

ABSTRACT
Lymphangioleiomyomatosis (LAM) is a rare cystic pulmonary disease that may occur in association with mutations in the tuberous sclerosis genes or arise sporadically. The histologic hallmark of the disease is the “LAM” cell, a spindled to epithelioid smooth muscle-like cell that bears morphologic and immunohistochemical resemblance to the perivascular epithelioid cell tumors (PEComas). The origin of the “LAM” cell is unknown; emerging theories suggest that a member of the PEComa family, the renal angiomyolipoma, may be the primary source and that both LAM and angiomyolipomas are associated with the genetic syndrome tuberous sclerosis. We present a young woman with LAM with an aggressive renal angiomyolipoma confirmed at autopsy.
KEYWORDS: Angiomyolipoma, lymphangioleiomyomatosis, perivascular epithelioid cell tumor, tuberous sclerosis
Lymphangioleiomyomatosis (LAM) is a rare pulmonary disease that has been found to primarily affect women of childbearing age.1 The disease is frequently associated with mutations in the tuberous sclerosis (TSC) genes TSC1 and TSC2, but other sporadic instances of LAM also occur in the absence of these mutations.2 Characteristic microscopic findings include cystic degeneration of the lung parenchyma associated with groups of smooth muscle-like spindle and epithelioid cells (or “LAM” cells); clinical findings (dyspnea, recurrent pneumothorax, and chylothorax) are sequelae of lymphovascular and airway obstruction by LAM cells.1,3 Renal angiomyolipomas are estimated to occur in 40% of women with the sporadic form of the disease, and emerging theories suggest that LAM cells may actually arise from these angiomyolipomas.2 Here, we present a woman with pulmonary LAM and infiltration of the right kidney and adrenal gland by an aggressive angiomyolipoma, which is the most likely source for the pulmonary LAM cells in this patient.
CASE DESCRIPTION
A 24-year-old woman on the lung transplant waiting list for suspected LAM presented to the emergency department for acute-onset dyspnea. She had had recurrent pneumothoraces, and computed tomography (CT) found diffuse cystic changes throughout both lungs that were highly suspicious and resulted in a presumptive diagnosis of LAM (Figure 1). Abdominal ultrasound found renal and hepatic tumors consistent with angiomyolipomas. On this admission, a chest radiograph also found a right-sided tension pneumothorax. The patient was not treated with sirolimus because of anticipated transplant in the near future and concerns about the impact of sirolimus on wound healing. Despite chest tube placement and mechanical ventilation, bilateral pneumothoraces persisted with increasing hypoxemia. Septic shock secondary to enterococcal and staphylococcal infection also ensued, precluding transplantation, and she died.
Figure 1.
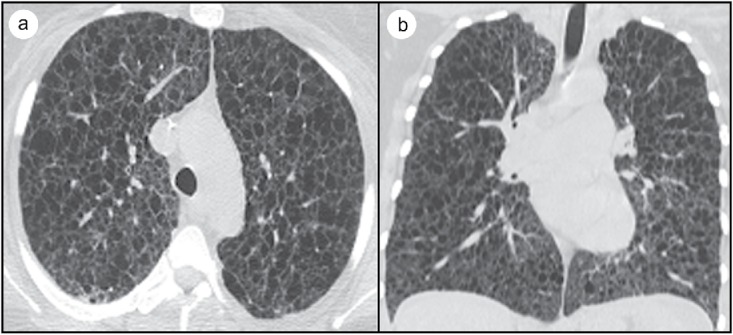
(a) Axial CT without contrast and (b) coronal CT without contrast highlighting innumerable thin-walled cysts and cystic spaces classically observed on CT scan with high suspicion for lymphangioleiomyomatosis.
The autopsy was limited to the chest and abdomen. The bilateral lung parenchyma was diffusely cystic with numerous hemorrhagic nodules (Figure 2). Multiple hepatic and renal cortical tumor nodules were also found, and the right kidney tumor extended through the renal capsule. Approximately 3200 mL of chylous fluid was present within the peritoneal cavity. Microscopic examination of the lung found cystic spaces with surrounding groups of spindled and epithelioid cells (Figure 3a) that had strong positive immunohistochemical staining for actin and desmin, as well as scattered cells staining positive for ER, PGR, and HMB45. The hepatic and renal nodules were composed of adipose tissue, blood vessels, and HMB45+ spindled and epithelioid cells, all consistent with angiomyolipomas. Sections of the right kidney angiomyolipoma showed increased cellularity with infiltration into the perinephric adipose tissue, lymphatic spaces, and adjacent adrenal gland (Figure 3b). Additional sections of lung hilar and paratracheal lymph nodes demonstrated metastatic involvement by HMB45+ spindled and epithelioid cells. A cardiac blood sample was submitted for genetic testing and was found to be negative for TSC1 and TSC2 gene mutations.
Figure 2.
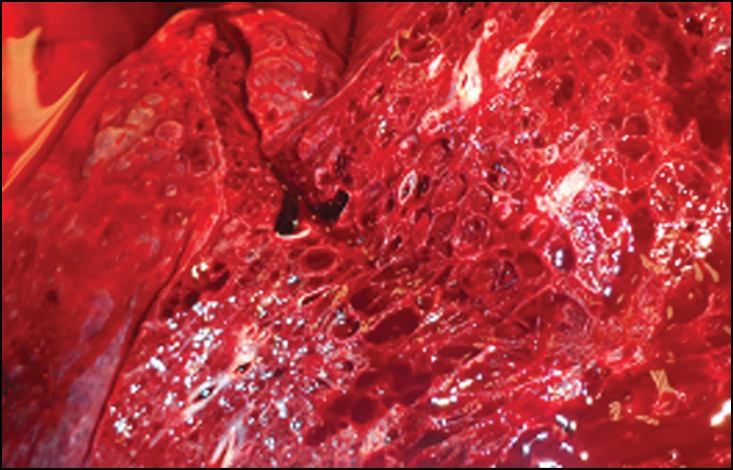
Lung parenchyma with multiple cystic spaces.
Figure 3.
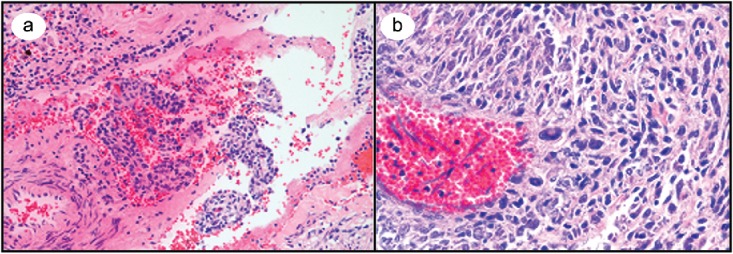
(a) Medium-power microscopic view of a hematoxylin and eosin–stained lung section containing a cyst with spindled and epithelioid cells. (b) High-magnification microscopic view of highly cellular kidney mass with multiple atypical cells.
DISCUSSION
LAM is a rare progressive lung disease affecting predominantly women, with a case prevalence of approximately 3.4 to 7.8 per million.4 The disease causes cystic destruction of lung parenchyma with smooth muscle cell infiltration by spindled “LAM” cells that results in dyspnea, pneumothorax, chylous pleural effusions, and ultimately respiratory failure. A genetic basis for the disease is suggested by its association with TSC, an autosomal dominant tumor suppressor syndrome, in which 26% of affected women also have LAM (TSC-LAM).5 In addition to TSC-LAM, there is a sporadic form of the disease (S-LAM), also linked to TSC-LAM. Further evidence for this link includes somatic inactivating mutations of the TSC2 gene6 in S-LAM and loss of heterozygosity at the TSC2 region of chromosome 16 in disseminated neoplastic cells in S-LAM.7 These findings provide a genetic basis for a common link between the two forms of the disease as well as evidence for the clonal nature of the neoplasms.
In addition to its association with TSC, LAM commonly demonstrates expression of the estrogen and progesterone receptors and is considered a sex steroid–dependent disease. Exactly how the receptor expression affects disease progression is unclear, but it has been suggested that estrogen enhances the neoplastic potential and survival of LAM cells. A research model utilizing xenograft TSC-LAM cells found that estradiol has a proliferative effect on TSC2-deficient cells.8 Other studies have shown that progesterone increases the metastatic potential of TSC2-deficient LAM patient-derived cells in vitro and lung metastasis in vivo.9 However, to date, there have been no randomized placebo-controlled trials studying the effects of hormonal therapy, and it is not currently recommended as a routine treatment.10
The ontogeny of LAM cells has not been clearly defined, but existing evidence suggests a relation to the perivascular epithelioid cell tumor (PEComa) family, which typically stains for HMB-45 and smooth muscle markers actin, vimentin, and desmin. This tumor family includes angiomyolipoma (renal and extrarenal) and clear cell “sugar” tumor (lung and extrapulmonary), which are both generally benign but can show aggressive behavior. It has been suggested that renal angiomyolipomas could be the primary source of LAM cells; interestingly, both LAM and angiomyolipomas are associated with TSC. A “benign” metastasis model of LAM pathogenesis was suggested by Henske in 2003,11 in which LAM cells spread from a peripheral source to the lungs. These peripheral “benign” cells were hypothesized to drive a lymphangiogenic program resulting in budding of LAM cells into the lymphatic system and pulmonary vasculature.12 The definitive treatment for LAM is lung transplantation; however, clinical trials suggest that the mTOR inhibitor sirolimus may be efficacious,3 and this was approved by the Food and Drug Administration in 2015.
ACKNOWLEDGMENTS
We gratefully acknowledge Dr. William Dockery for assistance with the CT image (Figure 1).
References
- 1.Hasegawa W, Yamauchi Y, Yasunaga H, et al.. Clinical features of 280 hospitalized patients with lymphangioleiomyomatosis in Japan. Respirology. 2015;20(1):160–165. doi: 10.1111/resp.12430. PMID:25385157. [DOI] [PubMed] [Google Scholar]
- 2.Henske EP, McCormack FX. Lymphangioleiomyomatosis—a wolf in sheep's clothing. J Clin Invest. 2012;122(11):3807–3816. doi: 10.1172/JCI58709. PMID:23114603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies DM, de Vries PJ, Johnson SR, et al.. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17(12):4071–4081. doi: 10.1158/1078-0432.CCR-11-0445. PMID:21525172. [DOI] [PubMed] [Google Scholar]
- 4.Harknett EC, Chang WY, Byrnes S, et al.. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM. 2011;104(11):971–979. doi: 10.1093/qjmed/hcr116. PMID:21764810. [DOI] [PubMed] [Google Scholar]
- 5.Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex Mayo. Clin Proc. 2000;75(6):591–594. doi: 10.4065/75.6.591. [DOI] [PubMed] [Google Scholar]
- 6.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2000;97(11):6085–6090. doi: 10.1073/pnas.97.11.6085. PMID:10823953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crooks DM, Pacheco-Rodriguez G, DeCastro RM, et al.. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2004;101(50):17462–17467. doi: 10.1073/pnas.0407971101. PMID:15583138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu JJ, Robb VA, Morrison TA, et al.. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci USA. 2009;106(8):2635–2640. doi: 10.1073/pnas.0810790106. PMID:19202070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Y, Zhang E, Lao T, et al.. Progesterone and estradiol synergistically promote the lung metastasis of tuberin-deficient cells in a preclinical model of lymphangioleiomyomatosis. Horm Cancer. 2014;5(5):284–298. doi: 10.1007/s12672-014-0192-z. PMID:25069840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson SR, Cordier JF, Lazor R, et al; Review Panel of the ERS LAM Task Force. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010;35(1):14–26. doi: 10.1183/09031936.00076209. PMID:20044458. [DOI] [PubMed] [Google Scholar]
- 11.Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38(4):376–381. doi: 10.1002/gcc.10252. PMID:14566858. [DOI] [PubMed] [Google Scholar]
- 12.Kumasaka T, Seyama K, Mitani K, et al.. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. 2005;29(10):1356–1366. doi: 10.1097/01.pas.0000172192.25295.45. PMID:16160479. [DOI] [PubMed] [Google Scholar]