

Abstract
Since the introduction of calcium imaging to monitor neuronal activity with single-cell resolution, optical imaging methods have revolutionized neuroscience by enabling systematic recordings of neuronal circuits in living animals. The plethora of methods for functional neural imaging can be daunting to the nonexpert to navigate. Here we review advanced microscopy techniques for in vivo functional imaging and offer guidelines for which technologies are best suited for particular applications.
Our understanding of the mammalian brain is still limited. One reason may be that most neural circuits are composed of hundreds of thousands of neurons that are interconnected in a highly distributed fashion. This organization makes it likely that nervous systems, rather than operating neuron by neuron, have emergent functional properties, similarly to the interacting pixels that produce an image on a television screen1. Thus one may need to record the activity of many, or most, neurons in a circuit in order to capture the functional properties built by the entire population. Access to such data in behaving animals or humans may enable ‘breaking the neural code’, i.e., the deciphering of how neural information is coded, stored and processed and how the brain generates behavior or mental states.
Calcium imaging with fluorescent indicators provides an optical approach to monitor action potentials2 and is being used systematically, complementing microelectrode recordings, to measure neuronal activity in vivo. This method opened the way to the reconstruction, albeit with low temporal resolution, of the activity of neuronal populations in entire brains of small model organisms3. The invention of two-photon microscopy4, and its combination with calcium imaging5, has allowed measuring activity of populations of deep neurons in vivo, either with synthetic6 or genetically engineered7,8 indicators.
Microscopists now have a multitude of methods9,10 to perform calcium imaging in vivo. An almost bewildering assortment of techniques and acronyms await the neophyte, making navigation of the relevant literature a challenge. But the physical problems that different microscopes are made to solve are the same: deliver photons effectively to the sample, minimize the dose of excitation light, collect emitted photons efficiently and perform measurements with high temporal and spatial resolution in as large and deep a territory as possible, all while preserving good signal-to-noise ratio (SNR). Here we review available optical systems to illustrate how different microscopes solve these problems, and we discuss their tradeoffs in different applications. To help readers, we provide a table summarizing these methods (Supplementary Table 1) and a flowchart as a guideline for choosing specific types of microscopes (Fig. 1). Our focus is on calcium imaging, but these considerations also apply to fluorescence imaging of living samples more generally.
Figure 1.
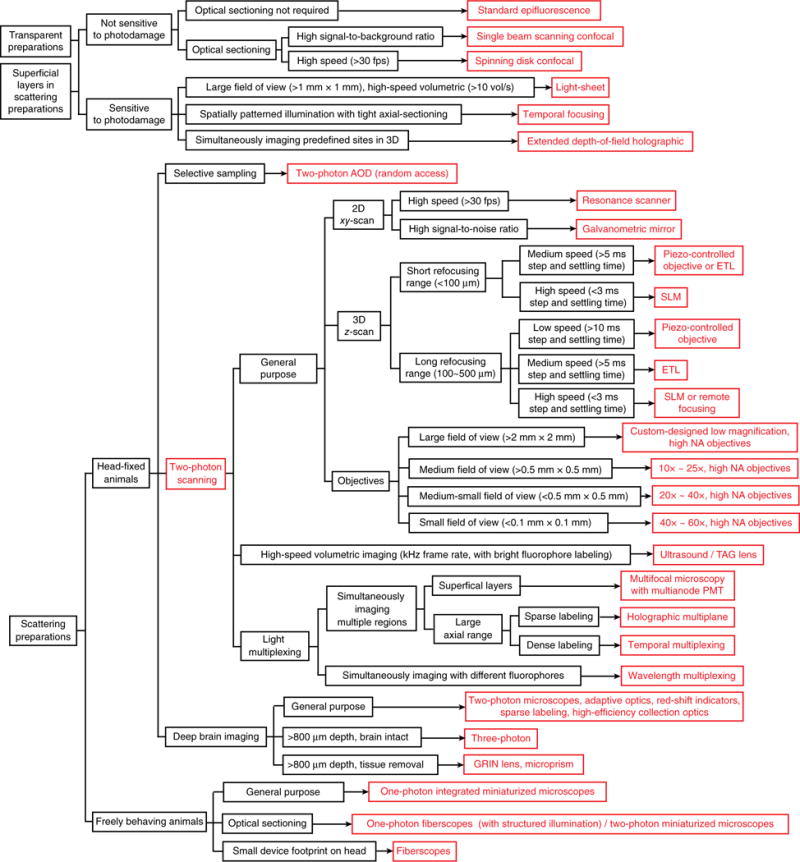
Flowchart to choose imaging platform.
Transparent samples
Neural activity in transparent organisms such as Hydra, worms, zebrafish and Drosophila larvae can be imaged by traditional wide-field microscopes. This technology offers high spatiotemporal resolution over a large area or volume, ideal for mapping neuronal activity across an entire brain region. In a wide-field microscope, the full-field signal is captured with a camera, and imaging speed is limited only by camera frame rate, which approaches 500 frames per second (fps) (512 × 512 pixels) for scientific complementary metal oxide semiconductor (sCMOS) cameras. But wide-field microscopes are sensitive to light scattering (deflection of a light ray from its original direction due to inhomogeneity in the tissue’s refractive index), which causes detrimental cross-talk between pixels in camera. Thus, they are most suitable for imaging transparent organisms, for superficial layers (<200 μm deep) of rodent cortex in vivo, or for fixed samples that have been turned transparent in vitro.
The basic wide-field fluorescence microscope (commonly termed epifluorescence microscope)11 illuminates the sample over an extended volume and captures the emitted fluorescence at the focal plane through the same objective lens. Under one-photon excitation conditions, the emission of the fluorophore increases linearly with excitation power until saturation. The out-of-focus fluorescence contributes to the image background, and can make in-focus features indiscernible in a thick sample with dense labeling. Nevertheless, this type of microscopy can still be used for meso-scale imaging (for example, across the whole neocortex of rodents) with a low-magnification objective. Though without single-cell resolution, this approach can map neural activity and functional connectivity across different brain regions12–14. To produce clearer images of focal planes, one can reject out-of-focus fluorescence by optical sectioning (Box 1). Confocal laser scanning microscopy15 achieves this goal by scanning the laser focal spot across the sample and using a pinhole to reject out-of-focus fluorescence. Spinning disk confocal microscopy15–17 parallelizes this strategy by simultaneously scanning multiple points. Although out-of-focus photons are not used for image formation in these approaches, they can nevertheless contribute to phototoxicity. Therefore, confocal microscopy is best suited for imaging fixed samples or those with low sensitivity to photodamage. For functional imaging in vivo, where phototoxicity must be minimized, methods using excitation photons more efficiently are preferred. This has led to methods that achieve optical sectioning by avoiding the generation of out-of-focus background in the first place.
BOX 1. POINT-SPREAD FUNCTION, SPATIAL RESOLUTION AND OPTICAL SECTIONING.
The point-spread function (PSF) characterizes how a point source appears upon detection in the microscope. The image of any object can be obtained by mathematically convoluting its intensity density with the microscope PSF. The smaller and sharper the PSF, the better the resolution of a microscope. For an aberration-free system, the PSF depends on wavelength (λ), refractive index (n) in the objective medium and the numerical aperture (NA), which is related to the microscope’s aperture shape. For laser scanning microscopy, an illumination PSF can also be defined to characterize the spatial profile of the excitation spot. The overall PSF is the product between the illumination PSF and the detection PSF convoluted with the 2D pixel area (D) of the detector projected into the object space.
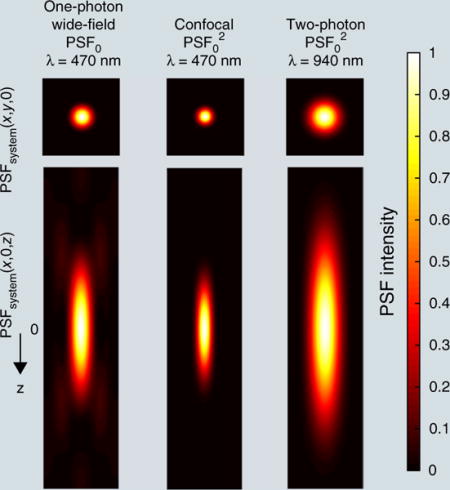
For a typical microscope with a circular aperture (assuming uniform illumination), the PSF is circular with a central bright disk and progressively weaker concentric dark and bright rings. The distance from its center to the first lateral minimum rxy and axial minimum rz is commonly quoted as the lateral and axial resolution (by Rayleigh criterion) of wide-field microscopes. The rz is proportional to the depth of field.
For confocal microscopes, the pinhole restricts the collection at the focus center, leading to a squared PSF (ignoring wavelength difference between illumination and emission), which provides a tighter confinement. Thus15
For standard two-photon microscopes, the excitation also scales as light intensity squared, so that resolution can also be expressed as above. Because the wavelength in two-photon microscopy is about twofold that used in confocal, its theoretical spatial resolution should be twofold as well. In practice they are similar, as the finite pinhole in confocal microscopy broadens its PSF.
A microscope capable of optical sectioning can discern the z location of a uniform thin fluorescence plane. The laterally integrated PSF describes this capability. In standard wide-field microscopy, the laterally integrated PSF is z independent and thus does not confer optical sectioning. But the squared PSF in confocal and two-photon renders a z dependency and thus optical sectioning.
Temporal focusing imposes additional nonlinearity at the focus and enables wide-field two-photon excitation. The calculation of its excitation PSF requires consideration of the spatial dependency of the laser pulse width24–27.
One such technique is light-sheet microscopy, which uses a thin sheet of light projected into the sample from the side, exciting only a two-dimensional (2D) section of the sample. The emitted fluorescence signal is then imaged in an orthogonal direction to the excitation plane (Fig. 2a)3,17–22. Light-sheet microscopy is particularly applicable for functional mapping of circuits across large brain volumes in transparent or semi-transparent samples and for measuring spatiotemporal responses to sensory, pharmacological or optogenetic stimuli; volumetric recording can be implemented with speeds of >10 vol/s (refs. 19–22). More detail on light-sheet microscopy is given in another Review in this issue23.
Figure 2.
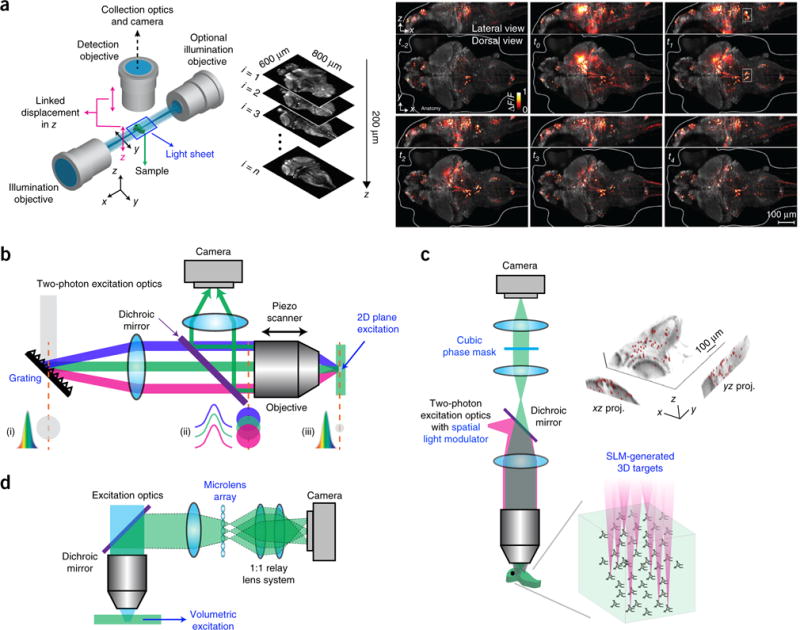
Wide-field imaging. (a) Left, a schematic of a light-sheet microscope. Right, whole-brain neuronal activity of a larval zebrafish recorded with a light-sheet microscope. Brighter hues represent active neurons. Reprinted and adapted from ref. 3, Macmillan Publishers Limited. (b) Schematic of a microscope using temporal focusing. (i–iii) Temporal and spatial cross-section profiles of the laser beam impinging on the grating (i), at the back aperture (ii) and focal plane of the objective (iii) are shown. Colors indicate different spectrum components. Adapted from ref. 28, Macmillan Publishers Limited. (c) Holographic microscope with extended depth of field. Right, calcium imaging of 49 neurons targeted simultaneously on a zebrafish. Image reprinted from ref. 34, Frontiers. (d) Schematic of a light-field microscope. Adapted from ref. 37, Macmillan Publishers Limited.
A different method for selective illumination of focal planes is two-photon excitation. Two-photon light sources are femtosecond lasers that emit periodic pulse trains with high peak power at infrared wavelengths. At these long wavelengths, a single photon’s energy is not enough to excite the fluorophore; it has to absorb two photons to emit fluorescence. Thus, the two-photon absorption rate is proportional to the light intensity squared, so only fluorophores in the focal spot receive enough photon densities to emit fluorescence, suppressing out-of-focus excitation and background, enabling optical sensing (Box 1). Although typical two-photon microscopes scan a focal spot across the sample and are often used for scattering samples (see below), wide-field two-photon imaging is also possible. However, the axial extent of the excitation volume depends on its lateral extent24. This can be solved by temporal focusing, which shapes the light pulse in time24–29 (Fig. 2b). This approach broadens the laser pulse with the help of a grating. The pulse is recovered at shortest temporal width and highest peak power at the focus, achieving simultaneous spatial and temporal focusing. This additional nonlinearity enhances the contrast between the focus and the background, leading to a tightly axially confined 2D excitation plane (for example, 60-μm diameter excitation area with ~1.9-μm axial confinement)28. Compared to light-sheet microscopy, temporal focusing requires a simpler optical setup for illumination. Moreover, when combined with a spatial light modulator (SLM), a device that can generate holographic light patterns30, arbitrary 2D excitation patterns can be projected to the sample24. This approach is well suited for imaging that requires high spatiotemporal resolution, such as imaging synaptic and dendritic activity in superficial cortical layers. Furthermore, temporal focusing is particularly applicable for photostimulation with optochemistry or optogenetics, as it generates a well-defined excitation pattern over the sample31,32. But because each point of the excitation pattern in the 2D plane needs to receive enough power for two-photon excitation, high-peak-power laser pulses (usually using regenerative amplifiers) are required, which necessitates caution owing to the potential for photodamage. Alternatively, a near-simultaneously excited 2D plane can be obtained by fast scanning of a 1D line26,29, which improves the sectioning capability and reduces the laser power.
Extended depth of field (EOF) holographic microscopy is another form of two-photon wide-field microscopy and one that we helped develop. Instead of exciting the sample with a whole 2D plane, a computer-generated hologram is built with an SLM leading to multiple focal excitation spots. A two-photon laser pulse is split into multiple beamlets33–35 that then target many neurons (>100) simultaneously in 3D (Fig. 2c). By placing a cubic phase mask (that modifies the spatial phase of the light) in the detection path, targeted neurons at different depths (which can span hundreds of micrometers)33,34 are focused on the camera, and their activity is recorded simultaneously34. By switching hologram patterns, different groups of neurons can be sequentially imaged35. EOF holography enables sparse excitation that reduces background, alleviating the effects of scattering and reducing photodamage. But if the sample has significant motion, the SLM pattern needs to be coordinated with such motion to avoid off-target excitation.
Finally, the development of computationally supported optics has made possible light-field microscopy, a type of wide-field imaging technology in which the whole sample is illuminated with one-photon light, and different imaging depths at the detection path are computationally extracted36,37 (Fig. 2d). A microlens array is set up in front of the camera, collecting different perspectives of the object. Both the spatial intensity of emitted light and its direction are recorded for the emitted light ‘field’, and then the sample can be reconstructed in 3D with a computational procedure. Light-field functional imaging of zebrafish larval brains spanning 700 × 700 × 200 μm has been demonstrated at 20 vol/s in vivo37.
Scattering samples: two-photon microscopy
Because of the deeper penetration in tissue of infrared light, two-photon laser scanning microscopy is ideally suited for imaging scattering tissues4,5,38,39 (Fig. 3a). In a typical two-photon microscope, a femtosecond laser beam is focused into a diffraction-limited spot and scanned across the sample. The emitted fluorescence is collected by a single photomultiplier tube (PMT), whose temporal signal is mapped to the corresponding pixel, eventually forming an image. Because the sample is excited and the signals are collected point-by-point, this approach overcomes pixel cross-talk seen in wide-field imaging of scattering tissues. Furthermore, the longer excitation wavelengths are more resistant to tissue scattering than those used for one-photon excitation. Two-photon microscopy is generally a good alternative to confocal microscopy for functional imaging owing to its higher light-collection efficiency (because there is no pinhole in front of the PMT), deeper penetration and reduced phototoxicity. It has enabled a wide range of neuroscience studies in rodents, such as analyses of functional connectivity between cortical layers, cortical response to various sensory stimulations, neural activity during behavior, neurovascular coupling and spine structural and functional studies. This approach is also suitable for imaging neural activity in retina, which naturally responds to visible light but not to the infrared light of the two-photon excitation40.
Figure 3.
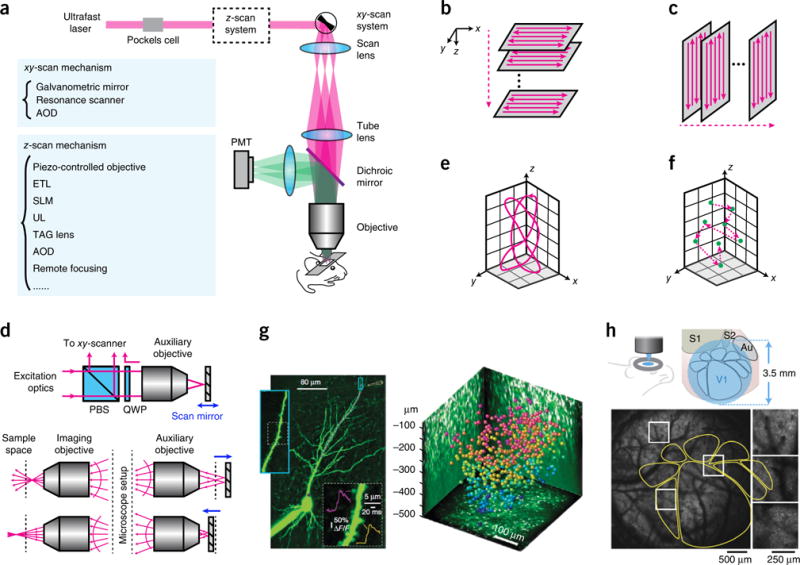
Two-photon microscopy. (a) Optical layout of a two-photon laser scanning microscope. (b) Traditional scan trajectory for volumetric imaging. (c) Scan trajectory enabled by ultrasound or TAG lens. (d) Principle of remote focusing. PBS, polarization beam splitter. QWP, quarter waveplate. Adapted from ref. 48 with permission from the National Academy of Sciences. (e) Custom scanning. Adapted from ref. 48 with permission from the National Academy of Sciences. (f) Random-access scanning in a 3D volume. (g) Examples of data taken with 3D random-access hopping microscope using AODs. Left, calcium imaging of a CA1 pyramidal cell; purple dots represent the scanning points in a dendrite. Right, neurons color coded to depth in mouse visual cortex, imaged with AODs. Reprinted from ref. 53, Macmillan Publishers Limited. (h) Large FOV microscope. Bottom, calcium imaging of a transgenic mouse expressing GCaMP6s. The yellow outlines indicate the anatomy mapped out by intrinsic signal optical imaging. Reprinted from ref. 57, Macmillan Publishers Limited.
But the Achilles’ heel of two-photon microscopy, or any laser scanning microscopy, is its slow speed, as the sample is imaged sequentially, pixel by pixel. This will be a fundamental roadblock to imaging the activity of larger neuronal circuits in the future. Various scanning methods are available to minimize the transition time from pixel to pixel (Fig. 3a). The most prevalent scanners are galvanometric mirrors (<10 fps) or resonance scanners (>30 fps) for xy scanning, and piezo-controlled objectives41 or electrically tunable lenses (ETL)42 for z scanning (Fig. 3b). Although piezos offer a traveling range as large as 400 μm, with a step and settling time <20 ms for large mass objectives (preferred for deep imaging in scattering tissue for their high N.A. and low magnification43), ETLs have a lower cost and shorter settling time (<10 ms).
For high-speed volumetric (3D) imaging, it is also desirable to reduce the transition time between illuminating different depths. A few technologies are promising to quickly change focal planes. SLMs can switch focus across >500-μm depth in <3 ms (ref. 44) and can be simultaneously used to implement beam multiplexing and adaptive optics, which corrects beam distortion from the optics and sample. Another z-scanning strategy employs an ultrasound lens (UL) or a tunable acoustic gradient index of refraction (TAG) lens, where each frame is taken at a yz plane (Fig. 3c). These lenses operate continuously and can scan in z with a resonance frequency >450 kHz. ULs require a phase-locked loop for high-precision control of oscillation to avoid image distortions, enabling a ~1-kHz frame rate45. Finally, remote focusing is another z-scanning strategy46–48 that depends on an auxiliary objective and a scan mirror at its image plane. Light enters the unit and is reflected by the mirror into the pupil plane of the imaging objective. Because the mirror at the auxiliary unit is light weight and can be rapidly moved, the focal spot can be scanned in z at high speed across the sample48 (Fig. 3d). With careful design, spherical aberrations can be avoided across different focal depths. Furthermore, kilohertz scan rates over arbitrary trajectories in 3D spanning hundreds of micrometers in each dimension (Fig. 3e) are possible48.
The scanning trajectories discussed so far are continuous at the scanned plane (Fig. 3b,c) or in space (Fig. 3e). This means that substantial imaging time is wasted if samples are sparsely labeled. Selective access to targeted regions of interest (sometimes referred to as random scanning) would thus significantly increase imaging speed (Fig. 3f). One solution employs acousto-optic deflectors (AODs)49–53. In microscopes using this technology, acoustic waves applied to the AODs direct light to different positions in 3D. During imaging, the target points (N in total) are sequentially visited, with a cycle rate >50/N kHz. Using AODs, fast calcium imaging was demonstrated in a 400 × 400 × 500 μm volume of mouse cortex in vivo53 (Fig. 3g). Owing to its fast random hopping capability, this type of imaging is suited for investigation of calcium dynamics in dendrite arbors across multiple cortical layers; it can also record neural activity at different spatial scales, offering an approach to study connectivity and interaction between local and global networks. But, as with any point-targeting strategy, sample motion needs to be minimal or closely monitored to update target coordinates. Further, the alignment of the AOD is sensitive to wavelength.
Besides scanning speed, an important parameter for imaging is the size of the field of view (FOV). The FOV can be increased by reducing the objective magnification, but this also reduces numerical aperture (NA) and thus resolution. Low-magnification, high-NA objectives are commercially available but are not designed for large-scale imaging. To obtain large NAs and FOVs, large beam diameters and high scan angles are required, which makes it challenging to maintain minimal aberration and uniform excitation over the whole FOV. To do so, special design of the scan lens, tube lens, aberration compensation optics54,55, scan engine56,57 and even objectives55–57 is required (Fig. 3h). These large FOV microscopes can be equipped with fast z-scanning modules such as an ETL or a remote focusing unit. Using such systems, researchers have demonstrated calcium imaging over a 3.5-mm-wide FOV57 and a 4.4-mm × 4.2-mm FOV56 with cellular resolution on mouse cortex in vivo. Large FOV microscopes are important for systems neuroscience, because they reveal interactions across different areas. But to maintain single-cell resolution and high temporal resolution, the whole FOV is typically not scanned at the same time. Multiplexing methods, described below, can maintain the temporal resolution while increasing the scanned area.
Multiplexing two-photon microscopy
Fast-scanning methods, in xy and z, can improve imaging speed, but the tradeoff is a decreased dwell time on each pixel and thus lower SNR. The SNR in turn can be boosted by increasing laser power, but fluorescence saturation and photodamage can then pose substantial problems. An alternative approach to solve the speed problem of laser scanning microscopy is to increase (multiplex) the number of laser beams, and de-multiplex the detected signals. This strategy helps minimize transit time between regions by directing the beams to each desired FOV. Multiplexing can be achieved in different ways. The typical repetition rate of a femtosecond laser for two-photon imaging is ~80 MHz, corresponding to ~12 ns per pulse, whereas a typical fluorophore’s decay time τ is a few nanoseconds. This opens the opportunity to pack several pulses into a time window, each targeting different locations, achieving temporal multiplexing58 (Fig. 4a). For example58, the laser pulse train can be separated into four copies, interleaved in ~3-ns intervals, enough for the fluorescence of each fluorophore to decay. These four beams are directed to scan different FOVs or depths, and their fluorescence can be distinguished and assigned to the corresponding region. This method has been combined with large FOV systems to increase the imaging areas57 (Fig. 3h); different cortical regions can be imaged simultaneously.
Figure 4.
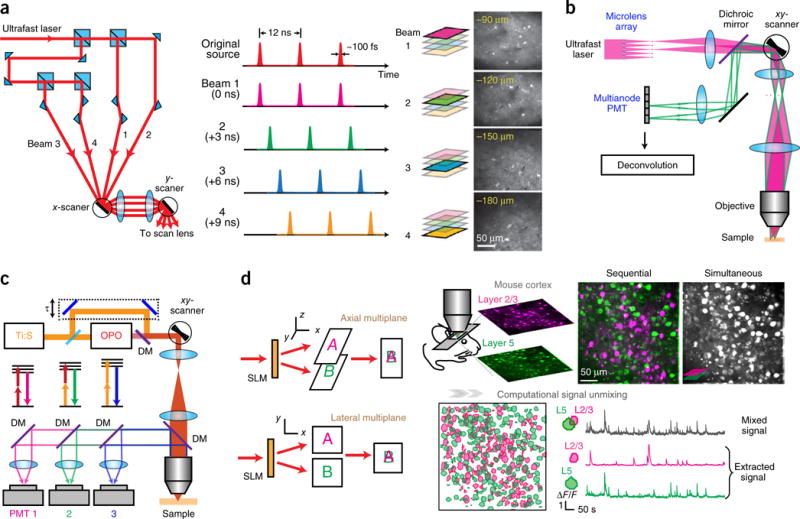
Multiplexing two-photon microscopy. (a) Temporal multiplexing. The single laser beam is split, and one beamlet is delayed such that the pulse trains are interleaved in time. Right, example of a four-plane calcium imaging on mouse brain. Reprinted from ref. 58, Macmillan Publishers Limited. (b) Multifocal multiphoton microscope. Two beamlets are ray-traced. Adapted from ref. 59 with permission from the Optical Society of America. (c) Wavelength multiplexing. Pulse trains from two lasers (Ti:S, Ti:Sapphire and OPO) target two fluorophores with different two-photon absorption spectra. DM, dichroic mirror. Adapted from ref. 62, Macmillan Publishers Limited. (d) Holographic simultaneous multiplane imaging. An SLM splits the laser beam to illuminate different axial planes simultaneously. The right panel shows simultaneous calcium imaging on layers 2/3 and 5 on a GCaMP6f-transfected mouse visual cortex in vivo and post hoc signal separation. Reprinted and adapted from ref. 44 with permission from Elsevier.
But the minimal time interval between different pulses is still limited by the fluorescence decay time τ to avoid cross-talk. This represents a fundamental barrier to the imaging speed of laser scanning microscopy, where the maximum pixel rate is set by τ–1 (~1/(3 ns) = 333.3 MHz, applies to most fluorophores), translating into a volumetric imaging rate of 12.7 vol/s for 512 × 512 × 100 pixels. In practice, the imaging rate is much lower when taking into account a reasonable pixel dwell time and the beam translation time between pixels.
Other multiplexing schemes allow truly simultaneous excitation of multiple spots in a sample. They are no longer limited by τ but instead by how well the demultiplexing scheme minimizes cross-talk and achieve high SNR. In a space multiplexing approach, N beams simultaneously scan N lateral regions, respectively, and pair themselves with N PMTs (multianode PMTs) for detection59 (Fig. 4b). This method is derived from a multifocal two-photon microscope, essentially a wide-field method using a camera60,61. Replacing the camera with a multianode PMT increases the effective detector area for the emission from each focal spot, resulting in a more efficient light collection and thus an increased imaging depth in scattering tissues.
Wavelength multiplexing (or multicolor two-photon imaging) is another strategy to increase speed (Fig. 4c). Here, multiple laser beams, each with a different wavelength, are used to image cells labeled with multiple fluorophores62. At the detection side, dichroic mirrors separate the fluorescence with different wavelengths and direct these fluorescent wavelengths to different PMTs. Although a single excitation beam can excite fluorophores that emit at different wavelengths63, additional excitation wavelengths provide more flexibility. Different fluorophores may be used to label different cell populations. This is useful for structural imaging and to distinguish activity between different cell types, particularly with dense labeling.
In the code-multiplexing64 strategy, an SLM splits the laser beam to target different neurons. Each beam is subsequently modulated by a digital micromirror device (DMD) to carry a unique temporal pattern (code), which is passed on to the fluorescence emitted by the corresponding target. A single PMT collects all the signals, and a decoding algorithm extracts signals for each target. Simultaneous imaging of more than five cells was carried out with submillisecond resolution64, which could be improved by a faster DMD. Imaging of larger cell populations with this method is yet to be demonstrated.
The above multiplexing schemes essentially tag each beam with a unique feature and use this feature to unmix the detected fluorescence. An alternative approach takes advantage of data statistics and computation to unmix the signals. With our recently developed holographic multiplane44 strategy (Fig. 4d), multiple focal spots created by an SLM are simultaneously scanned across different planes of the sample, forming images that overlap at a single PMT. Then, a computational algorithm (such as constrained non-negative matrix factorization)65 is used to assign the photons arriving simultaneously from different focal points to cells in each focal plane. The algorithm takes advantage of prior information from each imaged plane (for example, cell positions and their calcium kinetics obtained in conventional single-plane imaging). It can efficiently unmix signals from different planes, provided that the spatiotemporal content of the neural activity is sparse, which is often the case. Simultaneous three-plane in vivo calcium imaging across a 500-μm depth range in cortex was demonstrated at 10 fps using this approach44. This method, essentially a projective imaging modality, is particularly useful for sparsely labeled samples and studies that require simultaneous imaging of multiple cortical planes or layers.
A final class of multiplexing strategy goes beyond the concept of generating multiple beamlets; it instead relies on more sophisticated point-spread function (PSF) or wavefront engineering. For example, one can replace the diffraction-limited focal spot by a thin disk (for example, 5 × 5 × 10 μm) generated by temporal focusing and scan it over the sample66. This approach gains temporal resolution (160 fps over 500 × 500 μm) and can use statistical algorithms65,67,68 to extract the neural activity from the seemingly decreased spatial resolution images. Other examples include using Bessel beams, which do not spread out laterally over an extended depth of field and can thus image all dendrites or neurons that the beams intersect69,70. It aims to acquire 3D volumetric imaging by a 2D projection scan for sparsely labeled samples.
For all these approaches, the total power deposited into the brain is constrained by the thermal dissipation rate71. The full success of these strategies requires ongoing development of calcium indicators with increased sensitivity and brightness.
Deep brain imaging
In scattering tissue, the excitation beam gets distorted as it propagates in the sample, resulting in an larger PSF and a decreasing signal to background ratio (SBR) and limiting the imaging depth in two-photon microscopes39,72. Scattering is quantified by the ‘mean free path’ or ‘scattering length’ (ls), the average distance between scattering events, i.e., between collisions of photons and sample. For wavelengths (λ) ranging ~800–1,100 nm, commonly used for two-photon imaging, ls ranges from ~150 μm to ~250 μm72. Although increasing the laser power increases the signal at deep layers, it also increases background signal, which is generated mainly within one ls length from the surface. It is the SBR that ultimately determines how deep (z) the sample can be imaged. The SBR of two-photon microscopy can be expressed as SBR ≈[6NA2z2/(λls)]exp(−2z/ls)(V/Vs), where V/Vs is the inverse of the volume fraction of the sample that is fluorescence labeled72. For a SBR = 1, z is ~ 1 mm with λ = 940 nm (commonly used for GCaMP6 calcium imaging), NA = 1 and V/Vs = 1.
One could extend the depth by enhancing the fluorescence collection efficiency η, such as by using low magnification (M), high-NA objectives (as η scales as NA2/M2 (ref. 43), or implementing specially designed optics to collect the fluorescence that escapes from the collection cone of the objective73,74. However, such measures do not solve the distortion of the excitation beam. Adaptive-optics approaches can counteract scattering75. They apply a distortion to the light wavefront, of opposite sign to that generated by the scattering tissue. This is commonly done with an SLM or deformable mirror (Fig. 5a). To measure how much the wavefront is distorted by the sample, which is necessary to calculate the distortion pattern, one can use direct76,77 or indirect wavefront sensing78–80. Using adaptive optics, fine structures can be resolved in deep layers with an increased SBR81. The required laser power for imaging can also be reduced. A Perspective in this issue gives an in-depth discussion on adaptive optics82.
Figure 5.
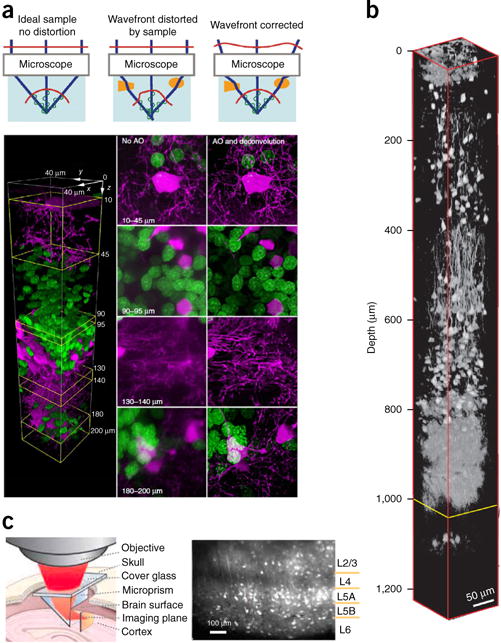
Deep brain imaging. (a) Adaptive optics (AO). Top, a simple model of optical focus formation for different situations. Reprinted from ref. 76, Macmillan Publishers Limited. Bottom, two-color confocal imaging in a zebrafish optic tectum in vivo with and without adaptive optics. Oligodendrocytes are shown in magenta; neuronal nuclei in green. Reprinted from ref. 77, Macmillan Publishers Limited. (b) In vivo three-photon imaging of red fluorescence protein labeled neurons in mouse brain. Reprinted from ref. 72, Macmillan Publishers Limited. (c) Microprism-assisted imaging. Reprinted from ref. 85 with permission from Elsevier.
As light scattering decreases with increasing wavelength, three-photon excitation, using infrared light, should in principle be better than two-photon excitation for deep tissue imaging. To avoid water absorption, one needs to use wavelengths in the 1,700-nm range for tissue penetration72. In three-photon microscopy, the third-order nonlinearity provides optical sectioning, reduces the out-of-focus background and photodamage and improves the SBR even beyond the performance of two-photon approaches. But because the efficiency of three-photon absorption is low, a higher laser power or longer integration time (and thus slower frame rates) is required to acquire images. Nevertheless, novel laser sources have made three-photon excitation practical for fast imaging of neuronal activity, enabling cellular fluorescence imaging up to ~1 mm in depth83 (Fig. 5b). This powerful method enables noninvasive imaging of neural activity at deeper brain structures (for example, cortical layers 5 and 6 and hippocampus) as well as generate less photodamage and better SBR even for superficial imaging.
Finally, other deep-brain imaging strategies involve the physical insertion into the brain of a gradient-index (GRIN) lens to directly access a deep layer84, or of a microprism, which provides views of a vertical cross section of the brain85,86 (Fig. 5c). Though invasive, these approaches provide direct access to the targeted deep brain region. The lengths of GRIN lenses can be customized, and brain regions under the cortex, such as hippocampus, thalamus and hypothalamus, and even fine structures such as dendritic spines in CA1 hippocampus, can be imaged87. Microprisms allow simultaneous imaging of neural activity across all cortical layers in a single FOV and can thus be used to study inter- and intralaminar cortical dynamics and information flow.
Functional imaging of freely behaving animals
The microscopes discussed so far require samples to be well secured without movement (for rodents, this requires anesthetization or head fixing). But many biological questions need freely behaving animals. To tackle this challenge, miniature microscopes and fiberscopes that can be mounted on the animal heads have been developed. The weight of such devices can be as low as a few grams, which, albeit nontrivial for a small animal, still enables measurements under relatively normal behavioral conditions.
Two types of wide-field epifluorescence miniature scopes exist. A light-weight miniaturized one-photon epifluorescence microscope uses optoelectronic components and a GRIN lens as an objective (Fig. 6a). In one implementation, the device weighs 1.9 g and provides an 800-μm × 600-μm FOV with 1.50-μm lateral resolution88. This enables probing the brain in freely behaving mice during sensory, cognitive and motor tasks89.
Figure 6.
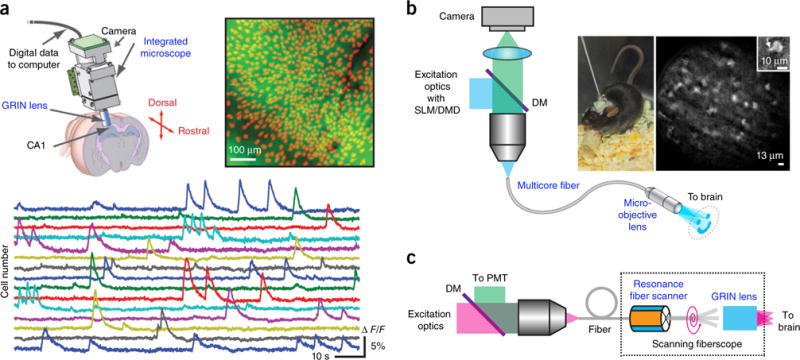
Imaging freely behaving animals. (a) Epifluorescence miniature microscope. Top right, mean fluorescence image from a head-mounted microscope in a behaving GCaMP3-transfected mouse. Identified cells are shown in red. Bottom, fluorescence signals for 15 cells. Reprinted and adapted from ref. 89, Macmillan Publishers Limited. (b) Epifluorescence fiberscope. Right, a freely behaving mouse with the fiberscope fixed to its skull and calcium imaging of molecular layer interneurons recorded with structured illumination. Inset, single interneuron with a portion of a dendrite (arrow). DM, dichroic mirror. Reprinted and adapted from ref. 91 with permission from Elsevier. (c) Two-photon scanning fiberscope. Adapted from ref. 94 with permission from the Optical Society of America.
Fiberscopes constitute a second type of miniature wide-field microscope, where a multicore fiber or fiber bundle (with GRIN lenses at one end) links between a bulk microscope and the animal’s brain90,91 (Fig. 6b). Though not as flexible as the first approach, a fiberscope occupies a smaller footprint on the animal’s head, enabling multiple fiberscopes to probe multiple brain regions simultaneously. Emitted light from the sample is collected through the same fiber and directed to a camera in the bulk microscope. Although at the cost of reduced resolution due to the pixilation of the multicore fiber, the light pattern is preserved at the two ends of the fiber, enabling advanced illumination patterns (for example, structured illumination) and targeted photostimulation patterns91. This has been used to study the influence of interneurons in local network during a behavior task in mice91. A drawback is that the FOV of fiberscopes is typically small.
Miniature two-photon microscopes require substantially more engineering owing to the necessary beam scanning mechanism, with the laser sources coupled externally through a fiber92. The PMT can be positioned at the proximal end of the fiber, and the scan engine at the distal end, followed by GRIN lenses interfacing with the brain93–95 (Fig. 6c). The scan engine can be placed at the proximal end as well, and a fiber bundle used to transmit the light pattern96. Further innovation to increase the FOV and imaging speed and optimize scanning could make miniaturized two-photon microscopes very useful for neuroscience research.
Outlook: into a hybrid future
Calcium imaging of neural activity in vivo has flourished with the rapid advancement of fluorescence microscopy in recent years. The enormous improvements in imaging speed, depth and volume open new avenues to tackle biological problems. It is important to choose the appropriate technologies for different sample preparations. It seems logical for different techniques to be combined to boost overall performance. For example, adaptive optics could generally improve the imaging of scattering tissues by high-speed wide-field microscopes. Holographic devices, which can mimic most optical transfer functions, are universal optics that could be widely used to enhance the flexibility of all laser microscopes. Likewise, combining multiplexing technologies could further improve the speed of two-photon laser scanning microscopy, particularly for large FOVs. All of this could be further improved with three-photon excitation. Miniature microscopes and fiberscopes with higher level of integration, perhaps with microelectromechanical system (MEMS) technologies, could widen their applications. Advances in computational optics and statistical methods should be combined with hardware innovations. The future of microscopy is likely to be hybrid, harnessing advantages from many different designs.
Novel fluorophores8,63,97,98 and data analysis algorithms65,67,99 are critical for in vivo fluorescence imaging, although they are not reviewed here. In addition, although focused on calcium imaging, our discussion also applies to the next generation of voltage imaging probes. The ongoing Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative100 and similar initiatives in other countries101 will provide the fuel to develop novel technologies. Optical methods will continue to revolutionize neuroscience and provide precise readout and manipulation of neural activity from microscale to macroscale, and are likely to help decipher the neural code used by different nervous systems. As in other fields of science, it is often new methods that first enable new discoveries and eventually new ideas102.
Supplementary Material
Acknowledgments
The authors thank D. Peterka and other members of R.Y.’s lab for fruitful discussions. W.Y. holds a Career Award at the Scientific Interface from Burroughs Wellcome Fund. Our work is supported by the National Eye Institute (NEI) under grants number DP1EY024503, R01EY011787 (R.Y.); National Institute of Mental Health (NIMH) under grants numbers R01MH101218, R01MH100561 (R.Y.) and the Defense Advanced Research Projects Agency (DARPA) under contracts number N66001-15-C-4032 (SIMPLEX) (R.Y.) and HR0011-17-C-0026 (R.Y.). This material is based upon work supported by, or in part by, the US Army Research Laboratory and the US Army Research Office under contract number W911NF-12-1-0594 (MURI) (R.Y.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Crick FHC. Thinking about the brain. Scientific American. 1979;241:219–232. doi: 10.1038/scientificamerican0979-219. [DOI] [PubMed] [Google Scholar]
- 2.Smetters D, Majewska A, Yuste R. Detecting action potentials in neuronal populations with calcium imaging. Methods. 1999;18:215–221. doi: 10.1006/meth.1999.0774. [DOI] [PubMed] [Google Scholar]
- 3.Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Methods. 2013;10:413–420. doi: 10.1038/nmeth.2434. Example of whole-brain functional imaging in vivo. [DOI] [PubMed] [Google Scholar]
- 4.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. Invention of two-photon microscopy. [DOI] [PubMed] [Google Scholar]
- 5.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 6.Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hasan MT, et al. Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol. 2004;2:e163. doi: 10.1371/journal.pbio.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuste R, editor. Imaging: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2011. Theory and practice of optical imaging methods. [Google Scholar]
- 10.Helmchen F, Konnerth A, editors. Imaging in Neuroscience: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2011. Review of optical imaging methods in neuroscience. [Google Scholar]
- 11.Lanni F, Keller HE. In: Imaging: A Laboratory Manual. Yuste R, editor. Cold Spring Harbor Laboratory Press; 2011. pp. 1–56. [Google Scholar]
- 12.Ferezou I, et al. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56:907–923. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Mohajerani MH, et al. Spontaneous cortical activity alternates between motifs defined by regional axonal projections. Nat Neurosci. 2013;16:1426–1435. doi: 10.1038/nn.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carandini M, et al. Imaging the awake visual cortex with a genetically encoded voltage indicator. J Neurosci. 2015;35:53–63. doi: 10.1523/JNEUROSCI.0594-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pawley JB. Handbook of Biological Confocal Microscopy. 3. Springer; 2006. [Google Scholar]
- 16.Petran M, Hadravsk M, Egger MD, Galambos R. Tandem-scanning reflected-light microscope. J Opt Soc Am. 1968;58:661–664. [Google Scholar]
- 17.Mertz J. Optical sectioning microscopy with planar or structured illumination. Nat Methods. 2011;8:811–819. doi: 10.1038/nmeth.1709. [DOI] [PubMed] [Google Scholar]
- 18.Chen BC, et al. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science. 2014;346:1257998. doi: 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahrbach FO, Voigt FF, Schmid B, Helmchen F, Huisken J. Rapid 3D light-sheet microscopy with a tunable lens. Opt Express. 2013;21:21010–21026. doi: 10.1364/OE.21.021010. [DOI] [PubMed] [Google Scholar]
- 20.Tomer R, et al. SPED light sheet microscopy: fast mapping of biological system structure and function. Cell. 2015;163:1796–1806. doi: 10.1016/j.cell.2015.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quirin S, et al. Calcium imaging of neural circuits with extended depth-of-field light-sheet microscopy. Opt Lett. 2016;41:855–858. doi: 10.1364/OL.41.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouchard MB, et al. Swept confocally-aligned planar excitation (SCAPE) microscopy for high speed volumetric imaging of behaving organisms. Nat Photonics. 2015;9:113–119. doi: 10.1038/nphoton.2014.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Power RM, Huisken J. A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat Methods. 2017;14:360–373. doi: 10.1038/nmeth.4224. [DOI] [PubMed] [Google Scholar]
- 24.Papagiakoumou E, de Sars V, Oron D, Emiliani V. Patterned two-photon illumination by spatiotemporal shaping of ultrashort pulses. Opt Express. 2008;16:22039–22047. doi: 10.1364/oe.16.022039. [DOI] [PubMed] [Google Scholar]
- 25.Oron D, Tal E, Silberberg Y. Scanningless depth-resolved microscopy. Opt Express. 2005;13:1468–1476. doi: 10.1364/opex.13.001468. [DOI] [PubMed] [Google Scholar]
- 26.Zhu G, van Howe J, Durst M, Zipfel W, Xu C. Simultaneous spatial and temporal focusing of femtosecond pulses. Opt Express. 2005;13:2153–2159. doi: 10.1364/opex.13.002153. [DOI] [PubMed] [Google Scholar]
- 27.Papagiakoumou E, de Sars V, Emiliani V, Oron D. Temporal focusing with spatially modulated excitation. Opt Express. 2009;17:5391–5401. doi: 10.1364/oe.17.005391. [DOI] [PubMed] [Google Scholar]
- 28.Schrödel T, Prevedel R, Aumayr K, Zimmer M, Vaziri A. Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nat Methods. 2013;10:1013–1020. doi: 10.1038/nmeth.2637. [DOI] [PubMed] [Google Scholar]
- 29.Dana H, et al. Hybrid multiphoton volumetric functional imaging of large-scale bioengineered neuronal networks. Nat Commun. 2014;5:3997. doi: 10.1038/ncomms4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nikolenko V, et al. SLM microscopy: scanless two-photon imaging and photostimulation with spatial light modulators. Front Neural Circuits. 2008;2:5. doi: 10.3389/neuro.04.005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rickgauer JP, Deisseroth K, Tank DW. Simultaneous cellular-resolution optical perturbation and imaging of place cell firing fields. Nat Neurosci. 2014;17:1816–1824. doi: 10.1038/nn.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernandez O, et al. Three-dimensional spatiotemporal focusing of holographic patterns. Nat Commun. 2016;7:11928. doi: 10.1038/ncomms11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quirin S, Peterka DS, Yuste R. Instantaneous three-dimensional sensing using spatial light modulator illumination with extended depth of field imaging. Opt Express. 2013;21:16007–16021. doi: 10.1364/OE.21.016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quirin S, Jackson J, Peterka DS, Yuste R. Simultaneous imaging of neural activity in three dimensions. Front Neural Circuits. 2014;8:29. doi: 10.3389/fncir.2014.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang SJ, et al. Extended field-of-view and increased-signal 3D holographic illumination with time-division multiplexing. Opt Express. 2015;23:32573–32581. doi: 10.1364/OE.23.032573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Broxton M, et al. Wave optics theory and 3-D deconvolution for the light field microscope. Opt Express. 2013;21:25418–25439. doi: 10.1364/OE.21.025418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prevedel R, et al. Simultaneous whole-animal 3D imaging of neuronal activity using light-field microscopy. Nat Methods. 2014;11:727–730. doi: 10.1038/nmeth.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zipfel WR, Williams RM, Webb WW. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21:1369–1377. doi: 10.1038/nbt899. Nonlinear microscopies and their applications in biological imaging. [DOI] [PubMed] [Google Scholar]
- 39.Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–940. doi: 10.1038/nmeth818. Review of two-photon microscopy. [DOI] [PubMed] [Google Scholar]
- 40.Briggman KL, Helmstaedter M, Denk W. Wiring specificity in the direction-selectivity circuit of the retina. Nature. 2011;471:183–188. doi: 10.1038/nature09818. [DOI] [PubMed] [Google Scholar]
- 41.Göbel W, Helmchen F. New angles on neuronal dendrites in vivo. J Neurophysiol. 2007;98:3770–3779. doi: 10.1152/jn.00850.2007. [DOI] [PubMed] [Google Scholar]
- 42.Grewe BF, Voigt FF, van ’t Hoff M, Helmchen F. Fast two-layer two-photon imaging of neuronal cell populations using an electrically tunable lens. Biomed Opt Express. 2011;2:2035–2046. doi: 10.1364/BOE.2.002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beaurepaire E, Mertz J. Epifluorescence collection in two-photon microscopy. Appl Opt. 2002;41:5376–5382. doi: 10.1364/ao.41.005376. [DOI] [PubMed] [Google Scholar]
- 44.Yang W, et al. Simultaneous multi-plane imaging of neural circuits. Neuron. 2016;89:269–284. doi: 10.1016/j.neuron.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong L, et al. Continuous volumetric imaging via an optical phase-locked ultrasound lens. Nat Methods. 2015;12:759–762. doi: 10.1038/nmeth.3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Botcherby EJ, Juskaitis R, Booth MJ, Wilson T. An optical technique for remote focusing in microscopy. Opt Commun. 2008;281:880–887. [Google Scholar]
- 47.Anselmi F, Ventalon C, Bègue A, Ogden D, Emiliani V. Three-dimensional imaging and photostimulation by remote-focusing and holographic light patterning. Proc Natl Acad Sci USA. 2011;108:19504–19509. doi: 10.1073/pnas.1109111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Botcherby EJ, et al. Aberration-free three-dimensional multiphoton imaging of neuronal activity at kHz rates. Proc Natl Acad Sci USA. 2012;109:2919–2924. doi: 10.1073/pnas.1111662109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan A, Friedman N, Davidson N. Acousto-optic lens with very fast focus scanning. Opt Lett. 2001;26:1078–1080. doi: 10.1364/ol.26.001078. [DOI] [PubMed] [Google Scholar]
- 50.Duemani Reddy G, Kelleher K, Fink R, Saggau P. Three-dimensional random access multiphoton microscopy for functional imaging of neuronal activity. Nat Neurosci. 2008;11:713–720. doi: 10.1038/nn.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grewe BF, Langer D, Kasper H, Kampa BM, Helmchen F. High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat Methods. 2010;7:399–405. doi: 10.1038/nmeth.1453. [DOI] [PubMed] [Google Scholar]
- 52.Kirkby PA, Nadella KMNS, Silver RA. A compact acousto-optic lens for 2D and 3D femtosecond based 2-photon microscopy. Opt Express. 2010;18:13720–13744. doi: 10.1364/OE.18.013720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katona G, et al. Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nat Methods. 2012;9:201–208. doi: 10.1038/nmeth.1851. [DOI] [PubMed] [Google Scholar]
- 54.Tsai PS, et al. Ultra-large field-of-view two-photon microscopy. Opt Express. 2015;23:13833–13847. doi: 10.1364/OE.23.013833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McConnell G, et al. A novel optical microscope for imaging large embryos and tissue volumes with sub-cellular resolution throughout. eLife. 2016;5:e18659. doi: 10.7554/eLife.18659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sofroniew NJ, Flickinger D, King J, Svoboda K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. eLife. 2016;5:e14472. doi: 10.7554/eLife.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stirman JN, Smith IT, Kudenov MW, Smith SL. Wide field-of-view, multi-region, two-photon imaging of neuronal activity in the mammalian brain. Nat Biotechnol. 2016;34:857–862. doi: 10.1038/nbt.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng A, Gonçalves JT, Golshani P, Arisaka K, Portera-Cailliau C. Simultaneous two-photon calcium imaging at different depths with spatiotemporal multiplexing. Nat Methods. 2011;8:139–142. doi: 10.1038/nmeth.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim KH, et al. Multifocal multiphoton microscopy based on multianode photomultiplier tubes. Opt Express. 2007;15:11658–11678. doi: 10.1364/oe.15.011658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bewersdorf J, Pick R, Hell SW. Multifocal multiphoton microscopy. Opt Lett. 1998;23:655–657. doi: 10.1364/ol.23.000655. [DOI] [PubMed] [Google Scholar]
- 61.Watson BO, et al. Two-photon microscopy with diffractive optical elements and spatial light modulators. Front Neurosci. 2010;4:29. doi: 10.3389/fnins.2010.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahou P, et al. Multicolor two-photon tissue imaging by wavelength mixing. Nat Methods. 2012;9:815–818. doi: 10.1038/nmeth.2098. [DOI] [PubMed] [Google Scholar]
- 63.Inoue M, et al. Rational design of a high-affinity, fast, red calcium indicator R-CaMP2. Nat Methods. 2015;12:64–70. doi: 10.1038/nmeth.3185. [DOI] [PubMed] [Google Scholar]
- 64.Ducros M, Goulam Houssen Y, Bradley J, de Sars V, Charpak S. Encoded multisite two-photon microscopy. Proc Natl Acad Sci USA. 2013;110:13138–13143. doi: 10.1073/pnas.1307818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pnevmatikakis EA, et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron. 2016;89:285–299. doi: 10.1016/j.neuron.2015.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prevedel R, et al. Fast volumetric calcium imaging across multiple cortical layers using sculpted light. Nat Methods. 2016;13:1021–1028. doi: 10.1038/nmeth.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukamel EA, Nimmerjahn A, Schnitzer MJ. Automated analysis of cellular signals from large-scale calcium imaging data. Neuron. 2009;63:747–760. doi: 10.1016/j.neuron.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friedrich J, et al. Multi-scale approaches for high-speed imaging and analysis of large neural populations. 2016 doi: 10.1371/journal.pcbi.1005685. Preprint at https://dx.doi.org/10.1101/091132. [DOI] [PMC free article] [PubMed]
- 69.Lu R, et al. Video-rate volumetric functional imaging of the brain at synaptic resolution. Nat Neurosci. 2017 doi: 10.1038/nn.4516. http://dx.doi.org/10.1038/nn.4516. [DOI] [PMC free article] [PubMed]
- 70.Song A, et al. Volumetric two-photon imaging of neurons using stereoscopy (vTwINS) Nat Methods. 2017;14:420–426. doi: 10.1038/nmeth.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Podgorski K, Ranganathan GN. Brain heating induced by near-infrared lasers during multi-photon microscopy. J Neurophysiol. 2016;116:1012–1023. doi: 10.1152/jn.00275.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Horton NG, et al. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nat Photonics. 2013;7:205–209. doi: 10.1038/nphoton.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Combs CA, et al. Optimizing multiphoton fluorescence microscopy light collection from living tissue by noncontact total emission detection (epiTED) J Microsc. 2011;241:153–161. doi: 10.1111/j.1365-2818.2010.03411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crosignani V, et al. Deep tissue fluorescence imaging and in vivo biological applications. J Biomed Optics. 2012;17:116023. doi: 10.1117/1.JBO.17.11.116023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Booth MJ. Adaptive optical microscopy: the ongoing quest for a perfect image. Light-Sci Appl. 2014;3:e165. [Google Scholar]
- 76.Ji N, Milkie DE, Betzig E. Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat Methods. 2010;7:141–147. doi: 10.1038/nmeth.1411. [DOI] [PubMed] [Google Scholar]
- 77.Wang K, et al. Rapid adaptive optical recovery of optimal resolution over large volumes. Nat Methods. 2014;11:625–628. doi: 10.1038/nmeth.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Débarre D, Booth MJ, Wilson T. Image based adaptive optics through optimisation of low spatial frequencies. Opt Express. 2007;15:8176–8190. doi: 10.1364/oe.15.008176. [DOI] [PubMed] [Google Scholar]
- 79.Débarre D, et al. Image-based adaptive optics for two-photon microscopy. Opt Lett. 2009;34:2495–2497. doi: 10.1364/ol.34.002495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Neil MAA, Booth MJ, Wilson T. New modal wave-front sensor: a theoretical analysis. J Opt Soc Am A Opt Image Sci Vis. 2000;17:1098–1107. doi: 10.1364/josaa.17.001098. [DOI] [PubMed] [Google Scholar]
- 81.Sun W, Tan Z, Mensh BD, Ji N. Thalamus provides layer 4 of primary visual cortex with orientation- and direction-tuned inputs. Nat Neurosci. 2016;19:308–315. doi: 10.1038/nn.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ji N. Adaptive optical fluorescence microscopy. Nat Methods. 2017;14:374–380. doi: 10.1038/nmeth.4218. [DOI] [PubMed] [Google Scholar]
- 83.Ouzounov DG, et al. In vivo three-photon imaging of activity of GCaMP6-labeled neurons deep in intact mouse brain. Nat Methods. 2017;14:388–390. doi: 10.1038/nmeth.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levene MJ, Dombeck DA, Kasischke KA, Molloy RP, Webb WW. In vivo multiphoton microscopy of deep brain tissue. J Neurophysiol. 2004;91:1908–1912. doi: 10.1152/jn.01007.2003. [DOI] [PubMed] [Google Scholar]
- 85.Andermann ML, et al. Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron. 2013;80:900–913. doi: 10.1016/j.neuron.2013.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Low RJ, Gu Y, Tank DW. Cellular resolution optical access to brain regions in fissures: imaging medial prefrontal cortex and grid cells in entorhinal cortex. Proc Natl Acad Sci USA. 2014;111:18739–18744. doi: 10.1073/pnas.1421753111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Attardo A, Fitzgerald JE, Schnitzer MJ. Impermanence of dendritic spines in live adult CA1 hippocampus. Nature. 2015;523:592–596. doi: 10.1038/nature14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghosh KK, et al. Miniaturized integration of a fluorescence microscope. Nat Methods. 2011;8:871–878. doi: 10.1038/nmeth.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ziv Y, et al. Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci. 2013;16:264–266. doi: 10.1038/nn.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Flusberg BA, et al. High-speed, miniaturized fluorescence microscopy in freely moving mice. Nat Methods. 2008;5:935–938. doi: 10.1038/nmeth.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szabo V, Ventalon C, De Sars V, Bradley J, Emiliani V. Spatially selective holographic photoactivation and functional fluorescence imaging in freely behaving mice with a fiberscope. Neuron. 2014;84:1157–1169. doi: 10.1016/j.neuron.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 92.Helmchen F, Fee MS, Tank DW, Denk W. A miniature head-mounted two-photon microscope. high-resolution brain imaging in freely moving animals. Neuron. 2001;31:903–912. doi: 10.1016/s0896-6273(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 93.Flusberg BA, et al. Fiber-optic fluorescence imaging. Nat Methods. 2005;2:941–950. doi: 10.1038/nmeth820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Myaing MT, MacDonald DJ, Li X. Fiber-optic scanning two-photon fluorescence endoscope. Opt Lett. 2006;31:1076–1078. doi: 10.1364/ol.31.001076. [DOI] [PubMed] [Google Scholar]
- 95.Rivera DR, et al. Compact and flexible raster scanning multiphoton endoscope capable of imaging unstained tissue. Proc Natl Acad Sci USA. 2011;108:17598–17603. doi: 10.1073/pnas.1114746108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Göbel W, Kerr JND, Nimmerjahn A, Helmchen F. Miniaturized two-photon microscope based on a flexible coherent fiber bundle and a gradient-index lens objective. Opt Lett. 2004;29:2521–2523. doi: 10.1364/ol.29.002521. [DOI] [PubMed] [Google Scholar]
- 97.Chen Z, Wei L, Zhu X, Min W. Extending the fundamental imaging-depth limit of multi-photon microscopy by imaging with photo-activatable fluorophores. Opt Express. 2012;20:18525–18536. doi: 10.1364/OE.20.018525. [DOI] [PubMed] [Google Scholar]
- 98.Akerboom J, et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci. 2013;6:2. doi: 10.3389/fnmol.2013.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Theis L, et al. Benchmarking spike rate inference in population calcium imaging. Neuron. 2016;90:471–482. doi: 10.1016/j.neuron.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Insel TR, Landis SC, Collins FS. The NIH BRAIN Initiative. Research priorities. Science. 2013;340:687–688. doi: 10.1126/science.1239276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yuste R, Bargmann C. Towards a global BRAIN Initiative. Cell. 2017 doi: 10.1016/j.cell.2017.02.023. http://dx.doi.org/10.1016/j.cell.2017.02.023. [DOI] [PubMed]
- 102.Robertson M. Biology in the 1980s, plus or minus a decade. Nature. 1980;285:358–359. doi: 10.1038/285358a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.