

Summary
Pathogenesis in tauopathies involves the accumulation of tau in the brain and progressive synapse loss accompanied by cognitive decline. Pathological tau is found at synapses, and it promotes synaptic dysfunction and memory deficits. The specific role of toxic tau in disrupting the molecular networks that regulate synaptic strength has been elusive. A novel mechanistic link between tau toxicity and synaptic plasticity involves the acetylation of two lysines on tau, K274 and K281, which are associated with dementia in Alzheimer’s disease (AD). We propose that an increase in tau acetylated on these lysines blocks the expression of long-term potentiation at hippocampal synapses leading to impaired memory in AD. Acetylated tau could inhibit the activity-dependent recruitment of postsynaptic AMPA-type glutamate receptors required for plasticity by interfering with the postsynaptic localization of KIBRA, a memory-associated protein. Strategies that reduce the acetylation of tau may lead to effective treatments for cognitive decline in AD.
Keywords: acetylation, Alzheimer’s disease, KIBRA, long-term potentiation, synaptic plasticity, tau
Graphical abstract
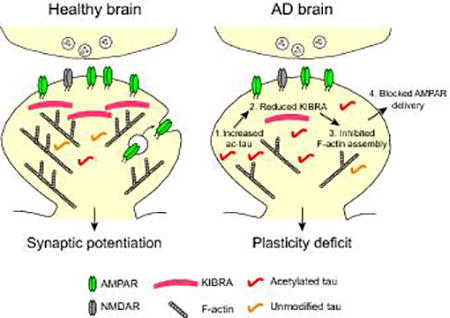
Taupathies are age-related neurodegenerative diseases, including Alzheimer’s disease, that are characterized by accumulation of tau protein in the brain and cognitive decline. We hypothesize that enhanced levels of abnormally acetylated tau in tauopathy disrupts the KIBRA-dependent mechanisms that regulate plasticity at synapses leading to memory loss.
Introduction
The sequence of molecular events in neurons that trigger progressive synaptic decline in Alzheimer’s disease (AD) is poorly understood. Aggregation of the microtubule-binding protein tau in the brain coincides with synapse and neuronal loss in tauopathy [1–3] and implicates tau in the disease progression. Tau is highly expressed in the axons of healthy neurons and has been found in both presynaptic and postsynaptic compartments in human brain [4,5]. In AD, tau aberrantly accumulates in dendrites [6] and affects postsynaptic function [4,7–11]. Interestingly, the spread of pathological tau across brain regions may occur at synaptic connections between neurons [12,13]. These studies highlight tau localization at synapses and support the notion that tau directly affects synaptic signaling networks, leading to synaptic dysfunction (Fig. 1).
Figure 1.
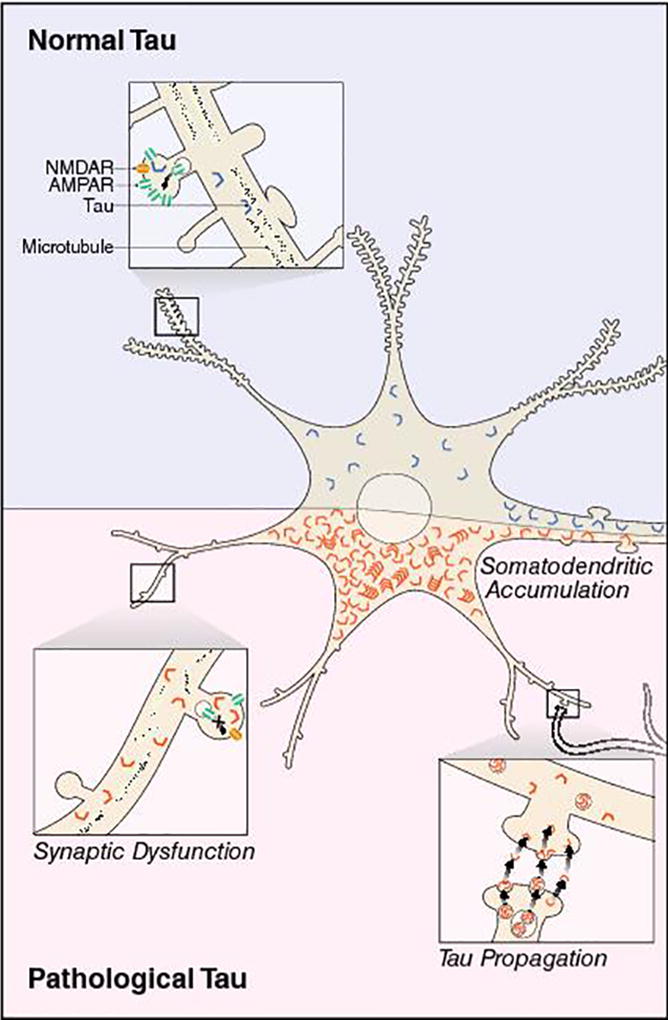
Pathological tau at synapses in AD. In normal conditions, tau is predominantly localized in axons, but it can also be found at synapses. In disease, pathogenic tau is missorted and accumulates in the somatodendritic compartment of neurons. The buildup of toxic tau can affect synaptic transmission by downregulating postsynaptic AMPA-type glutamate receptors and blocking the expression of synaptic plasticity. Pathogenic tau can also promote the loss of dendritic spines. The propagation of toxic tau among neurons could be trans-synaptic and induced by activity, but the mechanism for tau release is still unclear.
Recent research has focused on what makes tau toxic to neurons. Post-translational modifications are one possibility. Many residues in human tau are subject to post-translational modifications, such as phosphorylation [14], acetylation [9,15,16], ubiquitination [17,18], and methylation [19,20]. Increased levels of abnormally modified tau are a hallmark of AD pathogenesis. Neurofibrillary tangles, containing highly phosphorylated tau, form in parallel with the manifestation of cognitive impairments in AD [21,22], but tangles may not cause memory loss [23,24] and neurodegeneration [25]. To investigate the role of specific acetylated residues, lysine can be substituted with glutamine (KQ) to mimic the chemical structure and neutral charge of acetylated lysine [26–29]. We used KQ mutations to elucidate the role of acetylated tau (ac-tau) in neurodegeneration [30], cytoskeletal dysregulation [31], and synaptic dysfunction [9] related to AD pathogenesis. Our laboratory and others have shown that acetylation of tau is associated with tauopathy [9,15,16,30–32] and significantly affects tau function in neurons. Acetylation blocks tau degradation [15], inhibits tau microtubule binding [16,31], and promotes tau aggregation [16,30].
Synaptic transmission is altered in transgenic mice and cell cultures expressing human tau with familial mutations that cause frontotemporal lobar degeneration with tau inclusions (FTLD-tau) [8,33,34]. In fact, the FTLD-tau-induced impairment of glutamatergic synaptic transmission in transgenic mice triggers memory deficits before neurodegeneration begins [34], supporting the notion that tau affects synapses early in disease progression. Moreover, synaptic impairments are rescued by depleting tau in transgenic mice expressing mutant human amyloid precursor protein (hAPP) [35], which demonstrates that tau has a significant role in hAPP/Aβ-induced synaptotoxicity. In another AD mouse model, the postsynaptic missorting of tau recruits Fyn kinase. This action leads to NMDA-type glutamate receptor (NMDAR)-induced excitotoxicity [7], and raises the possibility of reducing Fyn-dependent NMDAR activity to treat AD. Targeting tau-dependent mechanisms that cause synaptic dysfunction at the early stages of AD pathogenesis could be an effective strategy to prevent memory loss.
Using transgenic mice that express human tau with KQ mutations to mimic acetylation, we found that ac-tau inhibits synaptic plasticity underlying impairments in hippocampal-dependent memory [9]. Acetyl-mimicking tau blocked activity-induced actin polymerization and AMPA-type glutamate receptor (AMPAR) trafficking in postsynaptic spines during long-term potentiation (LTP). The loss of postsynaptic KIBRA underlies the plasticity deficit caused by tau. Here we will discuss how ac-tau contributes to cognitive decline in AD and speculate about the postsynaptic mechanisms by which ac-tau affects synaptic plasticity. We propose to reduce ac-tau levels to treat cognitive dysfunction in AD. Salsalate, a drug that inhibits the acetylation of tau, restored memory deficits in a tauopathy mouse model [30]. Ongoing clinical studies are testing salsalate treatment as a new approach for therapeutic intervention in tauopathy. Further investigation of the mechanisms that promote tau acetylation and the downstream effects of ac-tau may uncover additional strategies to prevent cognitive decline.
Pathological enhancement of tau acetylation in neurodegenerative disease
We identified the acetylation of three lysines on tau, K174, K274 and K281, in a tauopathy mouse model and human AD brain [9,30]. While acetylated K174 levels were elevated at early Braak stages (see Box 1), higher levels of acetylated K274 (ac-K274) and acetylated K281 (ac-K281) were found in the brains of severely demented AD patients [9,30]. Transgenic mice expressing high levels of hAPP and Aβ had significantly increased ac-K274 and ac-K281 on tau [9]. Tau acetylation was also increased in cultured neurons treated with Aβ oligomers [15]. These results support that the link between hAPP/Aβ toxicity and the upregulation of ac-tau represents a novel mechanism that promotes cognitive decline in AD.
Box 1. Braak Stages.
Pathological tau gradually accumulates in AD brain forming neurofibrillary tangles (NFTs). The emergence of NFTs at specific locations in the brain is classified into six characteristic Braak stages during the progression of AD. Braak staging indicates the extent of tau pathology in the brain and the severity of the disease. In the early Braak stages I–II, pathological accumulates in the transentorhinal region. In Braak stages III–IV, NFTs are observed in limbic areas, including the hippocampus. In Braak stages V–VI, NFTs extend into neocortical areas.
How do high levels of hAPP/Aβ increase ac-tau? In neuroblastoma cells, expression of the Swedish hAPP mutant increased the acetyltransferase activity of p300 [36]. Tau is acetylated by p300 [15], and the expression and acetyltransferase activity of p300 is enhanced in AD brains [37,38]. The dysregulation of p300 could promote aberrant tau acetylation. Additional studies may uncover more acetyltransferases that modulate tau acetylation. Tau also has intrinsic auto-acetylation activity that could enhance pathological tau levels [39,40]. Tau is deacetylated by sirtuin 1 [15], and sirtuin 1 levels in the brain decrease during the progression of AD [41,42]. Treatment of neuroblastoma cells with the amyloid beta (Aβ) (25–35) peptide reduced the expression of sirtuin 1 [43], indicating that Aβ toxicity may reduce the sirtuin 1-mediated deacetylation of tau. Evidently, a combination of factors could enhance the ac-tau levels in neurons under pathological conditions.
Strategies that reduce tau acetylation could offer novel approaches to treat cognitive decline in tauopathies. Activators of sirtuin 1 could be explored as options to reduce ac-tau and protect against tau-mediated neurodegeneration. Importantly, salsalate, a prodrug of salicylate, inhibits p300 acetyltransferase activity, blocks the acetylation of tau and restores memory deficits in FTLD-tau mice [30], supporting the possibility of p300 as a therapeutic target. Another study showed that treatment with sodium salicylate restored Aβ-induced synaptic and memory deficits in rats [44], supporting the therapeutic potential of salsalate for AD. Salsalate did not prevent neurodegeneration in mice expressing an acetyl-tau mimic [30], suggesting that the reduction of ac-tau is an important mechanism in salsalate’s protective effect. In addition to reducing ac-tau levels, treatment with salsalate could alter other molecular pathways that ameliorate cognitive decline. Salsalate may restore synaptic deficits by inhibiting cyclooxygenase [44], a protein that is upregulated in AD [45]. Moreover, salsalate is a non-steroidal anti-inflammatory drug and may be protective due to its anti-inflammatory effects [46]. Salicylate could also have a beneficial effect by activating adenosine monophosphate-activated protein kinase (AMPK) and increasing autophagy [47]. Given that salsalate can target different pathogenic mechanisms, including lowering ac-tau levels by p300 inhibition, salsalate treatment may improve clinical outcomes in tauopathy patients due to multiple benefits.
The acetylation of each residue in AD could involve distinct mechanisms and functional outcomes. K274 acetylation was observed in neurofibrillary tangles and neurotic plaques in AD and other tauopathies [31,32]. It was also enhanced in AD brains at late Braak stages (5–6), compared to early stages (0–2) [31]. Whether acetylation of K281 is associated with pathological tau inclusions in human brain remains to be determined. Enhanced levels of K281 acetylation were associated with dementia [9], but there was no difference in ac-K281 levels across Braak stages [31]. Moreover, brains from mildly demented AD cases (clinical dementia rating, CDR=0.5) had higher ac-K281 levels than non-demented cases, but they had comparable ac-K274 levels [9]. Given these differences, ac-K281 could have a role in the early stages of cognitive decline, whereas K274 acetylation contributes later, coinciding with the progression of tau pathology. Full-length tau has four microtubule-binding repeats (4R), but alternatively spliced tau has only three repeats (3R) and contains K274, but not K281. Neurofibrillary tangles in AD contain equivalent amounts of 3R and 4R tau [48], indicating that 3R tau with ac-K274 could be characteristic of pathology independently of ac-K281.
Post-translational modifications have been identified on many of tau’s residues [49], including the acetylation of more than 20 lysines [15]. However, we found that the acetylation of K174, K274 and K281 is associated with tau pathology and dementia [9,30], indicating that they may play a key role in AD pathogenesis. K274 and K281 are located in the microtubule-binding domain, where most of the familial FTLD-tau mutations are located. Several studies suggest that the acetylation of specific lysines on tau can alter its function in different ways. The acetylation of K274 and K281 on tau triggers synaptic deficits and tau mislocalization into the somatodendritic compartment [9,31]. The acetylation of K274/K281 and K280 in the microtubule-binding domain can reduce the interaction of tau with microtubules [16,31], whereas K280 acetylation can enhance tau aggregation [16]. The acetylation of K174 regulates tau homeostasis and promotes neurodegeneration [30]. In contrast, the acetylation of RXGS motifs inhibits tau phosphorylation and aggregation [50]. Although many detailed underlying mechanisms remain to be determined, we speculate that the aberrant increase in the site-specific acetylation of tau marks a key event in AD associated with synaptic pathophysiology and cognitive decline.
Acetylation affects the buildup of distinct toxic tau species
Acetylation of tau could induce pathogenesis by promoting the accumulation of forms of tau that are known to be toxic, including phosphorylated or truncated tau. Mimicking the acetylation of K274 and K281 on human tau with glutamine (tauKQ) in transgenic mice did not alter the phosphorylation of several residues associated with tau pathology, but reduced phospho-serine 202/205 (AT8) levels in the hippocampus [9,31]. Mimicking the acetylation of other lysine residues led to either an increase or decrease in the level of tau phosphorylation [30,50,51]. Thus, the pathogenicity induced by acetylated tau is likely to be distinct from that induced by phosphorylated tau. How acetylation affects the phosphorylation status of tau and subsequent pathogenesis may depend on which lysine is acetylated. Caspase-cleaved tau fragments have been implicated in pathological tau accumulation in the brain [52]. However, we observed KQ-induced deficits in transgenic mice lacking caspase-cleaved tau, suggesting that caspase-cleaved tau fragments are unlikely involved in the synaptic and cognitive deficits caused by ac-K274 and ac-K281 [9].
The acetylation of tau may promote toxicity by enhancing tau oligomerization. Tau oligomers are upregulated in AD brain and may play a role in the early stages of pathogenesis [53,54]. Human tau oligomers injected into mice caused impaired memory and synaptic deficits [55], but the mechanism that initiates pathological tau oligomerization remains to be determined. MC1-positive pathological tau could contribute to the pathogenesis triggered by acetylation, because it was observed in the hippocampus of tauKQ-expressing mice [9]. The accumulation of the abnormal conformation of tau recognized by the MC1 antibody is an early event in Alzheimer’s disease [56]. This pathological tau species is positioned to alter synaptic circuits because MC1-positive tau has been implicated in the spread of tau from the entorhinal cortex to synaptically connected neurons in the hippocampus in aged transgenic mice [12]. Tau oligomers found within presynaptic and postsynaptic compartments may indicate the trans-synaptic spread of oligomers [5]. Whether ac-tau propagates from cell to cell in the brain is unknown. It is possible that the toxicity induced by enhanced levels of ac-tau at synapses is exacerbated by the trans-synaptic spreading of MC1-positive tau or tau oligomers.
Acetylated tau promotes AD-related memory loss
Early in the progression of AD, patients show symptoms of impaired episodic memory [57,58]. A crucial aspect of forming episodic memories is the ability to distinguish between similar experiences. Hippocampal neurons mediate this process of pattern separation that involves the encoding of sensory inputs as distinct neural representations in the brain [59,60]. Human behavioral studies showed that patients with mild cognitive impairment (MCI) or AD have impaired pattern separation, which likely interferes with their ability to form new memories [61–63]. Moreover, MCI patients displayed abnormal activation of their entorhinal cortex-dentate gyrus-CA3 circuitry during a behavioral task of pattern separation [63]. Mice lacking NMDA-type glutamate receptors exclusively in dentate granule cells have impaired dentate gyrus LTP and perform poorly in a test of context discrimination, indicating that dentate granule cell function is required for pattern separation [59]. TauKQ-expressing mice were similarly impaired in distinguishing between experiences, and LTP was inhibited in dentate granule cells [9], supporting that ac-tau affects pattern separation in the dentate gyrus.
Hippocampal synaptic dysfunction also underlies behavioral impairments in spatial memory in mouse models of tauopathy [8,35,64–66]. In the Morris water maze, tauKQ-expressing mice learned the location of the hidden platform, but could not retain the spatial memory of the platform location during a probe trial [9]. In another study, mice expressing K174Q human tau also demonstrated memory deficits in the Morris water maze [30]. These findings support that the enhanced acetylation of individual lysines on tau can have a significant effect on the encoding of hippocampal-dependent memory.
Tau mediates synaptic dysfunction
The activity-dependent modulation of synaptic strength is believed to enable the encoding of new memories. Synapses are enriched with a complex network of proteins that orchestrate the physiological and structural modifications needed to change synaptic efficacy. NMDAR-dependent LTP in the hippocampus involves the long-lasting strengthening of synaptic transmission when additional AMPARs are recruited to the surface of the postsynaptic membrane [67,68]. Hippocampal LTP deficits are observed in mouse models of tauopathy, including FTLD-tau mutant mice [8,34,69–72]. FTLD-tau mutants also affect basal glutamatergic synaptic transmission [8,34]. Furthermore, the amplitude and frequency of AMPAR-mediated miniature postsynaptic currents were reduced by expression of tau mutated to mimic the phosphorylation of 14 residues [8], indicating that phosphorylated tau affects postsynaptic receptor stability. The mechanism for the effect of phosphorylated tau on glutamate receptors is unknown. Recordings from tauKQ mice revealed that ac-tau inhibits LTP in the dentate gyrus, but basal synaptic transmission was not affected [9]. Unlike phosphorylated tau, the mechanism by which ac-tau causes synaptic dysfunction is specific to activity-induced changes in synaptic strength. Together, these findings suggest that distinct forms of toxic tau may affect synaptic properties in different ways. Neuronal pathways in the brain have diverse synaptic characteristics and express distinct types of plasticity [73]. Therefore, the impact of tau on synaptic transmission could also depend on the unique features of the particular synapse in the brain that is affected. To tease apart the pathogenic events at synapses in AD, it will be necessary to delineate the mechanisms by which tau affects synaptic machinery leading to dysfunction.
Acetylated tau disrupts activity-dependent synaptic signaling
We detected ac-K274 and ac-K281 on tau in the postsynaptic density fraction extracted from human AD brain [9]. How does ac-tau get to postsynaptic sites? Tau can be found in the nucleus [74], raising the possibility that tau is acetylated by p300 in the nucleus. On the other hand, p300 can also modify proteins in the cytosol [75,76]. The exact locations where tau is acetylated in neurons remain to be determined. Nevertheless, our study showed that ac-tau is missorted to the somatodendritic compartment [31]. One likely mechanism involves destabilization at the axon initial segment by ac-K274 and ac-K281 [31]. Another possibility is that ac-tau is transmitted from presynaptic to postsynaptic sites. The formation of neurofibrillary tangles in AD occurs in a hierarchical progression through brain regions, starting in the entorhinal cortex and appearing at later stages in the hippocampus and cortex. This observation suggests that tau propagates across synaptic circuits [77]. Studies on transgenic mice confirmed the spreading of pathological tau from entorhinal cortical neurons into the hippocampus [12,13,78]. Neurons secrete tau in response to enhanced network activity [79,80], and the depolarization of synaptosome preparations from human AD brains evoked more tau release than control brains [81]. In addition to acetylated tau missorted into dendrites [31], secreted tau may be acetylated, offering a potential mechanism by which acetylation affects the release and propagation of tau at synapses.
Filamentous actin (F-actin)-rich structures in spines dynamically modulate the strength of glutamatergic transmission and anchor signaling complexes in the postsynaptic density [82]. Actin polymerization in spines is required for the activity-dependent recruitment of synaptic AMPARs and the maintenance of LTP [83,84]. Our results suggest that ac-tau blocks the assembly of F-actin required for AMPAR trafficking during potentiation [9]. Spines contain specialized regulatory proteins that can modulate F-actin levels in response to synapse activation [85]. TauKQ did not affect basal actin polymerization in spines or glutamatergic transmission [9]; therefore, it probably affects the function of actin-binding regulatory proteins that specifically modulate F-actin during plasticity.
Tau can interact directly with F-actin [86,87] and induce actin filament organization [88,89]. The accumulation of F-actin caused by an FTLD-tau mutant expressed in Drosophila was linked to neurodegeneration [89]. Mimicking the phosphorylation of 14 residues was sufficient to induce abnormal actin polymerization and toxicity [89], suggesting that phosphorylated tau increases F-actin assembly. Mimicking ac-K274 and ac-K281 on tau weakened its interaction with F-actin and inhibited activity-dependent actin polymerization, indicating that ac-tau has a distinct effect on the cytoskeleton. These divergent effects may involve the subcellular localization of pathogenic tau. Ac-tau blocks actin dynamics in spines [9], whereas phosphorylated tau promotes the bundling of actin in the soma [89]. Indeed, a unique role for tau in the regulation of postsynaptic actin is supported by a study that showed that synaptic activity or Aβ oligomers trigger the movement of tau into spines where it interacts with F-actin [90]. The physiological significance of tau binding to F-actin in spines is unknown. In addition to tau translocation, synaptic activity can promote microtubule polymerization from the dendrite into F-actin rich spines [91,92]. In a cell-free system, tau binding to F-actin cross-linked the actin and microtubule cytoskeletons and enhanced their coordinated polymerization [93]. Tau may facilitate crosstalk between actin and microtubule networks in regulating synaptic strength.
Together with F-actin, many proteins are involved in the dynamic regulation of AMPARs during plasticity [94]. KIBRA is one of these postsynaptic proteins that is of particular interest: it has been linked to human memory performance [95] and to the risk for late-onset AD [96–98]. KIBRA is required for the expression of hippocampal LTP, and mice deficient in KIBRA have memory impairments [99,100]. TauKQ reduced KIBRA levels in spines, and elevating KIBRA expression in neurons restored the tau-mediated inhibition of postsynaptic actin polymerization and AMPAR delivery [9]. The inhibitory effect of tau on KIBRA signaling represents a novel mechanism underlying the tau-mediated impairment of synaptic plasticity (Fig. 2A). How ac-tau lowers the KIBRA levels in spines has not yet been established. Since tauKQ did not affect the overall expression of KIBRA in the hippocampus, ac-tau may block the trafficking of KIBRA into spines or compromise the stability of KIBRA in the postsynaptic milieu. The effect of postsynaptic ac-tau on KIBRA-dependent signaling and activity-dependent cytoskeletal dynamics supports the specific role of ac-tau in disrupting synaptic plasticity without affecting basal synaptic transmission.
Figure 2.
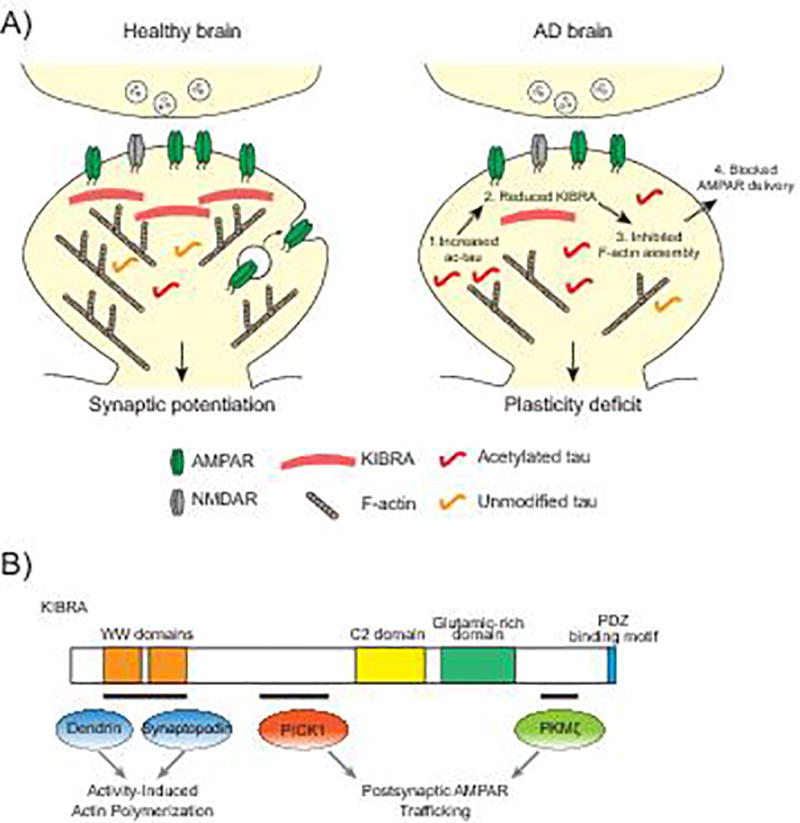
Acetylated tau blocks KIBRA-dependent signaling. A: Increased levels of ac-tau in AD brain causes a reduction in postsynaptic KIBRA. The KIBRA deficiency blocks activity-dependent actin polymerization and AMPAR insertion into the postsynaptic membrane during LTP. B: The functional domains of KIBRA include two WW domains, a C2 domain, a glutamic-rich domain and a PDZ binding motif. The tau-mediated loss of KIBRA could affect the function of proteins that bind to KIBRA at synapses including dendrin and synaptopodin, which regulate the actin cytoskeleton, and PICK1 and PKMζ, which regulate AMPARs.
KIBRA-dependent signaling is impaired in AD
Studies on individual genomic variation revealed that a single nucleotide polymorphism in the KIBRA gene is associated with a predisposition for AD [96–98]. The effect of this gene polymorphism on KIBRA function in neurons and how it may increase risk for AD is unknown. KIBRA mRNA levels were greater in neurons of the hippocampus and middle temporal gyrus from AD brains than in control brains [97]. We showed that levels of KIBRA protein were significantly decreased in the superior temporal gyrus in AD cases with severe dementia, and this decrease correlated with an increase in levels of ac-tau [9]. Evidence from tauKQ mice supported that ac-tau disrupts the postsynaptic localization of KIBRA. The upregulation of KIBRA mRNA in AD brain could be explained as a compensatory mechanism for the tau-induced loss of KIBRA in spines.
KIBRA is crucial for the expression of LTP and long-term depression (LTD) in adult mice, and it modulates activity-dependent AMPAR trafficking [99]. How KIBRA regulates the molecular events during plasticity expression is unclear. KIBRA could control synaptic signaling pathways through its different protein-interacting domains (Fig. 2B). KIBRA has two WW domains that bind to the postsynaptic proteins dendrin and synaptopodin, which are both involved in actin cytoskeleton regulation [101–104]. Similar to mice with the genetic deletion of KIBRA [99], synaptopodin deficient mice have impaired LTP [105]. Our data suggest that the tau-mediated KIBRA deficiency disrupts actin dynamics in spines [9]. The decrease in KIBRA may compromise the regulation of actin by synaptopodin during plasticity. In human podocytes, KIBRA interacts with synaptopodin, co-localizes with actin and tubulin accumulation at lamellipodia and regulates cell migration [102]. Dynein light chain 1 is another cytoskeleton-associated protein that interacts with KIBRA [106] and links motor proteins with the cytoskeleton and protein complexes in spines [107]. KIBRA may serve as a platform for the interaction of actin regulatory proteins and other postsynaptic signaling components to regulate activity-dependent actin polymerization.
KIBRA may regulate AMPAR trafficking through its interaction with protein kinase Mζ (PKMζ) [108,109] and PICK1 [99]. PKMζ is the catalytic domain of an atypical protein kinase C (PKC) that continually phosphorylates substrates because it lacks the PKC regulatory domain. KIBRA can be phosphorylated by PKCζ/PKMζ [108], but the effect of phosphorylation on KIBRA function is unknown. PKMζ mRNA is locally translated at synapses [110], and its protein synthesis is enhanced during LTP [111]. PKMζ activity increases AMPAR-mediated synaptic transmission [112] and may regulate LTP expression [112,113] and memory [114]. However, whether it is required for LTP and memory is controversial [115]. Tau-mediated downregulation of KIBRA in spines may affect PKMζ activity. Interestingly, increased PKMζ levels were observed in neurofibrillary tangles in the hippocampus of human AD brain [116], which supports the notion that tau affects PKMζ function in disease. The interaction of KIBRA with PICK1 in a postsynaptic complex with AMPARs could also regulate plasticity [99]. PICK1 binds to AMPARs and modulates their cycling to the postsynaptic membrane [117–119], and PICK1 depletion impairs hippocampal LTD and LTP [120]. Since KIBRA is involved in several pathways that regulate cytoskeletal dynamics and receptor trafficking, KIBRA deficiency in AD may disrupt synaptic plasticity by obstructing multiple postsynaptic mechanisms.
Conclusions
Tau plays a significant role in changing synapses during AD pathogenesis. The underlying mechanisms, however, are not well understood. We propose that increased levels of tau acetylated on K274 and K281 promotes hippocampal-dependent memory loss in AD by blocking postsynaptic signaling required for plasticity. Specifically, ac-tau reduces the postsynaptic localization of KIBRA, which blocks actin polymerization and AMPAR trafficking. Multiple lysines on tau have been identified that are hyperacetylated in tauopathy, and the acetylation of each lysine may have distinct effects on the brain and cognition. The success with salsalate in preclinical mouse studies supports the feasibility of partial reduction of p300 activity as a strategy to counteract tau-mediated neurodegeneration and cognitive decline. Replenishing KIBRA or restoring its downstream signaling at synapses could be another strategy for therapeutic intervention in AD. The prospect of restoring memory loss in AD will continue to improve as we further our understanding of how synaptic mechanisms are affected by tau in disease.
Acknowledgments
We thank Xu Chen and other members of the Gan lab for discussions, John Carroll for graphics assistance, Gary Howard for editorial review, and Erica Nguyen for administrative assistance. This work was supported by the NIH (1R01AG036884 and R01AG030207 to L.G; 5F32AG043301-02 to T.E.T.), the Tau Consortium (to L.G.), and the BrightFocus Foundation (to T.E.T).
Abbreviations
- ac-tau
acetylated tau
- AD
Alzheimer’s disease
- FTLD
frontotemporal lobar degeneration
- hAPP
human amyloid precursor protein
- LTD
long-term depression
- LTP
long-term potentiation.
Footnotes
The authors have declared no conflicts of interest.
References
- 1.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 2.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 3.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–8. [PubMed] [Google Scholar]
- 4.Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, et al. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–35. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tai HC, Wang BY, Serrano-Pozo A, Frosch MP, et al. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer's disease. Acta Neuropathol Commun. 2014;2:146. doi: 10.1186/s40478-014-0146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer's disease. Ann Neurol. 1987;22:639–43. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 7.Ittner LM, Ke YD, Delerue F, Bi M, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–97. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 8.Hoover BR, Reed MN, Su J, Penrod RD, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–81. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tracy TE, Sohn PD, Minami SS, Wang C, et al. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 2016;90:245–60. doi: 10.1016/j.neuron.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–50. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopeikina KJ, Polydoro M, Tai HC, Yaeger E, et al. Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. 2013;521:1334–53. doi: 10.1002/cne.23234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Drouet V, Wu JW, Witter MP, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Calignon A, Polydoro M, Suarez-Calvet M, William C, et al. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–97. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mair W, Muntel J, Tepper K, Tang S, et al. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Anal Chem. 2016;88:3704–14. doi: 10.1021/acs.analchem.5b04509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Min SW, Cho SH, Zhou Y, Schroeder S, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cripps D, Thomas SN, Jeng Y, Yang F, et al. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J Biol Chem. 2006;281:10825–38. doi: 10.1074/jbc.M512786200. [DOI] [PubMed] [Google Scholar]
- 18.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, et al. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–60. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 19.Funk KE, Thomas SN, Schafer KN, Cooper GL, et al. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem J. 2014;462:77–88. doi: 10.1042/BJ20140372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas SN, Funk KE, Wan Y, Liao Z, et al. Dual modification of Alzheimer's disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol. 2012;123:105–17. doi: 10.1007/s00401-011-0893-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–9. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 22.Guilloz AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–36. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 23.Santacruz K, Lewis J, Spires T, Paulson J, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, et al. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–23. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 25.Spires TL, Orne JD, SantaCruz K, Pitstick R, et al. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hecht A, Laroche T, Strahl-Bolsinger S, Gasser SM, et al. Histone H3 and H4 N-termini interact with SIR3 and SIR4 proteins: a molecular model for the formation of heterochromatin in yeast. Cell. 1995;80:583–92. doi: 10.1016/0092-8674(95)90512-x. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Peng L, Seto E, Huang S, et al. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J Biol Chem. 2012;287:29168–74. doi: 10.1074/jbc.M112.371120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scroggins BT, Robzyk K, Wang D, Marcu MG, et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell. 2007;25:151–9. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang YH, Tsay YG, Tan BC, Lo WY, et al. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem. 2003;278:25568–76. doi: 10.1074/jbc.M212574200. [DOI] [PubMed] [Google Scholar]
- 30.Min SW, Chen X, Tracy TE, Li Y, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21:1154–62. doi: 10.1038/nm.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sohn PD, Tracy TE, Son HI, Zhou Y, et al. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 2016;11:47. doi: 10.1186/s13024-016-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grinberg LT, Wang X, Wang C, Sohn PD, et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. 2013;125:581–93. doi: 10.1007/s00401-013-1080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalby NO, Volbracht C, Helboe L, Larsen PH, et al. Altered function of hippocampal CA1 pyramidal neurons in the rTg4510 mouse model of tauopathy. J Alzheimers Dis. 2014;40:429–42. doi: 10.3233/JAD-131358. [DOI] [PubMed] [Google Scholar]
- 34.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Roberson ED, Halabisky B, Yoo JW, Yao J, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–11. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X, Deng Y, Yu D, Cao H, et al. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer's disease. PLoS One. 2014;9:e103067. doi: 10.1371/journal.pone.0103067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aubry S, Shin W, Crary JF, Lefort R, et al. Assembly and interrogation of Alzheimer's disease genetic networks reveal novel regulators of progression. PLoS One. 2015;10:e0120352. doi: 10.1371/journal.pone.0120352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong HK, Veremeyko T, Patel N, Lemere CA, et al. De-repression of FOXO3a death axis by microRNA-132 and-212 causes neuronal apoptosis in Alzheimer's disease. Hum Mol Genet. 2013;22:3077–92. doi: 10.1093/hmg/ddt164. [DOI] [PubMed] [Google Scholar]
- 39.Cohen TJ, Constance BH, Hwang AW, James M, et al. Intrinsic Tau Acetylation Is Coupled to Auto-Proteolytic Tau Fragmentation. PLoS One. 2016;11:e0158470. doi: 10.1371/journal.pone.0158470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cohen TJ, Friedmann D, Hwang AW, Marmorstein R, et al. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat Struct Mol Biol. 2013;20:756–62. doi: 10.1038/nsmb.2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lutz MI, Milenkovic I, Regelsberger G, Kovacs GG. Distinct patterns of sirtuin expression during progression of Alzheimer's disease. Neuromolecular Med. 2014;16:405–14. doi: 10.1007/s12017-014-8288-8. [DOI] [PubMed] [Google Scholar]
- 42.Julien C, Tremblay C, Emond V, Lebbadi M, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lattanzio F, Carboni L, Carretta D, Candeletti S, et al. Treatment with the neurotoxic Abeta (25–35) peptide modulates the expression of neuroprotective factors Pin1, Sirtuin 1, and brain-derived neurotrophic factor in SH-SY5Y human neuroblastoma cells. Exp Toxicol Pathol. 2016;68:271–6. doi: 10.1016/j.etp.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Doost Mohammadpour J, Hosseinmardi N, Janahmadi M, Fathollahi Y, et al. Non-selective NSAIDs improve the amyloid-beta-mediated suppression of memory and synaptic plasticity. Pharmacol Biochem Behav. 2015;132:33–41. doi: 10.1016/j.pbb.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 45.Ho L, Purohit D, Haroutunian V, Luterman JD, et al. Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progression of Alzheimer disease. Arch Neurol. 2001;58:487–92. doi: 10.1001/archneur.58.3.487. [DOI] [PubMed] [Google Scholar]
- 46.in 't Veld BA, Launer LJ, Breteler MM, Hofman A, et al. Pharmacologic agents associated with a preventive effect on Alzheimer's disease: a review of the epidemiologic evidence. Epidemiol Rev. 2002;24:248–68. doi: 10.1093/epirev/mxf001. [DOI] [PubMed] [Google Scholar]
- 47.Hawley SA, Fullerton MD, Ross FA, Schertzer JD, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–22. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau) J Mol Neurosci. 2011;45:384–9. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morris M, Knudsen GM, Maeda S, Trinidad JC, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci. 2015 doi: 10.1038/nn.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cook C, Carlomagno Y, Gendron TF, Dunmore J, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. 2014;23:104–16. doi: 10.1093/hmg/ddt402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorsky MK, Burnouf S, Dols J, Mandelkow E, et al. Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Sci Rep. 2016;6:22685. doi: 10.1038/srep22685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gamblin TC, Chen F, Zambrano A, Abraha A, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032–7. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. Faseb j. 2012;26:1946–59. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maeda S, Sahara N, Saito Y, Murayama S, et al. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 55.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, et al. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–27. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 57.Collie A, Maruff P. The neuropsychology of preclinical Alzheimer's disease and mild cognitive impairment. Neurosci Biobehav Rev. 2000;24:365–74. doi: 10.1016/s0149-7634(00)00012-9. [DOI] [PubMed] [Google Scholar]
- 58.Bondi MW, Jak AJ, Delano-Wood L, Jacobson MW, et al. Neuropsychological contributions to the early identification of Alzheimer's disease. Neuropsychol Rev. 2008;18:73–90. doi: 10.1007/s11065-008-9054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McHugh TJ, Jones MW, Quinn JJ, Balthasar N, et al. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–9. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- 60.Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, et al. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell. 2012;149:188–201. doi: 10.1016/j.cell.2012.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ally BA, Hussey EP, Ko PC, Molitor RJ. Pattern separation and pattern completion in Alzheimer's disease: evidence of rapid forgetting in amnestic mild cognitive impairment. Hippocampus. 2013;23:1246–58. doi: 10.1002/hipo.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wesnes KA, Annas P, Basun H, Edgar C, et al. Performance on a pattern separation task by Alzheimer's patients shows possible links between disrupted dentate gyrus activity and apolipoprotein E in4 status and cerebrospinal fluid amyloid-beta42 levels. Alzheimers Res Ther. 2014;6:20. doi: 10.1186/alzrt250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yassa MA, Stark SM, Bakker A, Albert MS, et al. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. Neuroimage. 2010;51:1242–52. doi: 10.1016/j.neuroimage.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harris JA, Devidze N, Halabisky B, Lo I, et al. Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer's disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci. 2010;30:372–81. doi: 10.1523/JNEUROSCI.5341-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Polydoro M, Acker CM, Duff K, Castillo PE, et al. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–9. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sydow A, Van der Jeugd A, Zheng F, Ahmed T, et al. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011;31:2511–25. doi: 10.1523/JNEUROSCI.5245-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hayashi Y, Shi SH, Esteban JA, Piccini A, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–7. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 68.Shi SH, Hayashi Y, Petralia RS, Zaman SH, et al. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science. 1999;284:1811–6. doi: 10.1126/science.284.5421.1811. [DOI] [PubMed] [Google Scholar]
- 69.Crouzin N, Baranger K, Cavalier M, Marchalant Y, et al. Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer's disease: dissociation between somatosensory cortex and hippocampus. PLoS One. 2013;8:e74667. doi: 10.1371/journal.pone.0074667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palop JJ, Chin J, Roberson ED, Wang J, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun B, Zhou Y, Halabisky B, Lo I, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–57. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oddo S, Caccamo A, Shepherd JD, Murphy MP, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 73.Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- 74.Liu C, Gotz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS One. 2013;8:e84849. doi: 10.1371/journal.pone.0084849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi D, Pop MS, Kulikov R, Love IM, et al. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc Natl Acad Sci U S A. 2009;106:16275–80. doi: 10.1073/pnas.0904305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sebti S, Prebois C, Perez-Gracia E, Bauvy C, et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc Natl Acad Sci U S A. 2014;111:4115–20. doi: 10.1073/pnas.1313618111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 78.Harris JA, Koyama A, Maeda S, Ho K, et al. Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One. 2012;7:e45881. doi: 10.1371/journal.pone.0045881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pooler AM, Phillips EC, Lau DH, Noble W, et al. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–94. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamada K, Holth JK, Liao F, Stewart FR, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–93. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sokolow S, Henkins KM, Bilousova T, Gonzalez B, et al. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer's disease. J Neurochem. 2014 doi: 10.1111/jnc.12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–36. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, et al. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–60. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- 84.Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–61. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lei W, Omotade OF, Myers KR, Zheng JQ. Actin cytoskeleton in dendritic spine development and plasticity. Curr Opin Neurobiol. 2016;39:86–92. doi: 10.1016/j.conb.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Correas I, Padilla R, Avila J. The tubulin-binding sequence of brain microtubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem J. 1990;269:61–4. doi: 10.1042/bj2690061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.DuBoff B, Gotz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75:618–32. doi: 10.1016/j.neuron.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cunningham CC, Leclerc N, Flanagan LA, Lu M, et al. Microtubule-associated protein 2c reorganizes both microtubules and microfilaments into distinct cytological structures in an actin-binding protein-280-deficient melanoma cell line. J Cell Biol. 1997;136:845–57. doi: 10.1083/jcb.136.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, et al. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–48. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- 90.Frandemiche ML, De Seranno S, Rush T, Borel E, et al. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci. 2014;34:6084–97. doi: 10.1523/JNEUROSCI.4261-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Merriam EB, Millette M, Lumbard DC, Saengsawang W, et al. Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci. 2013;33:16471–82. doi: 10.1523/JNEUROSCI.0661-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shirao T, Gonzalez-Billault C. Actin filaments and microtubules in dendritic spines. J Neurochem. 2013;126:155–64. doi: 10.1111/jnc.12313. [DOI] [PubMed] [Google Scholar]
- 93.Elie A, Prezel E, Guerin C, Denarier E, et al. Tau co-organizes dynamic microtubule and actin networks. Sci Rep. 2015;5:9964. doi: 10.1038/srep09964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–17. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, et al. Common Kibra alleles are associated with human memory performance. Science. 2006;314:475–8. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- 96.Burgess JD, Pedraza O, Graff-Radford NR, Hirpa M, et al. Association of common KIBRA variants with episodic memory and AD risk. Neurobiol Aging. 2011;32:557, e1–9. doi: 10.1016/j.neurobiolaging.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Corneveaux JJ, Liang WS, Reiman EM, Webster JA, et al. Evidence for an association between KIBRA and late-onset Alzheimer's disease. Neurobiol Aging. 2010;31:901–9. doi: 10.1016/j.neurobiolaging.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rodriguez-Rodriguez E, Infante J, Llorca J, Mateo I, et al. Age-dependent association of KIBRA genetic variation and Alzheimer's disease risk. Neurobiol Aging. 2009;30:322–4. doi: 10.1016/j.neurobiolaging.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 99.Makuch L, Volk L, Anggono V, Johnson RC, et al. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron. 2011;71:1022–9. doi: 10.1016/j.neuron.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heitz FD, Farinelli M, Mohanna S, Kahn M, et al. The memory gene KIBRA is a bidirectional regulator of synaptic and structural plasticity in the adult brain. Neurobiol Learn Mem. 2016 doi: 10.1016/j.nlm.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 101.Kremerskothen J, Plaas C, Buther K, Finger I, et al. Characterization of KIBRA, a novel WW domain-containing protein. Biochem Biophys Res Commun. 2003;300:862–7. doi: 10.1016/s0006-291x(02)02945-5. [DOI] [PubMed] [Google Scholar]
- 102.Duning K, Schurek EM, Schluter M, Bayer M, et al. KIBRA modulates directional migration of podocytes. J Am Soc Nephrol. 2008;19:1891–903. doi: 10.1681/ASN.2007080916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kremerskothen J, Kindler S, Finger I, Veltel S, et al. Postsynaptic recruitment of Dendrin depends on both dendritic mRNA transport and synaptic anchoring. J Neurochem. 2006;96:1659–66. doi: 10.1111/j.1471-4159.2006.03679.x. [DOI] [PubMed] [Google Scholar]
- 104.Asanuma K, Kim K, Oh J, Giardino L, et al. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. 2005;115:1188–98. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deller T, Korte M, Chabanis S, Drakew A, et al. Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc Natl Acad Sci U S A. 2003;100:10494–9. doi: 10.1073/pnas.1832384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rayala SK, den Hollander P, Manavathi B, Talukder AH, et al. Essential role of KIBRA in co-activator function of dynein light chain 1 in mammalian cells. J Biol Chem. 2006;281:19092–9. doi: 10.1074/jbc.M600021200. [DOI] [PubMed] [Google Scholar]
- 107.Naisbitt S, Valtschanoff J, Allison DW, Sala C, et al. Interaction of the postsynaptic density-95/guanylate kinase domain-associated protein complex with a light chain of myosin-V and dynein. J Neurosci. 2000;20:4524–34. doi: 10.1523/JNEUROSCI.20-12-04524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buther K, Plaas C, Barnekow A, Kremerskothen J. KIBRA is a novel substrate for protein kinase Czeta. Biochem Biophys Res Commun. 2004;317:703–7. doi: 10.1016/j.bbrc.2004.03.107. [DOI] [PubMed] [Google Scholar]
- 109.Yoshihama Y, Hirai T, Ohtsuka T, Chida K. KIBRA Co-localizes with protein kinase Mzeta (PKMzeta) in the mouse hippocampus. Biosci Biotechnol Biochem. 2009;73:147–51. doi: 10.1271/bbb.80564. [DOI] [PubMed] [Google Scholar]
- 110.Muslimov IA, Nimmrich V, Hernandez AI, Tcherepanov A, et al. Dendritic transport and localization of protein kinase Mzeta mRNA: implications for molecular memory consolidation. J Biol Chem. 2004;279:52613–22. doi: 10.1074/jbc.M409240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hernandez AI, Blace N, Crary JF, Serrano PA, et al. Protein kinase M zeta synthesis from a brain mRNA encoding an independent protein kinase C zeta catalytic domain. Implications for the molecular mechanism of memory. J Biol Chem. 2003;278:40305–16. doi: 10.1074/jbc.M307065200. [DOI] [PubMed] [Google Scholar]
- 112.Ling DS, Benardo LS, Serrano PA, Blace N, et al. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–6. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- 113.Serrano P, Yao Y, Sacktor TC. Persistent phosphorylation by protein kinase Mzeta maintains late-phase long-term potentiation. J Neurosci. 2005;25:1979–84. doi: 10.1523/JNEUROSCI.5132-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Drier EA, Tello MK, Cowan M, Wu P, et al. Memory enhancement and formation by atypical PKM activity in Drosophila melanogaster. Nat Neurosci. 2002;5:316–24. doi: 10.1038/nn820. [DOI] [PubMed] [Google Scholar]
- 115.Volk LJ, Bachman JL, Johnson R, Yu Y, et al. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature. 2013;493:420–3. doi: 10.1038/nature11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Crary JF, Shao CY, Mirra SS, Hernandez AI, et al. Atypical protein kinase C in neurodegenerative disease I: PKMzeta aggregates with limbic neurofibrillary tangles and AMPA receptors in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:319–26. doi: 10.1097/01.jnen.0000218442.07664.04. [DOI] [PubMed] [Google Scholar]
- 117.Lin DT, Huganir RL. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization. J Neurosci. 2007;27:13903–8. doi: 10.1523/JNEUROSCI.1750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xia J, Zhang X, Staudinger J, Huganir RL. Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron. 1999;22:179–87. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 119.Perez JL, Khatri L, Chang C, Srivastava S, et al. PICK1 targets activated protein kinase Calpha to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci. 2001;21:5417–28. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Volk L, Kim CH, Takamiya K, Yu Y, et al. Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc Natl Acad Sci U S A. 2010;107:21784–9. doi: 10.1073/pnas.1016103107. [DOI] [PMC free article] [PubMed] [Google Scholar]