

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and typically fatal lung disease characterised by subpleural fibrosis, subepithelial fibroblast foci, and microscopic honeycombing. Although understanding of the pathogenic mechanisms continues to evolve, evidence indicates that distal airway and alveolar epithelial cells are central drivers of the disease. In this Viewpoint, we review the history of naming and classifications used to define the disease now referred to as IPF, in the context of understanding the clinical presentation, causes, and pathogenesis of the disease. We aim to generate discussion on whether, given the substantial progress made in understanding the clinical, genetic, cellular, and molecular mechanisms involved in the development of IPF, a change of name should be considered. To initiate this discussion, we offer new suggestions to update the name of this disease and new approaches to classify all forms of pulmonary fibrosis.
Disease characteristics
Idiopathic pulmonary fibrosis (IPF) is a progressive, irreversible, and usually fatal lung disease, for which the average life expectancy is 3–5 years after diagnosis. The histopathological hallmarks of the disease are subpleural fibrosis, subepithelial fibroblast foci, and microscopic honeycombing. Affected areas are adjacent to histopathologically normal regions, in a pattern termed usual interstitial pneumonia.1–4 Although understanding of the pathogenic mechanisms continues to evolve, strong evidence indicates that aberrantly activated lung epithelial cells secrete mediators, leading to fibroblast proliferation and differentiation to highly active myofibroblasts, which deposit excessive amounts of extracellular matrix and irreversibly destroy the lung architecture.5
Origin of term idiopathic pulmonary fibrosis
IPF has had multiple names. First recognition of the disease is attributed to D J Corrigan in 1838, who called it cirrhosis of the lung. The clinical and pathological features described were typical of what has since been recognised as a distinct fibrotic lung disease in adults. Corrigan’s descriptor prevailed until 1893, when, in the text Principles and Practice of Medicine, William Osler renamed it chronic interstitial pneumonia but kept cirrhosis of the lung as a subtitle.
In 1948, Robbins6 first used the term IPF to describe patients with interstitial opacities on chest radiographs that were suggestive of pulmonary fibrosis but had no identifiable cause. At that time, associations between pulmonary fibrosis and postinfection fibrosis, pneumoconiosis, radiation therapy, and autoimmune diseases, such as rheumatoid arthritis or systemic sclerosis, had been recognised. IPF was found to occur in families, which inferred a genetic component.7 However, the term IPF was used infrequently until a 1976 review article by Crystal and colleagues8 popularised the term. Acquisition of surgical lung biopsy samples, led to the recognition of dissimilar histopathological patterns of pulmonary fibrosis that were separated into subtypes, such as usual interstitial pneumonia, desquamative interstitial pneumonia, and giant-cell interstitial pneumonia, by Liebow3 and by others, with different causes implied for the specific subtypes. The causes of some subtypes are now known: giant-cell interstitial pneumonia is caused by exposure to hard-metal dust and desquamative interstitial pneumonia is strongly associated with smoking. Nevertheless, in most patients specific causes could not be identified and, therefore, IPF included several histopathological subtypes of interstitial lung disease, such as usual interstitial pneumonia and non-specific interstitial pneumonia.9 At the same time, other terms were used to describe these diseases, including idiopathic interstitial pneumonia and cryptogenic fibrosing alveolitis. With the introduction of high-resolution CT, radiological patterns could be identified that corresponded to the pathological features of usual interstitial pneumonia.10 Clinicians began to apply the name IPF more selectively to patients with a pattern of lung fibrosis indicative of usual interstitial pneumonia on high-resolution CT, relevant lung pathology, or both, without a known cause.
Two advances led to further phenotyping of IPF. First, clinicians and pathologists with expertise in interstitial lung disease recognised that pathological subtypes of IPF were clinically distinct.11 A consensus statement was published that defined specific clinical and histopathological features of IPF. Evidence that accumulated over the next decade led to guidelines that further refined the definition of IPF and established precise criteria for the radiological and histopathological features of usual interstitial pneumonia.12 The term IPF was reserved for patients with usual interstitial pneumonia patterns of lung disease in the absence of known secondary causes, such as autoimmune diseases or defined environmental exposures. Second, combined input of clinicians, radiologists, and pathologists was recognised as being the most accurate way to diagnose the disease.13 The diagnostic criteria might be viewed as restrictive because they led to a substantial proportion of patients being judged to have unclassifiable fibrosis,14 but they have been useful in defining a population of patients with a fairly uniform clinical and pathological pattern of lung disease.
Risk factors for idiopathic pulmonary fibrosis
Over the past decade, IPF has been revealed to be a complex, heterogeneous disorder associated with rare and common sequence variants in several genes (MUC5B, TERT, TERC, RTEL1, PARN, DKC1, TINF2, SFTPC, SFTPA2, and ABCA3)15–25 and in at least 11 loci26,27 associated with multiple emerging epigenetic28–32 and transcriptional31,33–36 profiles. The MUC5B promoter variant rs35705950 has been validated as a risk variant for IPF in 11 independent studies.26,37–46 This variant is the strongest known risk factor (odds ratio [OR] for carriers of the T (minor) allele 4·51, 95% CI 3·91–5·21),26 accounting for at least 30% of the overall risk of developing IPF,26,38–46 and might be useful to identify individuals early in the disease course.47,48 Family history of more than one case of IPF in the previous one or two generations and in biological siblings is an independent risk factor for this disease.49 Familial and sporadic IPF share many genetic risk factors,16,20,22–24,26,38–43,45,46 which suggests that family members of people with sporadic IPF could also represent an at-risk population.
Various exposures, such as microaspiration,50,51 metal and wood dust,52–54 viruses,55–57 and drugs, have been associated with the development of IPF, but the most important environmental risk factor is smoking cigarettes (of note, some of the cited studies were completed before the current classification criteria were established). Ever having smoked cigarettes remains an important risk factor for the development of sporadic IPF even many years after smoking cessation.58 Cigarette smoking is also a strong risk factor for the development of familial interstitial pneumonia (OR 3·6, 95% CI 1·3–9·8),49 which suggests that cigarette smoking contributes substantially to familial cases of IPF.
Primary role of lung epithelia in disease pathogenesis
An approach to understanding IPF pathogenesis is to consider it as a three-stage process: predisposition, activation, and progression (figure).4 With this approach, ageing, smoking, environmental exposures (eg, accumulation of environmental chitin59), and genetic background are predisposing risk factors.60,61 Epidemio logical studies confirm IPF as a disease of ageing, with interstitial lung abnormalities becoming increasingly prevalent with advancing age.47 IPF is unusual in individuals younger than 50 years, but prevalence nearly doubles with every decade of life thereafter.61 These intrinsic and extrinsic risk factors only increase an individual’s probability of developing IPF. That is, many individuals might have one or more risk factors but never develop the disease.
Figure. Three-stage description of the pathogenesis of idiopathic pulmonary fibrosis.
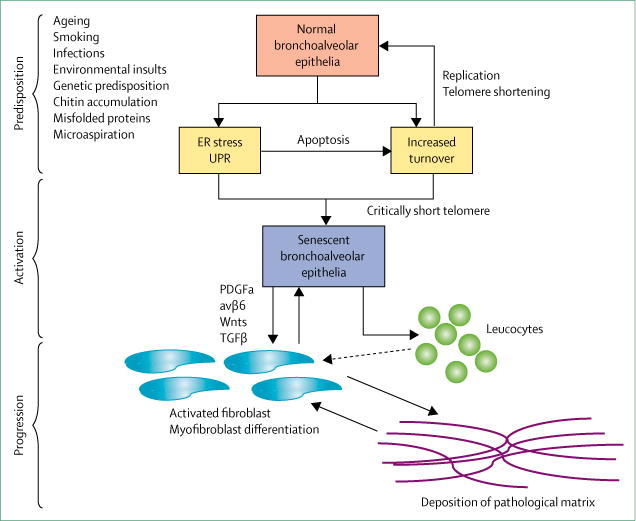
In the predisposition stage, recurrent environmental insults lead, in genetically predisposed individuals, to increased turnover of alveolar type II cells, ER-stress-mediated activation of UPR, apoptosis, and progressive telomere attrition. In the activation stage, accumulation of a lifetime insults leads to pathological alterations of the lung epithelium, such as senescence reprogramming, and release of profibrotic mediators (eg, TGFβ, Wnts, and PDGFβ) by the alveolar epithelium. These mediators, either directly or indirectly via leucocytes, activate fibroblasts to deposit pathological matrix. In the progression stage, the pathological matrix promotes additional differentiation of fibroblasts to myofibroblasts, which deposit more matrix and further activate fibroblasts in a feed-forward loop of lung remodelling. ER=endoplasmic reticulum. UPR=unfolded protein response. PDGF=platelet-derived growth factor. TGF=transforming growth factor.
During the activation phase, accumulated environmental exposures in a genetically predisposed individual lead to pathological alterations to the lung epithelium.5,62 One durable alteration is the critical shortening of telomeres in alveolar type II cells,63,64 which can lead to molecular changes within lung epithelial cells sufficient to promote lung remodelling and fibrosis.65 Other epithelial alter ations are activation of senescence pro gramming,66–68 accumulation of dysfunctional mitochondria,69 and activation of the unfolded protein response.70 The abnormal epithelium expresses numerous mediators that might lead to mesenchymal-cell activation and lung remodelling. Activation may be direct or indirect via immune cells, such as macrophages or lymphocytes,71 although the exact roles of immune cells in IPF remain unclear. Candidate mediators include transforming growth factor (TGF) β, its activating integrin αvβ6, platelet-derived growth factor β, and Wnts, which activate mesenchymal cells when expression is increased.4,5 Conversely, dysfunctional epithelium might reduce the expression of some mediators, such as prostaglandin E2, that under normal circumstances suppress mesenchymal cell expansion.72,73 Additionally, alveolar type II cells are thought to act as stem cells in adult lungs, and can form so-called alveolar organoids in vitro.74 Alveolar type II cells isolated from the lung tissue of individuals with IPF at the time of transplantation have impaired ability to form organoids, which suggests that alveolar stem-cell failure contributes to IPF pathogenesis.75,76 Thus, lung remodelling in IPF arises from alterations in epithelial-cell growth and repair and epithelial–mesenchymal crosstalk.
In the progression phase, the normal alveolar structure of the lung is lost and replaced by remodelled fibrotic tissue characterised by bronchiolised cystic airspaces, which might include continuous proliferation of bronchiolar epithelium to honeycomb cysts. During this phase, the pathological matrix might contribute to remodelling via mechanisms independent of epithelial-cell dysfunction. Examples include increased stiffness and stretch-induced activation of TGFβ by the remodelled lung.77–79 Remodelling of lung tissue in patients with IPF alters expression of multiple matrix molecules,80 many of which can activate profibrotic-signalling pathways in the mesenchymal cells that engage them.78 These IPF fibroblasts, which are potentially metabolically aberrant,81 can acquire destructive properties, such as the ability to invade matrix, which could contribute to chronic remodelling.82
This overview of the pathogenesis of IPF does not include all candidate cellular or molecular mediators, but biological findings can be incorporated in the organisational concepts of predisposition, activation, and progression as more is learned about their specific roles in the disease process. Our proposed organisational framework (figure) also identifies two potential feed-forward loops of disease activity that might explain the relentless progression of IPF. The first is senescence of alveolar type II cells. Because these cells act as the functional stem cells of the lung, when one becomes senescent, adjacent non-senescent alveolar type II cells must compensate, which leads to increased frequency of replication, accelerated telomere attrition, and a predisposition to senescence. A second feed-forward loop is triggered by matrix deposition, which stiffens lung tissue,80 and can lead to the conversion of fibroblasts to myofibroblasts79 and increased epithelial-cell activation, collagen and matrix deposition, and lung remodelling.
Overall, when the pathogenesis of IPF is considered as a continuum of predisposition, activation, and progression, distal bronchiolar and alveolar epithelial lung cells are shown to be the pathologically abnormal cells in IPF lungs, and fibrosis to be the consequence of epithelial-cell dysfunction. Therefore, IPF is a disease of lung epithelial cells that manifests as fibrosis rather than being an intrinsically fibrotic disease.
The case to rebrand idiopathic pulmonary fibrosis
With growing understanding of the causes and pathogenesis of IPF, the term idiopathic no longer seems to describe this progressive lung disease accurately. Furthermore, the term fibrosis limits consideration of the clinical and pathological attributes of this complex interstitial lung condition and does not highlight the primary role of lung epithelia in its pathogenesis. Because the name of a disease can affect a patient’s (or caregiver’s) under standing of their disease and expectations for its behaviour and management,83 we propose renaming IPF.
There is precedent for changing the name of diseases. For example, eponymous diseases named to honour the individual who first described them have been renamed when scientific understanding of the disease no longer reflects the first description. Diseases named initially as idiopathic, which implies a distinct, primary entity for which the cause is unknown, have been changed when causes are recognised. For instance, idiopathic thrombocytopenia purpura was renamed as immune-mediated thrombocytopenia to reflect the disease mechanism.84 The term idiopathic might falsely convey that there is little or no understanding for the cause or pathogenesis. For IPF, however, although uncertainties remain, genetic, environmental, cellular, and molecular mechanisms are now well recognised as being involved in its development.
The term fibrotic implicates only matrix accumulation, yet, although fibrosis is a major contributor to the disease process in IPF, it is only one component and is the consequence of dysfunctional epithelia. Moreover, IPF is a biologically and temporally heterogeneous process within and across patients. The most frequently identified pathological abnormalities in lung remodelling are subpleural fibrosis, subepithelial foci of fibroblasts and myofibroblasts, metaplastic and hyperplastic changes of epithelial cells lining the alveoli, re-epithelialised air spaces (microscopic honeycombing), lymphoid aggregates, and increased numbers and subtypes of haemopoietic cells, including macrophages, dendritic cells, and mast cells.4,65,85,86 Although none of these features occurs alone, transcriptional studies suggest that they can be grouped into at least two major categories: IPF that predominantly expresses genes associated with the respiratory bronchiolar epithelium (cilia and mucins) and is more likely to have histological honeycomb cysts in the lungs, and IPF that primarily expresses matrix-related genes.35 Moreover, the lung epithelia in IPF represent a broad biological phenotype in which markers of conducting airway cells and type I and type II alveolar epithelial cells are co-expressed.87
Use of the term fibrosis also implies that only the extracellular matrix components of the remodelled lung represent IPF and neglects the other elements. It further implies that the non-fibrotic changes must either directly contribute to development of or be a consequence of fibrosis. Bronchoalveolar epithelial cells, however, seem to be the most important dysfunctional cells in IPF, and in many cases the temporal components of cell–cell interactions are unknown. For example, although microscopic honeycombing and fibrosis are characteristic features of IPF, no specific interactions between these pathological processes have been described. Without this knowledge, it is equally conceivable that microscopic honeycombing evolves from mechanisms independent of the fibrotic process. It is time to recognise specific elements of remodelling in the IPF lung and to define whether they arise from separate pathological drivers or relate specifically to matrix deposition and fibrosis. Independently considering other elements of lung remodelling does not challenge the importance of fibrosis in IPF. Rather, it enhances understanding of the relationships between fibrosis and these elements.
Use of the name IPF potentially limits understanding of therapeutic advances. Use of the term antifibrotic therapy to describe pirfenidone and nintedanib is somewhat premature. Although these medications slow the loss of forced vital capacity (FVC) in patients with IPF,88,89 there is no evidence that they slow fibrosis (ie, extracellular matrix deposition and scar formation). Thus, describing them as antifibrotic potentially discourages investigation of their possible effects on elements of the activation phase or non-fibrotic elements of lung tissue remodelling that might explain the preservation of FVC. Embracing the pathogenic heterogeneity of IPF could lead to the development of drugs specifically effective against fibrotic or non-fibrotic elements in remodelled IPF lung tissue. Identifying differences in the contri butions of these elements between patients might lead to implementation of precision medicine approaches in IPF.
Suggestions for moving forwards
In view of the latest understanding of the clinical, aetiological, genetic, and molecular features of IPF, we propose that it is time to reconsider whether the name accurately represents the underlying disease. We suggest several approaches that could be used to rename IPF. These suggestions are intended to start discussions that will need input from the wider IPF community to develop further.
One approach would be to simply rename IPF consistent with its pathogenesis, for instance epithelial-driven pulmonary fibrosis, primary pulmonary fibrosis, or progressive age-dependent pulmonary fibrosis. Another approach would be to reclassify pulmonary fibrosis by clinical and aetiological criteria, using a format similar to that for pulmonary hypertension (table 1). We suggest separating the subtypes of pulmonary fibrosis into four groups. Group 1 could be pulmonary fibrosis driven by epithelial cell dysfunction, representing the disease currently known as IPF. Group 2 could be pulmonary fibrosis driven by inflammatory-cell dysfunction, representing diseases such as pulmonary fibrosis associated with connective-tissue diseases or hypersensitivity pneumonitis. Group 3 could include pulmonary fibrosis due to occupational exposures or medications, such as asbestosis or nitrofurantoin. Hypersensitivity pneumonitis could also be positioned in group 3 if a definite exposure is identified. Group 4 could include pulmonary fibrosis due to smoking-related diseases, such as desquamative interstitial pneumonitis. Each group could be further separated to distinguish between patients with known and no known genetic causes. Patients could be reclassified if causes are identified or they are found to share genetic risk with another category, such as IPF, which has been described for a subset of patients with rheumatoid arthritis or hypersensitivity pneumonitis.90,91
Table 1.
Proposed groups for subtypes of pulmonary fibrosis
| Disorders in classification | |
|---|---|
| Group 1: pulmonary fibrosis driven by epithelial cell dysfunction | IPF |
| Group 2: pulmonary fibrosis driven by inflammatory cell dysfunction | RA-ILD, scleroderma, MCTD, Sjogren’s syndrome, hypersensitivity pneumonitis, sarcoidosis, NSIP |
| Group 3: occupational or drug induced pulmonary fibrosis | Asbestosis, silicosis, medications |
| Group 4: pulmonary fibrosis due to smoking | RBILD, DIP, LCH |
RA-ILD=rheumatoid-arthritis-associated interstitial lung disease. IPF=idiopathic pulmonary fibrosis. MCTD=mixed connective-tissue disease. NSIP=non-specific interstitial pneumonitis. RBILD=respiratory bronchiolitis with interstitial lung disease. DIP=desquamative interstitial pneumonia. LCH=Langerhan’s cell histiocytosis.
An alternative reclassification could use a personalised medicine approach to distribute pulmonary fibrosis into two classes. Class 1 could include cases of pulmonary fibrosis of unknown cause and class 2 those with a known cellular or molecular cause (table 2). These two classes would have clinical subcategories, such as autoimmune diseases and known environmental exposures, with further separation of patients by known cellular or molecular causes. Categories to consider include pulmonary fibrosis driven by epithelial-cell dysfunction, telomere dysfunction, or known gene mutations (eg, surfactant protein C). This approach would enable dynamic categorisation of pulmonary fibrosis that could be adapted with advances in cellular and molecular understanding of disease pathogenesis.
Table 2.
Proposed reclassification of pulmonary fibrosis
| Considerations for classification | |
|---|---|
| Class 1: no evidence of molecular markers | Autoimmune disease Known environmental exposures No autoimmune disease or environmental exposure |
| Class 2: genetic, transcriptomic, or proteomic explanation for pulmonary fibrosis* | Autoimmune disease Known environmental exposures No autoimmune disease or environmental exposure |
For example, carriers of MUC5B minor allele or mutations in TERT, TERC, RTEL1, SFTPC, and other genes.
To reach a consensus for renaming IPF or reclassifying all forms of pulmonary fibrosis, we propose that a committee of stakeholders that includes experienced clinicians and scientists, as well as patients and their advocates, assembles to review the literature with the objective of answering the following questions: whether IPF is idiopathic; whether definable subgroups exist; and whether the term fibrosis mischaracterises a disease that primarily involves abnormal lung epithelia. If the consensus to these questions is yes, the committee should be charged with deriving a new name or classification of pulmonary fibrosis that is less confining than IPF and is sufficiently broad to incorporate advances in the clinical, aetiological, and molecular understanding of the disease.
Key messages.
Idiopathic pulmonary fibrosis (IPF) is a disease primarily of epithelial-cell dysfunction, the consequence of which is fibrosis
The IPF lung contains elements of remodelling that might occur independently of fibrosis
Two unique feed-forward loops might explain the relentless progression of IPF
The name IPF no longer accurately represents understanding of the disease pathogenesis
It is time to consider renaming IPF in a way that reflects the disease process
Search strategy and selection criteria.
We searched PubMed with the search terms “lung”, “fibrosis”, “idiopathic pulmonary fibrosis”, “pathogenesis”, “interstitial”, “genetic”, “epithelium”, “cryptogenic fibrosing alveolitis”, “fibroblast”, “senescence”, “stem cell”, “telomere”, “macrophage”, “lymphocyte”, and “inflammation” for articles published from 1898 onwards. We searched the references of retrieved articles for further articles, and additional citations were suggested during peer review. We selected articles for citation if they reviewed or presented human data relating to pulmonary fibrosis or established the importance of specific pathological mechanisms.
Acknowledgments
PJW is supported by the Nina Ireland Program for Lung Health. NK is supported by the National Institutes of Health National Heart, Lung, and Blood Institute ([NIH NHLBI] grants U01 HL112707, R01 HL127349, U01 HL108642, and U54HG008540). TMM is supported by the National Institute for Health Research Clinician Scientist Fellowship (CS-2013-13-017) and the British Lung Foundation Chair in Respiratory Research (C17-3). DAS is supported by the NIH NHLBI (grants R01 HL097163, R33 HL120770, UH3 HL123442, P01 HL 092870, and X01 HL124585), the US Department of Defense (grant W81XWH-17-1-0597), and a VA Merit Award.
PJW has received grants from Genentech and MedImmune and personal fees from Roche. NK has received grants and personal fees from Biogen Idec, personal fees from Boehringer Ingelheim, Moerae Matrix, Pliant, Samumed, and Third Rock, and non-financial support from Actelion and Miragen. NK also has a patent for new therapies in pulmonary fibrosis licensed to Quitsa/SLI and a patent on peripheral blood gene expression. GJ has received grants and personal fees from Biogen, GlaxoSmithKline, InterMune, and MedImmune, personal fees and non-financial support from Boehringer Ingelheim, grants from Galecto Biotech, and personal fees from PharmAkea and Roche, and is a Trustee of the charity Action for Pulmonary Fibrosis and the British Thoracic Society. TMM has, via his institution, received industry-academic funding from GlaxoSmithKline R&D, Novartis, and UCB Pharma, and has received consultancy or speakers’ fees from Apellis, Astra Zeneca, Bayer, Biogen Idec, Boehringer Ingelheim, GlaxoSmithKline R&D, InterMune, ProMetic, Roche, Sanofi-Aventis, and UCB Pharma. GR has received financial support from Biogen, Bristol-Myers Squibb, Fibrogen, Roche-Genentech, and Sanofi, grants and other financial support from Boehringer Ingelheim, Gilead, and Promedior, and personal fees from Patara, UCB Pharma, and Veracyte. LR has received grants and personal fees from Boehringer Ingelheim and InterMune, personal fees from Biogen Idec, ImmuneWorks, MedImmune, Sanofi-Aventis, Roche, Shionogi, and Takeda. WAW has received grants from Boehringer Ingelheim and Roche. DAS has financial support from Eleven P15, a company involved in early detection of and intervention for idiopathic pulmonary fibrosis.
Footnotes
Contributors
PJW and DAS did the literature searches and wrote the first draft of the paper. All authors contributed to and revised drafts and approved the final version.
Declaration of interests
The other authors declare no competing interests.
Contributor Information
Paul J Wolters, Division of Pulmonary and Critical Care Medicine, Department of Medicine, University of California, San Francisco, CA, USA.
Timothy S Blackwell, Department of Medicine, Vanderbilt University, Nashville, TN, USA.
Oliver Eickelberg, Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado Denver, Aurora, CO, USA.
James E Loyd, Department of Medicine, Vanderbilt University, Nashville, TN, USA.
Naftali Kaminski, Section of Pulmonary, Critical Care, and Sleep Medicine, Yale University School of Medicine, New Haven, CT, USA.
Gisli Jenkins, Division of Respiratory Medicine, University of Nottingham, Nottingham University Hospitals, Nottingham, UK.
Toby M Maher, Fibrosis Research Group, National Heart and Lung Institute, Imperial College London, London, UK.
Maria Molina-Molina, Department of Pneumology, Unit of Interstitial Lung Diseases, University Hospital of Bellvitge Institute for Biomedical Research (IDIBELL), Barcelona, Spain.
Paul W Noble, Department of Medicine, Cedars-Sinai Medical Center, Los Angeles, CA, USA.
Ganesh Raghu, Center for Interstitial Lung Disease, University of Washington, Seattle, WA, USA.
Luca Richeldi, Division of Pulmonary Medicine, A Gemelli University Hospital, Catholic University of the Sacred Heart, Rome, Italy.
Marvin I Schwarz, Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado Denver, Aurora, CO, USA.
Moises Selman, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, Mexico City, Mexico.
Wim A Wuyts, Department of Pulmonary Medicine, Unit for Interstitial Lung diseases. University Hospitals Leuven, Leuven, Belgium.
David A Schwartz, Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado Denver, Aurora, CO, USA.
References
- 1.Jones MG, Fabre A, Schneider P, et al. Three-dimensional characterization of fibroblast foci in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e86375. doi: 10.1172/jci.insight.86375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–61. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 3.Liebow AA. Definition and classification of interstitial pneumonias in human pathology. Prog Respir Res. 1975;8:1–31. [Google Scholar]
- 4.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–79. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis an integral model. Am J Respir Crit Care Med. 2014;189:1161–72. doi: 10.1164/rccm.201312-2221PP. [DOI] [PubMed] [Google Scholar]
- 6.Robbins LL. Idiopathic pulmonary fibrosis: roentgenologic findings. Radiology. 1948;51:459–67. doi: 10.1148/51.4.459. [DOI] [PubMed] [Google Scholar]
- 7.Peabody JW, Peabody JW, Jr, Hayes EW, Hayes EW., Jr Idiopathic pulmonary fibrosis; its occurrence in identical twin sisters. Dis Chest. 1950;18:330–44. doi: 10.1016/s0096-0217(15)34710-5. [DOI] [PubMed] [Google Scholar]
- 8.Crystal RG, Fulmer JD, Roberts WC, Moss ML, Line BR, Reynolds HY. Idiopathic pulmonary fibrosis Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspects. Ann Intern Med. 1976;85:769–88. doi: 10.7326/0003-4819-85-6-769. [DOI] [PubMed] [Google Scholar]
- 9.Katzenstein ALA, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol. 1994;18:136–47. [PubMed] [Google Scholar]
- 10.Hunninghake GW, Lynch DA, Galvin JR, et al. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest. 2003;124:1215–23. doi: 10.1378/chest.124.4.1215. [DOI] [PubMed] [Google Scholar]
- 11.Katzenstein ALA, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–15. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 12.Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence–based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flaherty KR, King TE, Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med. 2004;170:904–10. doi: 10.1164/rccm.200402-147OC. [DOI] [PubMed] [Google Scholar]
- 14.Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J. 2013;42:750–57. doi: 10.1183/09031936.00131912. [DOI] [PubMed] [Google Scholar]
- 15.Alder JK, Stanley SE, Wagner CL, Hamilton M, Hanumanthu VS, Armanios M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. 2015;147:1361–68. doi: 10.1378/chest.14-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–26. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 17.Campo I, Zorzetto M, Mariani F, et al. A large kindred of pulmonary fibrosis associated with a novel ABCA3 gene variant. Respir Res. 2014;15:43. doi: 10.1186/1465-9921-15-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cogan JD, Kropski JA, Zhao M, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. 2015;191:646–55. doi: 10.1164/rccm.201408-1510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kropski JA, Mitchell DB, Markin C, et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest. 2014;146:e1–7. doi: 10.1378/chest.13-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawson WE, Grant SW, Ambrosini V, et al. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59:977–80. doi: 10.1136/thx.2004.026336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. 2015;47:512–17. doi: 10.1038/ng.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas AQ, Lane K, Phillips J, 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–28. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 23.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104:7552–57. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Moorsel CH, van Oosterhout MF, Barlo NP, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182:1419–25. doi: 10.1164/rccm.200906-0953OC. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fingerlin TE, Zhang W, Yang IV, et al. Genome-wide imputation study identifies novel HLA locus for pulmonary fibrosis and potential role for auto-immunity in fibrotic idiopathic interstitial pneumonia. BMC Genet. 2016;17:74. doi: 10.1186/s12863-016-0377-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandit KV, Corcoran D, Yousef H, et al. Inhibition and role of let–7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:220–29. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabinovich EI, Kapetanaki MG, Steinfeld I, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e33770. doi: 10.1371/journal.pone.0033770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders YY, Ambalavanan N, Halloran B, et al. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186:525–35. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang IV, Burch LH, Steele MP, et al. Gene expression profiling of familial and sporadic interstitial pneumonia. Am J Respir Crit Care Med. 2007;175:45–54. doi: 10.1164/rccm.200601-062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang IV, Pedersen BS, Rabinovich E, et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am J Respir Critical Care Med. 2014;190:1263–72. doi: 10.1164/rccm.201408-1452OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DePianto DJ, Chandriani S, Abbas AR, et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax. 2015;70:48–56. doi: 10.1136/thoraxjnl-2013-204596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selman M, Pardo A, Barrera L, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2006;173:188–98. doi: 10.1164/rccm.200504-644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang IV, Coldren CD, Leach SM, et al. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68:1114–21. doi: 10.1136/thoraxjnl-2012-202943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zuo FR, Kaminski N, Eugui E, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99:6292–97. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen RJ, Porte J, Braybrooke R, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med. 2017;5:869–80. doi: 10.1016/S2213-2600(17)30387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borie R, Crestani B, Dieude P, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8:e70621. doi: 10.1371/journal.pone.0070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horimasu Y, Ohshimo S, Bonella F, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology. 2015;20:439–44. doi: 10.1111/resp.12466. [DOI] [PubMed] [Google Scholar]
- 40.Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–17. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peljto AL, Selman M, Kim DS, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest. 2015;147:460–64. doi: 10.1378/chest.14-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–12. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stock CJ, Sato H, Fonseca C, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68:436–41. doi: 10.1136/thoraxjnl-2012-201786. [DOI] [PubMed] [Google Scholar]
- 44.van der Vis JJ, Snetselaar R, Kazemier KM, ten Klooster L, Grutters JC, van Moorsel CH. Effect of Muc5b promoter polymorphism on disease predisposition and survival in idiopathic interstitial pneumonias. Respirology. 2016;21:712–17. doi: 10.1111/resp.12728. [DOI] [PubMed] [Google Scholar]
- 45.Wei R, Li C, Zhang M, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res. 2014;163:494–502. doi: 10.1016/j.trsl.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–77. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araki T, Putman RK, Hatabu H, et al. Development and progression of interstitial lung abnormalities in the Framingham heart study. Am J Respir Crit Care Med. 2016;194:1514–22. doi: 10.1164/rccm.201512-2523OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunninghake GM, Hatabu H, Okajima Y, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. 2013;368:2192–200. doi: 10.1056/NEJMoa1216076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172:1146–52. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tobin RW, Pope CE, Pellegrini CA, Emond MJ, Sillery J, Raghu G. Increased prevalence of gastroesophageal reflux in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;158:1804–08. doi: 10.1164/ajrccm.158.6.9804105. [DOI] [PubMed] [Google Scholar]
- 51.Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:1390–94. doi: 10.1164/rccm.201101-0138OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baumgartner KB, Samet JM, Coultas DB, et al. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol. 2000;152:307–15. doi: 10.1093/aje/152.4.307. [DOI] [PubMed] [Google Scholar]
- 53.Hubbard R, Lewis S, Richards K, Johnston I, Britton J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet. 1996;347:284–89. doi: 10.1016/s0140-6736(96)90465-1. [DOI] [PubMed] [Google Scholar]
- 54.Miyake Y, Sasaki S, Yokoyama T, et al. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann Occup Hyg. 2005;49:259–65. doi: 10.1093/annhyg/meh090. [DOI] [PubMed] [Google Scholar]
- 55.Lawson WE, Crossno PF, Polosukhin VV, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–26. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 56.Stewart JP, Egan JJ, Ross AJ, et al. The detection of Epstein–Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1999;159:1336–41. doi: 10.1164/ajrccm.159.4.9807077. [DOI] [PubMed] [Google Scholar]
- 57.Tang YW, Johnson JE, Browning PJ, et al. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41:2633–40. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–8. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 59.Van Dyken SJ, Liang HE, Naikawadi RP, et al. Spontaneous chitin accumulation in airways and age–related fibrotic lung disease. Cell. 2017;169:497–509.e13. doi: 10.1016/j.cell.2017.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ley B, Collard HR. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol. 2013;5:483–92. doi: 10.2147/CLEP.S54815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174:810–16. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 62.Selman M, King TE, Pardo A, et al. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–51. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 63.Alder JK, Chen JJ, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105:13051–56. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kropski JA, Pritchett JM, Zoz DF, et al. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am J Respir Crit Care Med. 2015;191:417–26. doi: 10.1164/rccm.201406-1162OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Naikawadi RP, Disayabutr S, Mallavia B, et al. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight. 2016;1:e86704. doi: 10.1172/jci.insight.86704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Selman M, Lopez–Otin C, Pardo A. Age-driven developmental drift in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J. 2016;48:538–52. doi: 10.1183/13993003.00398-2016. [DOI] [PubMed] [Google Scholar]
- 67.Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Disayabutr S, Kim EK, Cha SI, et al. miR-34 miRNAs regulate cellular senescence in type II alveolar epithelial cells of patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0158367. doi: 10.1371/journal.pone.0158367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bueno M, Lai YC, Romero Y, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015;125:521–38. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Korfei M, Ruppert C, Mahavadi P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–46. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–50. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lama V, Moore BB, Christensen P, Toews GB, Peters-Golden M. Prostaglandin E-2 synthesis and suppression of fibroblast proliferation by alveolar epithelial cells is cyclooxygenase-2-dependent. Am J Respir Cell Mol Biol. 2002;27:752–58. doi: 10.1165/rcmb.4857. [DOI] [PubMed] [Google Scholar]
- 73.Epa AP, Thatcher TH, Pollock SJ, et al. Normal human lung epithelial cells inhibit transforming growth factor-β induced myofibroblast differentiation via prostaglandin E2. PLoS One. 2015;10:e0135266. doi: 10.1371/journal.pone.0135266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barkauskas CE, Cronce MJ, Rackley CR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–36. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alder JK, Barkauskas CE, Limjunyawong N, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112:5099–104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liang J, Zhang Y, Xie T, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med. 2016;22:1285–93. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Froese AR, Shimbori C, Bellaye PS, et al. Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 78.Parker MW, Rossi D, Peterson M, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124:1622–35. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tschumperlin DJ. Matrix, mesenchyme, and mechanotransduction. Ann Am Thorac Soc. 2015;12(suppl 1):S24–29. doi: 10.1513/AnnalsATS.201407-320MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Booth AJ, Hadley R, Cornett AM, et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med. 2012;186:866–76. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bernard K, Logsdon NJ, Ravi S, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem. 2015;290:25427–38. doi: 10.1074/jbc.M115.646984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Y, Jiang D, Liang J, et al. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208:1459–71. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scherer LD, Finan C, Simancek D, Finkelstein JI, Tarini BA. Effect of “pink eye” label on parents’ intent to use antibiotics and perceived contagiousness. Clin Pediatr (Phila) 2016;55:543–48. doi: 10.1177/0009922815601983. [DOI] [PubMed] [Google Scholar]
- 84.Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–93. doi: 10.1182/blood-2008-07-162503. [DOI] [PubMed] [Google Scholar]
- 85.Cha SI, Chang CS, Kim EK, et al. Lung mast cell density defines a subpopulation of patients with idiopathic pulmonary fibrosis. Histopathology. 2012;61:98–106. doi: 10.1111/j.1365-2559.2012.04197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Greer AM, Matthay MA, Kukreja J, et al. Accumulation of BDCA1+ dendritic cells in interstitial fibrotic lung diseases and Th-high asthma. PLoS One. 2014;9:e99084. doi: 10.1371/journal.pone.0099084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu Y, Mizuno T, Sridharan A, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 89.Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 90.Juge PA, Borie R, Kannengiesser C, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J. 2017;49:1602314. doi: 10.1183/13993003.02314-2016. [DOI] [PubMed] [Google Scholar]
- 91.Ley B, Newton CA, Arnould I, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5:639–47. doi: 10.1016/S2213-2600(17)30216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]