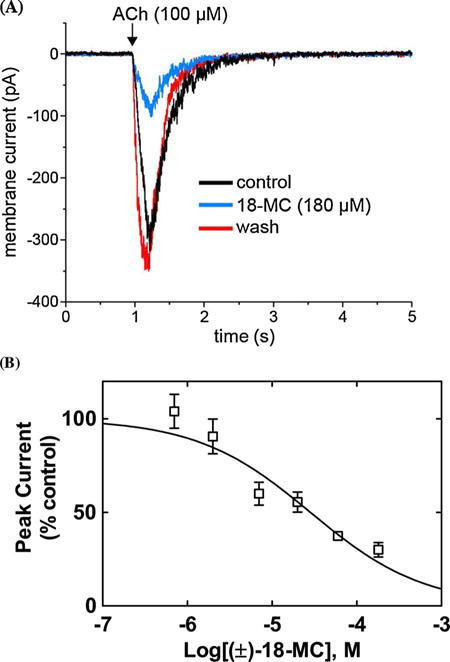
Inhibitory potency of (±)-18-MC on ACh-evoked currents from MHb (VI) neurons. (A) ACh puffer (100 μM)-evoked currents from MHb (VI) neurons are decreased by 180 μM (±)-18-MC. The puffer was performed for 250 ms at a pressure of 12 psi. After washing (13 min), the peak amplitude reached the same levels as that observed in control neurons, indicating a reversible inhibition. (B) Concentration-response relationship for the inhibitory activity of (±)-18-MC on ACh-evoked currents from MHb (VI) neurons. Response was normalized to the maximal ACh response which was set as 100%. The plot (r2 = 0.80) is representative of 5–7 determinations, where the error bars are the S.D. The calculated IC50 and nH values are summarized in Table 2.
Fig. 5.
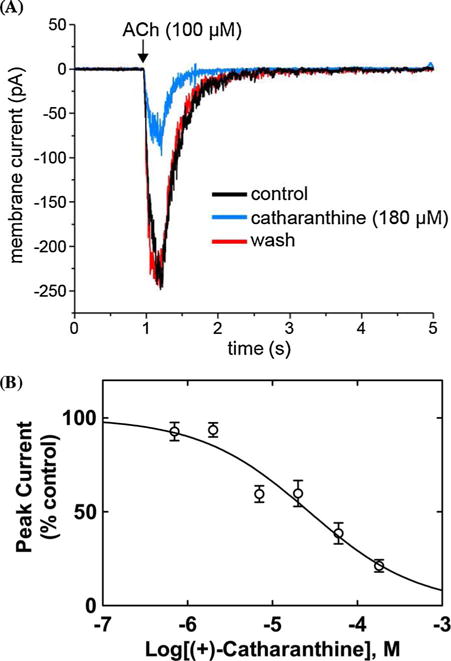
Inhibitory potency of (+)-catharanthine on ACh-evoked currents from MHb (VI) neurons. (A) ACh puffer (100 μM)-evoked currents from MHb (VI) neurons are decreased by 180 μM (+)-catharanthine. The puffer was performed for 250 ms at a pressure of 12 psi. After washing (10 min), the peak amplitude reached the same levels as that observed in control neurons, indicating a reversible inhibition. (B) Concentration-response relationship for the inhibitory activity of (+)-catharanthine on MHb (VI) neurons. Response was normalized to the maximal ACh response which was set as 100%. The plot (r2 = 0.87) is representative of 4–7 determinations, where the error bars are the S.D. The calculated IC50 and nH values are summarized in Table 2.
The observed inward currents elicited by ACh were reduced by superfusion of 180 μM (±)-18-MC (Fig. 4A) or 180 μM (+)-catharanthine (Fig. 5A), and drug washout for 13 and 10 min, respectively, resulted in complete recovery of the original response amplitude. The fact that the neuronal AChR activity was fully recovered after washing suggests that the recording remained stable and the reduction in current following coronaridine congener application was due neither to a large change in the seal quality nor input resistance but rather to the intrinsic antagonistic activity of these ligands. A concentration-dependent inhibition was determined for 18-MC (Fig. 4B) and (+)-catharanthine (Fig. 5B) by using a wide range of concentrations (i.e., 0.07–180 μM). The single exponential fit gave r2 (goodness-of-fit) values of 0.80 and 0.87, respectively. Although biphasic fitting slightly increased the r2 values, suggesting two AChR populations, the accuracy of the IC50 values was lost. Thus, the inhibitory potency for (+)-catharanthine (27 ± 4 μM) and 18-MC (28 ± 6 μM) (Table 2) was calculated considering single exponential fit. The observed nH values are lower than unity (Table 2), but still higher than 0.5, indicating that the inhibitory mechanism is mainly non-cooperative. These values only intend to make a comparison between both compounds, but due to different methodological approaches, they cannot be used to make a comparison with the nH values obtained by Ca2+ influx.
Table 2.
Inhibitory potency (IC50) of (+)-catharanthine and (±)-18-MC at ACh-evoked currents on HMb (VI) neurons determined by patch-clamp recording.
3.3. Binding affinity of bupropion for the [3H]ibogaine sites at hα3β4 AChRs in different conformational states
To determine whether bupropion binds to the [3H]ibogaine binding sites at hα3β4 AChRs, the effect of this antidepressant on [3H]ibogaine binding was determined on resting (i.e., in the absence of any agonist) and desensitized [i.e., in the presence of (−)-nicotine] hα3β4 AChRs (Fig. 6). The results indicated that the binding affinity of bupropion depends on the hα3β4 AChR conformational state. More specifically, bupropion interacts with the [3H]ibogaine sites at desensitized hα3β4 AChRs with higher affinity (Ki = 1.1 ± 0.1 μM) than that at resting receptors (2.4 ± 0.4 μM) (Student t-test p = 0.001) (Table 3). The observed nH values (close to unity) (Table 3) indicate that bupropion inhibits [3H]ibogaine binding to hα3β4 AChRs in different conformational states by non-cooperative mechanisms. This suggest, in turn, that there is potentially only one binding site or several sites with similar binding affinity in the AChR.
Fig. 6.
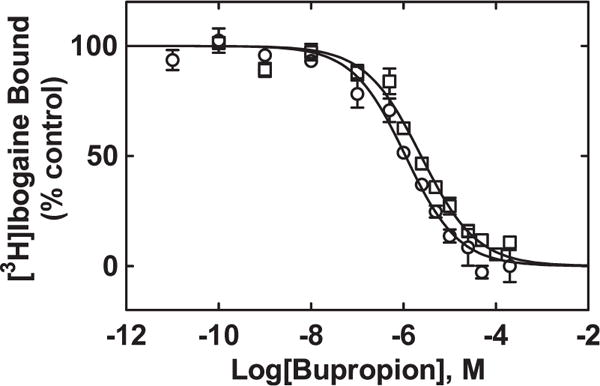
Bupropion-induced inhibition of [3H]ibogaine binding to hα3β4 AChRs in the resting (□) and desensitized (○) states, respectively. hα3β4 AChR membranes (1.5 mg/mL) were pre-incubated (30 min) with 16.6 nM [3H]ibogaine in the absence (receptors are mainly in the resting state) and presence of 1 μM (−)-nicotine (receptors are mainly in the desensitized state), and then equilibrated with increasing concentrations of bupropion. Nonspecific binding was determined at 100 μM (−)-ibogaine. The plots are combinations of two experiments, each performed in triplicate, where the error bars are the S.D. The IC50 and nH values were obtained by nonlinear least-squares fit of the plots (r2 = 0.95 for both). The Ki values, calculated using eq. 1 and summarized in Table 3, indicated that bupropion binds to desensitized hα3β4 AChRs with higher affinity than that at resting receptors (Student t-test p = 0.001).
Table 3.
Binding affinity of bupropion for the [3H]ibogaine sites at hα3β4 AChRs in the resting and desensitized states.
4. Discussion
This work seek to demonstrate the AChR selectivity for coronaridine congeners, the inhibitory activity on native α3β4* AChRs expressed in MHb (VI) neurons, and whether (−)-ibogaine, the archetype of these congeners (Arias et al., 2010b), and bupropion share the same binding site(s).
Combining the present Ca2+ influx results with previous data on hα3β4 AChRs (Arias et al., 2015), the following neuronal AChR selectivity was obtained for the studied coronaridine congeners: hα3β4 > hα4β2 > hα7. The selectivity for hα1β1γd AChRs was in general between that for hα4β2 and hα7 AChRs, except (±)-18-MAC and (±)-18-MC, which showed a higher and similar potency, respectively, compared to that for hα4β2 AChRs (Arias et al., 2011). Particularly, the selectivity of (±)-18-MC for hα3β4 AChRs is 4-fold compared to the hα4β2 AChR (Table 1), a value that agrees with that estimated by Glick’s laboratory using electrophysiological recordings (Glick et al., 2002a). Interestingly, the selectivity of other coronaridine congeners for the hα3β4 AChR was even higher. For instance, the selectivity for (+)-catharanthine was 18-, 32-, and 29-fold higher than that for the respective hα4β2, hα7 (this paper), and α1β1γδ AChRs (Arias et al., 2011), suggesting that this congener may produce less side effects compared to that for 18-MC, especially at higher doses. This is the first time of a quantitative estimation of the receptor selectivity for coronaridine congeners, an important pharmacological feature related to their anti-addictive properties (Glick et al., 2002a).
An important feature of (−)-ibogaine and (±)-18-MC is that their anti-addictive effects last for prolonged time (reviewed in Glick et al., 2000). In this regard, 18-MC is rapidly metabolized (i.e., half-life ∼100 min) to 18-HC (Zhang et al., 2002; Maissonneuve and Glick, 2003), which also has higher selectivity for hα3β4 AChRs (this paper), suggesting a potential role in the behavioral effects mediated by 18-MC. However, only small amounts of this metabolite were found in tissues, indicating that it may account for only a minimal activity (Maissonneuve and Glick, 2003). Alternatively, the observed deposition of 18-MC in fat tissues (e.g., brain) may account for its prolonged duration of action (Maissonneuve and Glick, 2003).
The results indicating that other NCAs with anti-addictive properties such as bupropion (i.e., antidepressant that decreases withdrawal and craving effects during smoking cessation; reviewed in Arias et al., 2014) and mecamylamine (e.g., decreases alcohol and nicotine rewarding effects in men; Chi and de Wit, 2003) show higher selectivity for α3β4 AChRs (Arias et al., 2014; Papke et al., 2001) are in agreement with the observed synergistic effects between each one of these drugs and 18-MC (Glick et al., 2002a,b; Maisonneuve and Glick, 2003). More specifically, inactive doses of bupropion (or mecamylamine) and 18-MC produce the same anti-addictive activity as 18-MC at higher doses. These correlations support the idea that this receptor subtype is an important target for the development of pharmacotherapies for drug addiction. The observed anti-addictive activity of AT-1001, a competitive antagonist with high affinity and selectivity for α3β4 AChRs (Toll et al., 2012), also supports this concept.
The Ca2+ influx results also showed the following inhibitory potency (in μM) sequence for the hα4β2 AChR: (±)-18-MC (6.3 ± 1.3) > (+)-catharanthine (12.6 ± 1.8) > (±)-18-MAC (20.7 ± 3.0) ∼ (±)-18-HC (24.8 ± 1.2), which is similar as that for hα3β4 AChRs (Arias et al., 2015), but slightly different to that for hα7 AChRs, and very different to that for muscle-type AChRs (Arias et al., 2011). This altered sequence could be ascribed to subtle structural differences among AChR subtypes. For example, (−)-18-MC (Arias et al., 2015) and (−)-ibogaine (Arias et al., 2010b) interact with the hα3β4 AChR at a luminal site formed between the phenylalanine/valine (position 13′) and serine (position 6′) rings. Fig. 7 illustrates the location of the ibogaine binding site in the lumen of the hα3β4 AChR ion channel (modified from Arias et al., 2010b). In particular, the aromatic rings of each ligand form π-π interactions with β4-Phe255 residues at position 13′ (Arias et al., 2010b; Arias et al., 2015). Since a valine ring (position 13′) is instead found at the hα4β2 and hα7 AChRs (Arias et al., 2016; Vázquez-Gómez et al., 2014; reviewed in Arias et al., 2014), the Phe residues are not present, decreasing ligand (i.e., π-π) interaction, and thus inhibitory activity, as observed in our results.
Fig. 7.

Model of the hα3β4 AChR transmembrane domain showing the ion channel lumen (modified from Arias et al., 2010b). (−)-Ibogaine (cyan) interacts with a luminal site formed between the phenylalanine/valine (position 13′) and serine (position 6′) rings (Arias et al., 2010b), which overlaps the site for (−)-bupropion (magenta) (Arias et al., submitted manuscript). The AChR and ligands are depicted by its solvent accessible surface.
Our competition binding results indicated that bupropion inhibits [3H]ibogaine binding to desensitized hα3β4 AChRs with 2-fold higher affinity than that for resting AChRs. The observed affinity difference and concentration range is similar to that observed for (−)-ibogaine itself (1.05 and 0.37 μM, respectively; Arias et al., 2010b). Since the [3H]ibogaine binding competition is mediated by a non-cooperative mechanism (i.e., nH ∼ 1), bupropion is probably interacting with only one [3H]ibogaine site or few sites with similar affinities at hα3β4 AChRs. Previous molecular docking results suggested that coronaridine congeners interact with a luminal site located between the phenylalanine/valine and serine rings of the hα3β4 AChR ion channel (Arias et al., 2010b; Arias et al., 2015), overlapping the bupropion site (Arias et al., submitted manuscript) as well as the site for other structurally-different antidepressants, including imipramine (Arias et al., 2010d) and fluoxetine (Arias et al., 2010c). Fig. 7 illustrates the hα3β4 AChR model with overlapping binding sites for (−)-ibogaine (cyan) (Arias et al., 2010b) and (−)-bupropion (magenta) (Arias et al., submitted manuscript).
The patch-clamp results showed, for the first time, that coronaridine congeners inhibit ACh-evoked currents from MHb (VI) neurons. Interestingly, the neuronal AChR activity was fully recovered after washing, suggesting that the ligands interact with portions of the AChR that are in contact with an aqueous environment such as the ion channel. The majority (∼75%) of the current response at MHb (VI) neurons has been ascribed to α3β4* AChRs (Quick et al., 1999). Although MHb neurons also express α4-containing AChRs, including α4β4, α4β2, and α4β3β2 subtypes (Grady et al., 2009; Scholze et al., 2012), they are preferably concentrated in MHb (VL) neurons (Shih et al., 2014). Based on our results using heterologous cells, we expected a potent blockade of α3β4* AChRs by (+)-catharanthine and (±)-18-MC. Interestingly, the observed IC50 values for (+)-catharanthine (27 ± 4 μM) and (±)-18-MC (28 ± 6 μM) are much higher than those observed at hα3β4 AChRs (0.68 ± 0.10 and 1.47 ± 0.21 μM, respectively) (Arias et al., 2015). This is not surprising, as drug potency may be diminished in a tissue slice. Nevertheless, it is possible to suggest that the observed potency differences could be due to the expression of a variety of α3β4* AChRs in MHb neurons whereas HEK293-hα3β4 cells express only hα3β4 AChRs. Supporting this assumption, there is evidence of MHb neurons expressing α3-containing AChRs, including α3β4, α3β2, α3β4β2, α3β4β3, α3α4β4, α3α6β4, and α3α5β4β2 subtypes (Quick et al., 1999; Grady et al., 2009; Scholze et al., 2012; Shih et al., 2014, 2015). Based on this variety of receptors, it is plausible that some of them, including their respective stoichiometric variants such as (α3)2(β4)3 and (α3)3(β4)2, may have lower sensitivity to coronaridine congeners, as observed with functionally-different ligands (Ochoa et al., 2016; Shih et al., 2015; Krashia et al., 2010). In this regard, future work will determine the activity of coronaridine congeners on specific α3β4-containing AChRs and soichiometries using heterologous cells and knockout mice.
Our findings clearly demonstrate that coronaridine congeners present higher selectivity for hα3β4 AChRs compared to other AChR subtypes. These compounds block the hα3β4 AChR in a noncompetitive fashion by interacting with the bupropion (luminal) site. The results showing that coronaridine congeners inhibit MHb (VI) neurons support that concept that native α3β4* AChRs are targets for the pharmacological and anti-addictive activity of these compounds.
Acknowledgments
This work was supported by grants from NIH (DA040626) (to R.M.D.), and California Northstate University College of Medicine (to H.R.A.). The authors thank to National Institute on Drug Addiction (NIDA, NIH, Bethesda, Maryland, USA) for the gift of ibogaine, to Dr. Kuehne (University of Vermont, VT, USA) for the gift of coronaridine congeners, to Dr. Targowska-Duda (Medical University of Lublin, Poland) for providing the hα3β4 AChR model with the overlapping sites for ibogaine and bupropion, and to Dr. Scholze (University of Vienna, Austria) for her fruitful comments.
Abbreviations
- AChR
nicotinic acetylcholine receptor
- ACh
acetylcholine
- NCA
noncompetitive antagonist
- RT
room temperature
- MHb
medial habenula
- VI
ventral inferior
- Ki
inhibition constant
- Kd
dissociation constant
- IC50
ligand concentration that produces 50% inhibition of binding (or of agonist activation)
- EC50
agonist concentration that produces 50% receptor activation
- nH
Hill coefficient
- r2
goodness-of-fit for the linear regression
- DMEM
Dulbecco’s Modified Eagle Medium
- BSA
bovine serum albumin
- HBSS
HEPES-buffered salt solution
- (−)-ibogaine
12-methoxyibogamine
- (±)-18-MC
(±)-18-methoxycoronaridine
- (±)-18-MAC
(±)-18-methylaminocoronaridine
- (±)-18-HC
(±)-18-hydroxycoronaridine
- (+)-catharanthine
(+)-3,4-didehydrocoronaridine
References
- Alper KR, Lotsof HS, Kaplan CD. The ibogaine medical subculture. J Ethnopharmacol. 2008;115:9–24. doi: 10.1016/j.jep.2007.08.034. [DOI] [PubMed] [Google Scholar]
- Arias HR, Rosenberg A, Feuerbach D, Targowska-Duda KM, Maciejewski R, Moaddel R, Glick SD, Wainer IW. Interaction of 18-methoxycoronaridine with nicotinic acetylcholine receptors in different conformational states. Biochem Biophys Acta. 2010a;1798:1153–1163. doi: 10.1016/j.bbamem.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR, Rosenberg A, Targowska-Duda KM, Feuerbach D, Yuan XJ, Jozwiak K, Moaddel R, Wainer IW. Interaction of ibogaine with human α3β4-nicotinic acetylcholine receptors in different conformational states. Int J Biochem Cell Biol. 2010b;42:1525–1535. doi: 10.1016/j.biocel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR, Feuerbach D, Targowska-Duda KM, Russell M, Jozwiak K. Interaction of selective serotonin reuptake inhibitors with neuronal nicotinic acetylcholine receptors. Biochemistry. 2010c;49:5734–5742. doi: 10.1021/bi100536t. [DOI] [PubMed] [Google Scholar]
- Arias HR, Targowska-Duda KM, Feuerbach D, Sullivan CJ, Maciejewski R, Jozwiak K. Different interaction between tricyclic antidepressants and mecamylamine with the human α3β4 nicotinic acetylcholine receptor ion channel. Neurochem Int. 2010d;56:642–649. doi: 10.1016/j.neuint.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Arias HR, Feuerbach D, Targowska-Duda KM, Jozwiak K. Structure-activity relationship of ibogaine analogs interacting with nicotinic acetylcholine receptors in different conformational states. Int J Biochem Cell Biol. 2011;43:1330–1339. doi: 10.1016/j.biocel.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Arias HR, Biała G, Kruk-Słomka M, Targowska-Duda KM. Interaction of nicotinic receptors with bupropion: structural functional, and pre-clinical perspectives. Recept Clin Invest. 2014;1:30–45. [Google Scholar]
- Arias HR, Feuerbach D, Targowska-Duda KM, Jozwiak K. Coronaridine congeners inhibit human α3β4 nicotinic acetylcholine receptors by interacting with luminal and non-luminal sites. Int J Biochem Cell Biol. 2015;65:81–90. doi: 10.1016/j.biocel.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Arias HR, Feuerbach D, Ortells MO. Bupropion and its photoreactive analog (±)-SADU-3-72 interact with luminal and non-luminal sites at human α4β2 nicotinic acetylcholine receptors. Neurochem Int. 2016;100:67–77. doi: 10.1016/j.neuint.2016.08.013. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chi H, de Wit H. Mecamylamine attenuates the subjective stimulant-like effects of alcohol in social drinkers. Alcohol Clin Exp Res. 2003;27:780–787. doi: 10.1097/01.ALC.0000065435.12068.24. [DOI] [PubMed] [Google Scholar]
- Glick SD, Maisonneuve IM, Szumlinski KK. 18-Methoxycoronaridine (18-MC) and ibogaine: comparison of antiaddictive efficacy, toxicity, and mechanisms of action. Ann NY Acad Sci. 2000;914:369–386. doi: 10.1111/j.1749-6632.2000.tb05211.x. [DOI] [PubMed] [Google Scholar]
- Glick SD, Maisonneuve IM, Kitchen BA, Fleck MW. Antagonism of α3β4 nicotinic receptors as a strategy to reduce opioid and stimulant self-administration. Eur J Pharmacol. 2002a;438:99–105. doi: 10.1016/s0014-2999(02)01284-0. [DOI] [PubMed] [Google Scholar]
- Glick SD, Maisonneuve IM, Kitchen BA. Modulation of nicotine self-administration in rats by combination therapy with agents blocking α3β4 nicotinic receptors. Eur J Pharmacol. 2002b;448:185–191. doi: 10.1016/s0014-2999(02)01944-1. [DOI] [PubMed] [Google Scholar]
- Glick SD, Sell EM, McCallum SE, Maisonneuve IM. Brain regions mediating α3β4 nicotinic antagonist effects of 18-MC on nicotine self-administration. Eur J Pharmacol. 2011;669:71–75. doi: 10.1016/j.ejphar.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, Clementi F, Gotti C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the α3β4* and α3β3β4* subtypes mediate acetylcholine release. J Neurosci. 2009;29:2272–2282. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashia P, Moroni M, Broadbent S, Hofmann G, Kracun S, Beato M, Groot-Kormelink PJ, Sivilotti LG. Human α3β4 neuronal nicotinic receptors show different stoichiometry if they are expressed in Xenopus oocytes or mammalian HEK293 cells. PLoS One. 2010;5:e13611. doi: 10.1371/journal.pone.0013611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve IM, Glick SD. Anti-addictive actions of an iboga alkaloid congener: a novel mechanism for a novel treatment. Pharmacol Biochem Behav. 2003;75:607–618. doi: 10.1016/s0091-3057(03)00119-9. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Cowe MA, Lewis SW, Glick SD. α3β4 nicotinic acetylcholine receptors in the medial habenula modulate the mesolimbic dopaminergic response to acute nicotine in vivo. Neuropharmacology. 2012;3:434–440. doi: 10.1016/j.neuropharm.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa V, George AA, Nishi R, Whiteaker P. The prototoxin LYPD6B modulates heteromeric α3β4-containing nicotinic acetylcholine receptors but not α7 homomers. FASEB J. 2016;30:1109–1119. doi: 10.1096/fj.15-274548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortells MO, Arias HR. Neuronal networks of nicotine addiction. Int J Biochem Cell Biol. 2010;42:1931–1935. doi: 10.1016/j.biocel.2010.08.019. [DOI] [PubMed] [Google Scholar]
- Pace CJ, Glick SD, Maisonneuve IM, He LW, Jokiel PA, Kuehne ME, Fleck MW. Novel iboga alkaloid congeners block nicotinic receptors and reduce drug self-administration. Eur J Pharmacol. 2004;492:159–167. doi: 10.1016/j.ejphar.2004.03.062. [DOI] [PubMed] [Google Scholar]
- Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine steroisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- Quick MW, Ceballos RM, Kasten M, McIntosh JM, Lester RA. α3β4 subunit-containing nicotinic receptors dominate function in rat medial habenula neurons. Neuropharmacology. 1999;38:769–783. doi: 10.1016/s0028-3908(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Scholze P, Koth G, Ort-Urtreger A, Huck S. Subunit composition of α5–containing nicotinic receptors in the rodent habenula. J Neurochem. 2012;12:551–560. doi: 10.1111/j.1471-4159.2012.07714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PY, Engle SE, Oh G, Deshpande P, Puskar NL, Lester HA, Drenan RM. Differential expression and function of nicotinic acetylcholine receptors in subdivisions of medial habenula. J Neurosci. 2014;34:9789–9802. doi: 10.1523/JNEUROSCI.0476-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PY, McIntosh JM, Drenan RM. Nicotine dependence reveals distinct responses from neurons and their resident nicotinic receptors in medial habenula. Mol Pharmacol. 2015;88:1035–1044. doi: 10.1124/mol.115.101444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L, Zaveri NT, Polgar WE, Jiang F, Khroyan LV, Zhou W, Xie X, Stauber GB, Costello MR, Leslie FM. AT-1001: A high affinity and selective α3β4 nicotinic acetylcholine receptor antagonist blocks nicotine self-administration in rats. Neuropsychopharmacology. 2012;37:1367–1376. doi: 10.1038/npp.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Gómez E, Arias HR, Feuerbach D, Miranda-Morales M, Mihailescu S, Targowska-Duda KM, Jozwiak K, García-Colunga J. Bupropion-induced inhibition of α7 nicotinic acetylcholine receptors expressed in heterologous cells and neurons from dorsal raphe nucleus and hippocampus. Eur J Pharmacol. 2014;740:103–111. doi: 10.1016/j.ejphar.2014.06.059. [DOI] [PubMed] [Google Scholar]
- Zhang W, Ramamoorthy Y, Tyndale RF, Glick SD, Maisonneuve IM, Kuehne ME, Sellers EM. Metabolism of 18-methoxycoronaridine an ibogaine analog, to 18-hydroxycoronaridine by genetically variable CYP2C19. Drug Metab Dispos. 2002;30:663–669. doi: 10.1124/dmd.30.6.663. [DOI] [PubMed] [Google Scholar]