

Summary
Dendritic cell sarcomas (DCS) are rare tumours of antigen presenting cells. Data regarding their biology, management and outcomes are sparse. We analysed 66 patients with follicular dendritic cell sarcoma (FDCS). Six patients also had Castleman disease, 9 had another malignancy and 13 had an autoimmune disease. Fifty-four per cent of patients presented with localized disease and 46% with systemic involvement. The median progression-free (PFS) and overall survival (OS) following frontline therapy was 21 months and 50 months, respectively. Survival outcomes were significantly inferior in patients with extranodal, bulky or intra-abdominal disease at presentation. Stage was not associated with survival. Management approaches were heterogeneous. Patients who underwent an upfront gross total resection (GTR) experienced better PFS and OS (both P<0.0001). In patients who underwent a GTR, consolidative radiotherapy was associated with improved local control (P = 0.03), PFS (P = 0.04) and OS (P = 0.05). In patients with measureable disease, gemcitabine with a taxane yielded an overall response rate of 80%. The pattern of relapse was predominantly locoregional. Salvage rates after recurrence were poor. Studies are underway at our institution to define the genomic profile in FDCS and identify potential novel therapeutic targets.
Keywords: Dendritic cell sarcoma, DCS, Follicular dendritic cell sarcoma, FDCS
Introduction
Dendritic cell sarcomas are rare tumours derived from antigen-presenting cells.(Kairouz, et al 2007) The most common subtype is follicular dendritic cell sarcoma (FDCS), which arises from cells that normally reside in nodal germinal centres and support humoral immunity. (Krautler, et al 2012) (Heesters, et al 2014) Management of FDCS is challenging for multiple reasons. First, it is a rare condition, comprising <0.4% of soft tissue sarcomas(Perkins and Shinohara 2013), so data are limited, with information derived primarily from case reports and series(Chan, et al 1997, Conry 2014, Duan, et al 2010, Fonseca, et al 1998, Perez-Ordonez, et al 1996, Wang, et al 2015), pooled analyses,(Saygin, et al 2013) and single centre experiences with small patient numbers(Dalia, et al 2014, Gounder, et al 2015, Soriano, et al 2007). Second, it is difficult to diagnose and may be confused with other neoplasms, such as lymphoma, sarcoma, meningioma and melanoma (Ohtake and Yamakawa 2013, Pai, et al 2015). Lastly, the clinical presentation of patients with FDCS is highly variable: disease can involve nodal and extranodal sites throughout the body and can follow a heterogeneous course.
The optimal management strategy remains to be defined; however, a combined modality approach, consisting of surgery, chemotherapy and/or radiotherapy (RT), is most commonly used (Gounder, et al 2015, Saygin, et al 2013). Historically, chemotherapy regimens used for aggressive lymphomas, such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) or ICE (ifosfamide, carboplatin, etoposide), have been employed. Recently, promising results with gemcitabine and docetaxel were published(Conry 2014). Due to limited data on cytogenetic (Perry, et al 2013) and molecular characteristics (Starr, et al 2015), few reports have demonstrated efficacy of targeted agents (Azim, et al 2007). Potential targets have been proposed, such as epidermal growth factor receptor (EGFR).(Sun, et al 2003) Additionally, recurrent loss of function alterations in NFKBIA and CDKN2A and copy number gains in chromosome 9p24 (site of CD274 [also termed PD-L1] and PDCD1LG2 [also termed PD-L2, CD273]) have been identified, indicating potential molecular targets.(Griffin, et al 2016) The association of FDCS with Epstein–Barr virus (Ge, et al 2014), Castleman disease(Chan, et al 2001, Cokelaere, et al 2002, Pauwels, et al 2000, Sun, et al 2003) and autoimmune conditions(Wang, et al 2005) also may provide insights into pathophysiology and therapeutic strategies.
In this report, we describe the clinical, pathological, treatment and outcome data of patients with FDCS who presented to our institution.
Materials and Methods
Patients
All patients with a confirmed diagnosis of FDCS evaluated at MD Anderson Cancer Center (MDACC) from 1995 to 2015 were included in this update(Soriano, et al 2007). Patients with histiocytic sarcoma, interdigitating dendritic cell sarcoma (IDCS) or Langerhans cell histiocytosis were excluded. Following institutional review board approval, clinical, pathological, treatment and outcome data were collected. Response assessment in the evaluable patients was done according to the lymphoma response criteria.(Cheson, et al 2007) All specimens were reviewed by haematopathologists at our institution, who confirmed the diagnosis of FDCS. In addition, next generation sequencing with a 50-gene panel was performed on 2 specimens.
Statistical analysis
Categorical variables were compared using Fisher’s exact test, and continuous variables were compared using the t-test. Progression-free survival (PFS) was measured from the date of treatment initiation to the date of progression or last follow-up. Overall survival (OS) was measured from the date of initial diagnosis to the date of death or date of last follow-up. Survival probabilities were estimated by the Kaplan-Meier method and compared using the log-rank test and univariate Cox proportion hazard model. Due to the retrospective nature of the study, only those patients with available covariate and outcome information could be included in the survival analyses, therefore there is a variability in number of patients in the survival analyses and figures (Figures 1-4). A P-value of <0.05 was considered significant. Statistical analyses were carried out using statistical software SAS 9.3 (SAS, Cary, NC) and S-Plus 8.2 (TIBCO Software Inc., Palo Alto, CA).
Figure 1. Progression-free survival (PFS) and overall survival (OS) according to extranodal disease involvement.
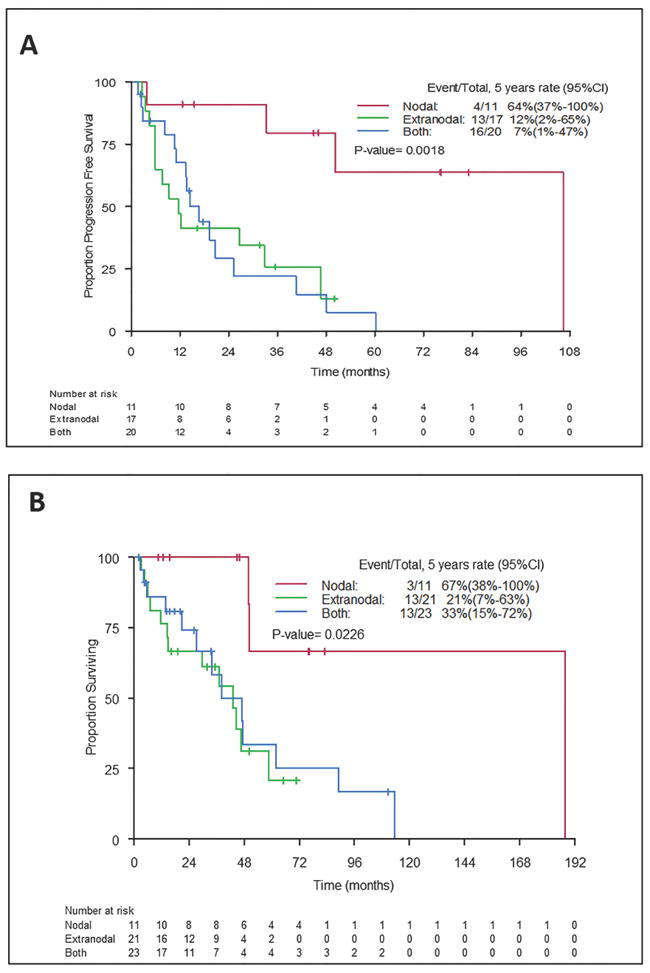
PFS (A) and OS (B) were significantly worse in patients with extranodal disease (P=0.0018 for PFS and P=0.023 for OS). Median PFS was 106 months for patients with nodal disease only, 12 months for patients with extranodal disease only and 17 months for patients with nodal and extranodal disease (log-rank P = 0.0018). Median OS was 188 months, 43 months and 43 months, respectively (log-rank P = 0.023). 95% CI, 95% confidence interval
Figure 4. Progression-free survival (PFS) and overall survival (OS) in patients who underwent a gross total resection (GTR) according to consolidation radiotherapy.
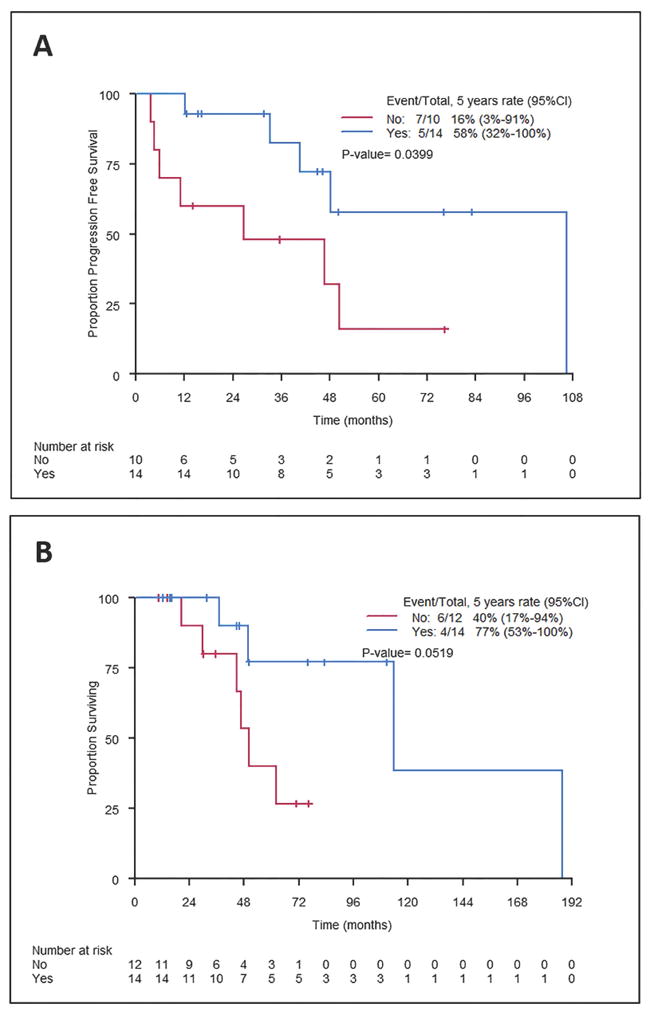
PFS (A) and OS (B) were better in patients who received consolidative RT after a GTR (log-rank P=0.04 for PFS and P=0.05 for OS). 95% CI, 95% confidence interval
Results
Patient characteristics
A total of 66 patients with FDCS were included. Patient and disease characteristics are summarized in Table I. The median age at initial presentation was 49 years (range, 16-78), and most patients were Caucasian (68%). Females were slightly more commonly affected than males (n=36 vs. 30). Six patients had hyaline vascular variant Castleman disease: 2 patients were diagnosed with Castleman disease prior to FDCS, and 4 patients received both diagnoses concurrently. Other cancers were observed in 9 patients before or after their diagnosis with FDCS, including prostate cancer (n=2), melanoma (n=1), chronic myeloid leukaemia (n=1), T cell-large granular lymphocytic leukaemia (n=1), giant cell tumour of the bone (n=1), colorectal cancer (n=1), cervical dysplasia (n=1) and meningioma (n=1). Thirteen patients (20%) had pre-existing or subsequent autoimmune disease. The distribution of autoimmune disease included pemphigus (n=4), myasthenia gravis (n=3), polyglandular autoimmune endocrinopathy (n=2), Grave disease (n=1), Sjogren syndrome (n=1), primary biliary cirrhosis (n=1), and Still disease (n=1). In this cohort, one patient had all 3 conditions: FDCS, Castleman disease and an autoimmune disease (pemphigus).
Table– I.
Baseline clinical, histopathological and disease characteristics of patients with follicular dendritic cell sarcoma
| Patients (n) | 66 |
|---|---|
|
| |
| Median age, years (range) | 49 (16-78) |
|
| |
| Gender (male:female), n | 30:36 |
|
| |
| Ethnicity n (%) | |
| Caucasian | 45 (68%) |
| African-American | 9 (14%) |
| Asian | 3 (5%) |
| Hispanic | 6 (9%) |
|
| |
| Initial site of involvement | |
| Nodal | 11 (20%) |
| Extranodal | 21 (37%) |
| Both nodal and extranodal | 24 (43%) |
|
| |
| Initial Stage | |
| Localized | 30 (54%) |
| Advanced | 26 (46%) |
|
| |
| Bulky disease (n, %) | 41 (73%) |
|
| |
| Castleman disease (n, %) | 6 (9%) |
|
| |
| Autoimmune disease (n, %) | 13 (20%) |
|
| |
| Feature | n (%) |
|
| |
| Classic morphology+ | 51 (88%) |
|
| |
| Giant cells + | 35 (69%) |
|
| |
| Mitosis >5-10/high powered field | 18 (55%) |
|
| |
| Immunohistochemistry | |
|
| |
| Clusterin | 22/24 (92%) |
|
| |
| Epidermal growth factor receptor | 25/30 (83%) |
|
| |
| CD21 | 59/63 (89%) |
|
| |
| CD35 | 45/55 (18%) |
|
| |
| CD23 | 32/49 (63%) |
|
| |
| S-100 | 7/62 (11%) |
|
| |
| CD68 | 12/45(27%) |
|
| |
| Epstein–Barr virus-encoded small RNA | 2/22 (9%) |
|
| |
| Vimentin | 32/36 (89%) |
|
| |
| Desmin | 3/30 (10%) |
|
| |
| Fascin | 11/15 (73%) |
Disease characteristics
Baseline staging information was available for 56 patients. Of these, 30 patients (54%) presented with limited stage disease and 26 (46%) with advanced stage disease. Bulk (>5 cm) was noted in 41 patients (73%). Twenty-four patients (43%) presented with both nodal and extranodal involvement, 21 (37%) with extranodal disease only and 11 (20%) with nodal disease only. The most commonly involved nodes were in the neck (15/35 patients with nodal involvement). Extranodal sites (with/without nodal involvement) included the liver (n=9), mediastinum (n=6), lung (n=6), spleen (n=6), gastrointestinal tract (n=5), bone/spine (n=5), brain (n=2), pancreas (n=2), mesentery (n=2), nasopharynx (n=2), adrenal/kidneys (n=2), retroperitoneal soft tissue (n=2), ovary (n=1), thyroid (n=1) and pleura (n=1). Intra-abdominal involvement, which may be associated with poor prognosis (Saygin, et al 2013), was observed in 30 patients (53%).
Pathological features
All cases met the criteria for FDCS, as defined in the current World Health Organization classification system (Swerdlow et al 2008). The pathological features are summarized in Table I. Electron microscopy was performed in 3 cases and revealed neoplastic cells with well-formed junctions and prominent desmosomes. Next generation sequencing with a 50-gene panel was performed for two patients. Both tumours contained TP53 mutations (c.743G>A p.R248Q, exon 7 and c.839G>A p.R280K, exon 8). One sample also contained a PTEN mutation (c.388C>T p.R130, exon 5).
Treatment information
Treatment information was available for 54 patients. Upfront management included surgery alone (n=14); surgery and chemotherapy (n=11); surgery and RT (n=7); RT and chemotherapy (n=4); chemotherapy alone (n=6); RT alone (n=2) and surgery, chemotherapy with RT (n=10).
Patient outcomes
Survival information was available for 60 patients, who were eligible for inclusion in OS analyses. Of these, 29 (48%) were alive at the time of last follow-up (median follow-up 35.5 months). The cause of death was known for 22/31 deceased patients, of whom 19 (87%) died of progressive disease. Three patients died of other causes while in remission. Disease status at the time of last follow-up was known for 50 patients; these patients were eligible for PFS analyses. Of the 29 patients alive at last follow-up, 15 were free of disease. The median PFS and OS times were 21 months and 47 months, respectively.
We analysed the impact of initial disease site and extent on survival in patients with available upfront staging and survival information. PFS and OS were significantly shorter in patients with extranodal disease (Figure 1). Additionally, bulky disease and intra-abdominal disease were associated with inferior PFS (log rank P=0.01 and P = 0.11, respectively) and OS (log rank P=0.01 and P = 0.01, respectively) (Figure 2A-D). Conversely, survival outcomes were not significantly different for patients with localized vs. advanced stage disease. As an exploratory analysis, we compared patients with localized nodal disease vs all others (i.e., extranodal and/or advanced stage disease). Patients with localized nodal disease had better PFS (log rank P=0.0031) and OS (log rank P = 0.035) (Figure 2E-F).
Figure 2. Progression-free survival (PFS) and overall survival (OS) according to the disease bulk, intra-abdominal disease site, and extent of involvement at diagnosis.
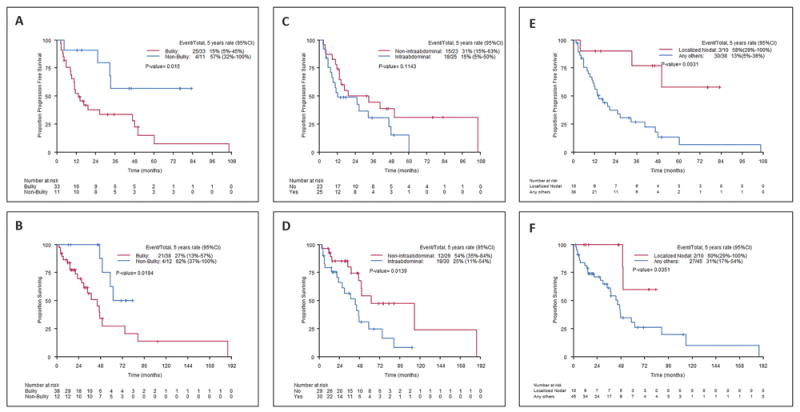
PFS (A) and OS (B) were significantly inferior in patients with bulky disease (log-rank P=0.02 for both). PFS (C) and OS (D) were inferior in patients with intra-abdominal involvement (log-rank P=0.11 for PFS and P=0.01 for OS). PFS (E) and OS (F) were better in patients with localized nodal disease vs. other presentations (advanced stage nodal or any stage extranodal disease) (log-rank P = 0.003 for PFS and P=0.035 for OS). 95% CI, 95% confidence interval
Pattern of relapse
The pattern of failure after frontline therapy was known for 27 patients. Locoregional relapse was the most common (n=19; 70%), followed by combined locoregional and distant (n=5; 19%), and then distant (n=3; 11%).
Impact of upfront therapy on survival
We assessed the association of initial treatment type with outcome in patients with available therapy and survival data. At a median follow-up of 33 months (range 4-114), there were 36 PFS events and 33 deaths. Patients who underwent an upfront gross total resection (GTR; n=27) experienced significantly longer PFS and OS (Figure 3A-B). Among the 27 patients who underwent GTR, margins were involved in 5 patients, negative in 7 patients, and margin status was unknown in 15 patients.
Figure 3. Progression-free survival (PFS) and overall survival (OS) according to upfront surgical resection.
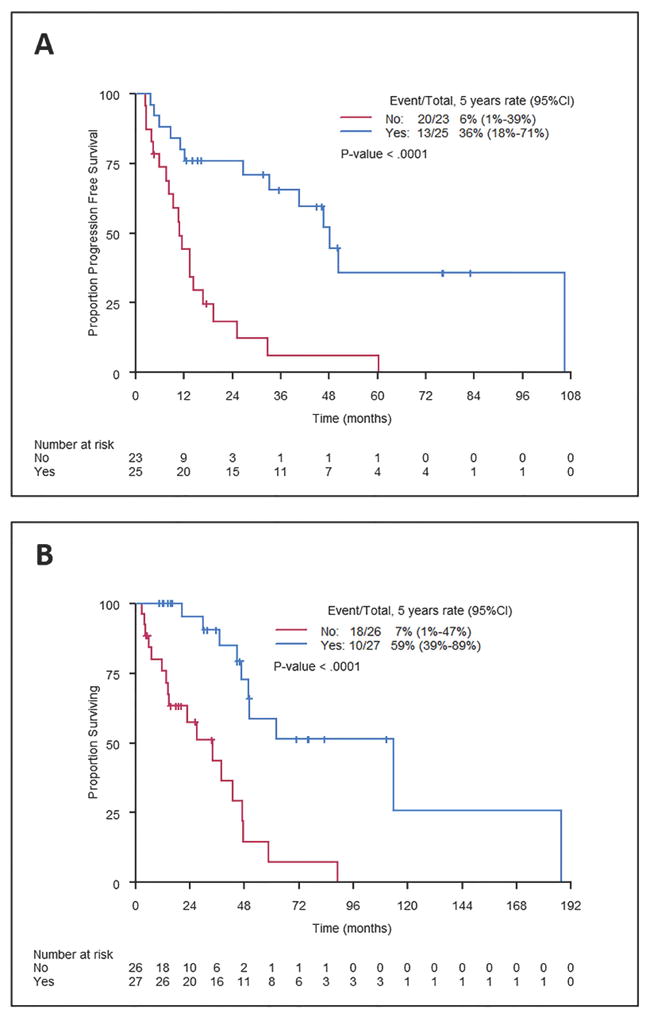
PFS (A) and OS (B) were significantly better in patients who underwent a gross total resection (both log-rank P<0.0001). 95% CI, 95% confidence interval
We also assessed the effect of RT and chemotherapy on survival. Given the profound influence of surgical resection on outcomes, we separately analysed patients who did vs. did not undergo a GTR. Of the patients who underwent an upfront GTR (n=27), 14 (52%) received consolidative RT. In this subgroup, RT was associated with improved local control (89% vs. 53% at 3 years; log rank P = 0.03), which translated into improved PFS and OS (Figure 4A-B). Fourteen patients (52%) received chemotherapy after a GTR. PFS and OS were not significantly different for patients who received adjuvant chemotherapy vs. those who did not. Among patients who did not undergo an upfront GTR (n = 23), the initial treatment strategy comprised chemotherapy and RT in 3 (14%), chemotherapy in 16 (69%) and RT in 4 (17%). There was no significant difference in PFS or OS in these groups. Univariate survival analyses are summarized in Table II.
Table II.
Univariate analyses of covariate associations with progression-free and overall survival
| Progression-Free Survival | |||||||
|---|---|---|---|---|---|---|---|
| Covariate | HR | 95% CI for HR | P-value | Event | Censored | Total | |
| Extranodal vs Nodal | 7.47 | 1.98 | 28.07 | 0.002 | 17 | 11 | 28 |
| Extranodal and Nodal vs. Nodal Only | 7.33 | 2.05 | 26.19 | 0.002 | 20 | 11 | 31 |
| Tumour Size | 3.48 | 1.19 | 10.16 | 0.02 | 29 | 15 | 44 |
| Intra-abdominal involvement | 1.75 | 0.86 | 3.57 | 0.11 | 33 | 15 | 48 |
| Gross Surgical Resection | 0.22 | 0.10 | 0.49 | 0.0001 | 33 | 15 | 48 |
| *Consolidation radiotherapy | 0.29 | 0.08 | 1.01 | 0.05 | 12 | 12 | 24 |
| Overall Survival | |||||||
| Extranodal vs Nodal | 6.45 | 1.43 | 29.14 | 0.015 | 16 | 16 | 32 |
| Extranodal and Nodal vs. Nodal Only | 5.43 | 1.21 | 24.24 | 0.026 | 16 | 18 | 34 |
| Tumour Size | 3.45 | 1.15 | 10.35 | 0.02 | 25 | 25 | 50 |
| Intra-abdominal involvement | 2.53 | 1.17 | 5.47 | 0.01 | 31 | 28 | 59 |
| Gross Surgical Resection | 0.18 | 0.07 | 0.43 | 0.0001 | 28 | 25 | 53 |
| *Consolidation radiotherapy | 0.23 | 0.04 | 1.15 | 0.07 | 10 | 16 | 26 |
Consolidation radiotherapy in patients with gross total resection
95% CI, 95% confidence interval; HR, Hazard ratio
Chemotherapy
Overall, 28 patients received upfront chemotherapy, either alone or as a part of multimodality management. The chemotherapy regimens were gemcitabine with a taxane (n=10); CHOP (n=11; with rituximab in 2); ifosfamide and doxorubicin (n=3); gemcitabine (n=1); ifosfamide, cyclophosphamide and etoposide (n=1); imatinib (n=1), and rituximab (n=1). Sixteen patients had measurable disease at the initiation of frontline chemotherapy and were assessed for response. In this subset, the most commonly used chemotherapy regimens were gemcitabine with a taxane (n=10), ifosfamide-based (n=3), and CHOP-based (n=2). The overall response rate (ORR) associated with each regimen was 80% (8/10; 2 complete response [CR], 6 partial response [PR], 2 non-responders), 100% (3/3; all PR) and 50% (1/2; 1 PR), respectively. The median duration of response with gemcitabine and docetaxel was 13.4 months (range 3-83 months).
Radiation therapy
Radiotherapy was used to treat 34 sites in 31 patients at any time during management and was part of frontline therapy in 17 patients. In patients with available RT records (n=21), the technique was intensity modulated RT (n=12), proton therapy (n=4), 3D-conformal RT (n=4) and intra-operative RT (n=1). In general, the radiographically apparent tumour, resection bed or nodal region was expanded by 1-2 cm to create a clinical target volume accounting for microscopic extension. In the setting of relapse within a surgical site, the entire pre-operative tumour bed was targeted, with a boost to gross disease.
In 14 cases, RT was used to treat the resection bed after an upfront GTR (“consolidative RT”). Thirteen of these patients were treated with external beam RT (median 50.4 Gy, range 35 - 66) and 1 with intraoperative RT (10 Gy at 2.7 cm depth). At a median follow-up of 48 months (range 13-188), 2 patients experienced disease relapse within the irradiated area. These recurrences occurred at 2.5 years and 8 years after completion of RT to a total dose of 39.6 Gy and 45 Gy, respectively. After a GTR and consolidative RT, local control was 89% (95% confidence interval [CI] 68-100%) at 3 years after completion of RT. Conversely, in patients who underwent a GTR but did not receive adjuvant RT (n=10), local control within the surgical bed was 53% (95% CI 16-90%) at 3 years post-operatively, which was significantly inferior (log rank P=0.03). This difference translated into an improvement in PFS (83% vs. 48% at 3 years, P = 0.04) and in OS (100% vs. 80% at 3 years, P = 0.05), as shown in Figure 4.
Three other patients received RT as a part of upfront therapy to address gross, surgically unresectable disease (60 Gy). In addition, 16 courses of RT were given for salvage of relapsed/refractory disease (median 50.4 Gy; range 40-70) and 1 for palliation (35 Gy). These 20 courses of RT were analysed together to assess the efficacy of RT in treating gross disease. Four patients received concurrent chemotherapy (2 gemcitabine, 1 paclitaxel, 1 cisplatin). At a median follow-up of 12 months (range 1-63), disease progressed within the irradiated area in 9 cases. Local progression occurred after a median dose of 60 Gy (range 40-70). At one year after completion of RT, freedom from local progression was 54% (95% CI 29-79%). Three of 4 patients treated with concurrent chemotherapy experienced local progression.
Salvage systemic therapy and stem cell transplantation
Multiple approaches were attempted in the salvage setting. Second- and third-line chemotherapy included gemcitabine with/without taxanes (n=9); CHOP (n=5); lenalidomide (n=3); ifosfamide-based (n=1); R-CHOP (n=1); bortezomib, lenalidomide and dexamethasone (n=1); pazopanib (n=1); and hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamthason, methotrexate, cytarabine; n=1). The ORR to second- and third-line systemic therapy was 16%, with no patient achieving a CR. Three patients underwent salvage allogeneic stem cell transplantation (SCT). Of these, 2 patients experienced disease relapse at 4 months and 6 months post-SCT. One patient died of graft-versus-host disease at 5 months after SCT, without evidence of disease.
Discussion
Follicular dendritic cell sarcoma (FDCS) is a rare tumour, and data regarding its biology, management and outcomes is derived largely from anecdotal case reports and series with limited patient numbers(Dalia, et al 2014, Gounder, et al 2015). The current study is the largest single institutional series. We reported on 66 patients with FDCS, who were managed with various approaches. Overall, the median PFS and OS times were 21 months and 50 months, respectively; however, patient outcomes were heterogeneous. Several disease-specific factors were associated with outcome, including limited nodal disease, non-bulky tumour, and extra-abdominal site. Patients treated with a combined modality approach, including GTR, experienced the best outcomes. In Figure 5, we have proposed an algorithm to manage these patients that we follow at our centre.
Figure 5. Proposed algorithm for management of patients with follicular dendritic cell sarcoma.
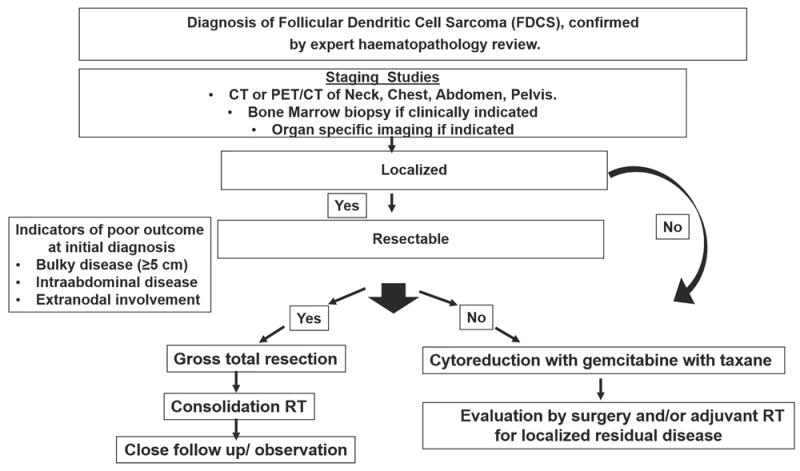
CT, computed tomography; PET, positron-emission tomography; RT, radiotherapy.
The baseline characteristics of our cohort were similar to those observed in other series, with a median age at diagnosis of 49 years and slight predilection towards female gender (Gounder, et al 2015, Saygin, et al 2013). Six patients had Castleman disease, which is associated with FDCS (Gounder, et al 2015, Saygin, et al 2013). The development of FDCS in patients with Castleman disease may be secondary to dysplastic follicular dendritic cells, which are thought to be clonally related to lymphocytes. Researchers have suggested that EGFR expression may link these two conditions (Sun, et al 2003). Also, it has been suggested that other neoplasms are more common in patients with FDCS (Gounder, et al 2015). In our cohort, 9 patients (14%) had another cancer. This frequency is lower than that reported in a recent series, in which 29% of patients with FDCS had other solid tumours (Gounder, et al 2015). Lastly, an association of FDCS with autoimmune conditions has been described (Saygin, et al 2013). In the present series, 13 patients (19%) had autoimmune disease; the most common autoimmune condition was pemphigus, consistent with other reports(Saygin, et al 2013). The mechanistic link of FDCS with autoimmune diseases is unknown; however, it may be hypothesized that neoplastic follicular dendritic cells secrete proinflammatory cytokines, which can trigger auto-immunity. In this study, one patient had FDCS, Castleman disease and an autoimmune condition (pemphigus).
In our cohort, several disease-specific factors were prognostically significant. First, extranodal disease portended significantly inferior PFS and OS, suggesting that extranodal FDCS may have distinct biological features. Additionally, as in other reports, patients with bulky or intra-abdominal disease experienced worse outcomes(Saygin, et al 2013). Conversely, PFS and OS were not statistically different in patients who presented with early vs. advanced stage disease. Other groups have also found no significant association between stage and survival (Perkins and Shinohara 2013, Saygin, et al 2013). On the other hand, a retrospective study of 31 patients with FDCS reported better OS in patients with localized vs. advanced stage disease (Gounder, et al 2015). A possible explanation for these discrepant findings is confounding factors. Although there was no association between stage and survival in our cohort, in an exploratory analysis, we found that patients with localized nodal disease had significantly longer PFS and OS than those with advanced stage and/or extranodal disease.
In patients whose disease progressed, the pattern of failure was predominantly locoregional. Consistent with this finding, treatment strategies that maximize local control, including surgery and RT, were associated with improved survival. Patients who underwent upfront GTR experienced significantly longer PFS and OS, consistent with previous reports(Saygin, et al 2013). Furthermore, in the subset of patients who underwent a GTR, consolidative RT was associated with improved local control (log rank P=0.03), which translated into improved PFS and OS. Two disease relapses occurred within the irradiated area following consolidative RT of 39.6 Gy and 45 Gy, respectively. The low number of relapses precluded the identification of a dose response. It is notable, however, that both recurrences arose after doses below the median, suggesting that patients may benefit from dose escalation above 45 Gy. The local control benefit associated with adjuvant RT has been described in the sarcoma literature (Pisters, et al 1996, Yang, et al 1998). However, previously published studies have not identified a benefit associated with adjuvant RT in FDCS (Gounder, et al 2015, Saygin, et al 2013). These discrepant findings may be due to selection bias, as patients with higher risk disease may be treated with adjuvant RT preferentially. Another caveat regarding these findings could be that initial therapy must have been efficacious in patients who received consolidation treatments and responded, therefore, these comparisons raise the possibility of a selection bias.
In this cohort, the most commonly used upfront systemic chemotherapy regimen in patients with measurable disease was gemcitabine and a taxane. In 10 patients, it yielded an ORR of 80%. This finding corroborates a recent report of favourable outcomes with this combination(Conry 2014). Common features of patients who did not respond to chemotherapy were bulky and/or intra-abdominal disease.
In our cohort, salvage rates after disease relapse were poor. The ORR to second- and third-line systemic therapy was 16%, with no CR. Likewise, RT did not effectively control gross tumour, and local progression occurred even after treatment with high doses (median 60 Gy) and administration of concurrent radiosensitizing chemotherapy. These findings support aggressive frontline management in FDCS.
Targeted agents may improve outcomes in DCS. We identified TP53 and PTEN gene mutations in 2 patients with FDCS. Further study is underway to define the genomic profile of DCS and identify molecular therapeutic targets.
In summary, FDCS is a rare malignancy with heterogeneous outcomes. Our findings support a role for aggressive local treatment with surgery and consolidative RT. Patients with extranodal, bulky, or intra-abdominal disease experience poor outcomes and may derive particular benefit from intensification of therapy. Ongoing efforts to identify genomic aberrations may result in novel treatment strategies. Multi-institutional collaboration for prospective studies is encouraged to define the optimal management of this rare disease.
Acknowledgments
None of the authors are employed by the National Institutes of Health.
Footnotes
Authorship Contributions –
P.J. S.M. and N.F. designed the study.
P.J, S.M., J.V. and N.F. analysed results.
P.J. S.M., and N.F. wrote the paper.
K.P., S.H., R.K.S., L.J.M., B.C.R. performed the pathology review.
N.F., L.N., M.W., L.F., Y.O., S.M., C.P, G.S, B.D. treated these patients.
All authors reviewed and gave the final approval for the paper.
Conflicts of Interest Disclosures: None of the authors declare any competing financial interests.
References
- Azim HA, Elsedewy E, Azim HA., Jr Imatinib in the treatment of follicular dendritic sarcoma: a case report and review of literature. Onkologie. 2007;30:381–384. doi: 10.1159/000103586. [DOI] [PubMed] [Google Scholar]
- Chan AC, Chan KW, Chan JK, Au WY, Ho WK, Ng WM. Development of follicular dendritic cell sarcoma in hyaline-vascular Castleman’s disease of the nasopharynx: tracing its evolution by sequential biopsies. Histopathology. 2001;38:510–518. doi: 10.1046/j.1365-2559.2001.01134.x. [DOI] [PubMed] [Google Scholar]
- Chan JK, Fletcher CD, Nayler SJ, Cooper K. Follicular dendritic cell sarcoma. Clinicopathologic analysis of 17 cases suggesting a malignant potential higher than currently recognized. Cancer. 1997;79:294–313. [PubMed] [Google Scholar]
- Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, Coiffier B, Fisher RI, Hagenbeek A, Zucca E, Rosen ST, Stroobants S, Lister TA, Hoppe RT, Dreyling M, Tobinai K, Vose JM, Connors JM, Federico M, Diehl V for the International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- Cokelaere K, Debiec-Rychter M, De Wolf-Peeters C, Hagemeijer A, Sciot R. Hyaline vascular Castleman’s disease with HMGIC rearrangement in follicular dendritic cells: molecular evidence of mesenchymal tumorigenesis. Am J Surg Pathol. 2002;26:662–669. doi: 10.1097/00000478-200205000-00013. [DOI] [PubMed] [Google Scholar]
- Conry RM. Response of follicular dendritic cell sarcoma to gemcitabine and docetaxel: report of two cases and literature review. Clin Sarcoma Res. 2014;4:6. doi: 10.1186/2045-3329-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalia S, Jaglal M, Chervenick P, Cualing H, Sokol L. Clinicopathologic characteristics and outcomes of histiocytic and dendritic cell neoplasms: the moffitt cancer center experience over the last twenty five years. Cancers (Basel) 2014;6:2275–2295. doi: 10.3390/cancers6042275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan GJ, Wu F, Zhu J, Guo DY, Zhang R, Shen LL, Wang SH, Li Q, Xiao HL, Mou JH, Yan XC. Extranodal follicular dendritic cell sarcoma of the pharyngeal region: a potential diagnostic pitfall, with literature review. Am J Clin Pathol. 2010;133:49–58. doi: 10.1309/AJCP7U8YISBUAVNW. [DOI] [PubMed] [Google Scholar]
- Fonseca R, Yamakawa M, Nakamura S, van Heerde P, Miettinen M, Shek TW, Myhre Jensen O, Rousselet MC, Tefferi A. Follicular dendritic cell sarcoma and interdigitating reticulum cell sarcoma: a review. Am J Hematol. 1998;59:161–167. doi: 10.1002/(sici)1096-8652(199810)59:2<161::aid-ajh10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Ge R, Liu C, Yin X, Chen J, Zhou X, Huang C, Yu W, Shen X. Clinicopathologic characteristics of inflammatory pseudotumor-like follicular dendritic cell sarcoma. Int J Clin Exp Pathol. 2014;7:2421–2429. [PMC free article] [PubMed] [Google Scholar]
- Gounder M, Desai V, Kuk D, Agaram N, Arcila M, Durham B, Keohan ML, Dickson MA, D’Angelo SP, Shukla N, Moskowitz C, Noy A, Maki RG, Herrera DA, Sanchez A, Krishnan A, Pourmoussa A, Qin LX, Tap WD. Impact of surgery, radiation and systemic therapy on the outcomes of patients with dendritic cell and histiocytic sarcomas. Eur J Cancer. 2015;51:2413–2422. doi: 10.1016/j.ejca.2015.06.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin GK, Sholl LM, Lindeman NI, Fletcher CD, Hornick JL. Targeted genomic sequencing of follicular dendritic cell sarcoma reveals recurrent alterations in NF-kappaB regulatory genes. Mod Pathol. 2016;29:67–74. doi: 10.1038/modpathol.2015.130. [DOI] [PubMed] [Google Scholar]
- Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. 2014;14:495–504. doi: 10.1038/nri3689. [DOI] [PubMed] [Google Scholar]
- Kairouz S, Hashash J, Kabbara W, McHayleh W, Tabbara IA. Dendritic cell neoplasms: an overview. Am J Hematol. 2007;82:924–928. doi: 10.1002/ajh.20857. [DOI] [PubMed] [Google Scholar]
- Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, Schwarz P, Armulik A, Browning JL, Tallquist M, Buch T, Oliveira-Martins JB, Zhu C, Hermann M, Wagner U, Brink R, Heikenwalder M, Aguzzi A. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell. 2012;150:194–206. doi: 10.1016/j.cell.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake H, Yamakawa M. Interdigitating dendritic cell sarcoma and follicular dendritic cell sarcoma: histopathological findings for differential diagnosis. J Clin Exp Hematop. 2013;53:179–184. doi: 10.3960/jslrt.53.179. [DOI] [PubMed] [Google Scholar]
- Pai VD, Desai S, Desouza A, Saklani AP. Extranodal follicular dendritic cell sarcoma: a frequently misdiagnosed entity. J Postgrad Med. 2015;61:55–56. doi: 10.4103/0022-3859.147058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels P, Dal Cin P, Vlasveld LT, Aleva RM, van Erp WF, Jones D. A chromosomal abnormality in hyaline vascular Castleman’s disease: evidence for clonal proliferation of dysplastic stromal cells. Am J Surg Pathol. 2000;24:882–888. doi: 10.1097/00000478-200006000-00016. [DOI] [PubMed] [Google Scholar]
- Perez-Ordonez B, Erlandson RA, Rosai J. Follicular dendritic cell tumor: report of 13 additional cases of a distinctive entity. Am J Surg Pathol. 1996;20:944–955. doi: 10.1097/00000478-199608000-00003. [DOI] [PubMed] [Google Scholar]
- Perkins SM, Shinohara ET. Interdigitating and follicular dendritic cell sarcomas: a SEER analysis. Am J Clin Oncol. 2013;36:395–398. doi: 10.1097/COC.0b013e31824be22b. [DOI] [PubMed] [Google Scholar]
- Perry AM, Nelson M, Sanger WG, Bridge JA, Greiner TC. Cytogenetic abnormalities in follicular dendritic cell sarcoma: report of two cases and literature review. In Vivo. 2013;27:211–214. [PubMed] [Google Scholar]
- Pisters PW, Harrison LB, Leung DH, Woodruff JM, Casper ES, Brennan MF. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol. 1996;14:859–868. doi: 10.1200/JCO.1996.14.3.859. [DOI] [PubMed] [Google Scholar]
- Saygin C, Uzunaslan D, Ozguroglu M, Senocak M, Tuzuner N. Dendritic cell sarcoma: a pooled analysis including 462 cases with presentation of our case series. Crit Rev Oncol Hematol. 2013;88:253–271. doi: 10.1016/j.critrevonc.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Soriano AO, Thompson MA, Admirand JH, Fayad LE, Rodriguez AM, Romaguera JE, Hagemeister FB, Pro B. Follicular dendritic cell sarcoma: a report of 14 cases and a review of the literature. Am J Hematol. 2007;82:725–728. doi: 10.1002/ajh.20852. [DOI] [PubMed] [Google Scholar]
- Starr JS, Attia S, Joseph RW, Menke D, Casler J, Smallridge RC. Follicular Dendritic Cell Sarcoma Presenting As a Thyroid Mass. J Clin Oncol. 2015;33:e74–76. doi: 10.1200/JCO.2013.49.3213. [DOI] [PubMed] [Google Scholar]
- Sun X, Chang KC, Abruzzo LV, Lai R, Younes A, Jones D. Epidermal growth factor receptor expression in follicular dendritic cells: a shared feature of follicular dendritic cell sarcoma and Castleman’s disease. Hum Pathol. 2003;34:835–840. doi: 10.1016/s0046-8177(03)00356-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO classification of tumours of haematopoietic and lymphoid tissues. 4. International Agency for Research on Cancer Press; Lyon, France: 2008. [Google Scholar]
- Wang J, Bu DF, Li T, Zheng R, Zhang BX, Chen XX, Zhu XJ. Autoantibody production from a thymoma and a follicular dendritic cell sarcoma associated with paraneoplastic pemphigus. Br J Dermatol. 2005;153:558–564. doi: 10.1111/j.1365-2133.2005.06599.x. [DOI] [PubMed] [Google Scholar]
- Wang RF, Han W, Qi L, Shan LH, Wang ZC, Wang LF. Extranodal follicular dendritic cell sarcoma: A clinicopathological report of four cases and a literature review. Oncol Lett. 2015;9:391–398. doi: 10.3892/ol.2014.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Chang AE, Baker AR, Sindelar WF, Danforth DN, Topalian SL, DeLaney T, Glatstein E, Steinberg SM, Merino MJ, Rosenberg SA. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998;16:197–203. doi: 10.1200/JCO.1998.16.1.197. [DOI] [PubMed] [Google Scholar]