

Abstract
Cochlear hair cells are vulnerable to a variety of insults like acoustic trauma and ototoxic drugs. Such injury can also lead to degeneration of spiral ganglion neurons (SGNs), but this occurs over a period of months to years. Neuronal survival is necessary for the proper function of cochlear prosthetics, therefore it is of great interest to understand the mechanisms that regulate neuronal survival in deaf ears. We have recently demonstrated that selective hair cell ablation is sufficient to attract leukocytes into the spiral ganglion (SG), and that fractalkine signaling plays a role in macrophage recruitment and in the survival of auditory neurons. Fractalkine (CX3CL1), a chemokine that regulates adhesion and migration of leukocytes is expressed by SGNs and signals to leukocytes via its receptor CX3CR1. The present study has extended the previous findings to more clinically relevant conditions of sensorineural hearing loss by examining the role of fractalkine signaling after aminoglycoside ototoxicity or acoustic trauma. Both aminoglycoside treatment and acoustic overstimulation led to the loss of hair cells as well as prolonged increase in the numbers of cochlear leukocytes. Lack of CX3CR1 did not affect macrophage recruitment after injury, but resulted in increased loss of SGNs and enhanced expression of the inflammatory cytokine interleukin-1β, when compared to mice with intact CX3CR1. These data indicate that the dysregulation of macrophage response caused by the absence of CX3CR1 may contribute to inflammation-mediated neuronal loss in the deafened ear, suggesting a key role for inflammation in the long-term survival of target-deprived afferent neurons.
Keywords: Hearing Loss, Spiral Ganglion Neurons, Inflammation, Macrophages, Fractalkine, Interleukin-1β
1. Introduction
Sensorineural hearing loss (SNHL) is caused by damage to the sensory receptors of the cochlea and/or their afferent neurons. Such pathology can occur after exposure to loud sounds, treatment with ototoxic medications, inner ear infections, or as a part of normal aging. It has generally been assumed that the loss of hair cells was the primary cause of SNHL, and that the degeneration of sensory neurons occurred as a secondary consequence of hair cell death (Bohne and Harding, 2000; Johnsson, 1974). However, results of more recent studies have suggested that the loss of cochlear hair cells does not necessarily lead to the rapid degeneration of SGNs (Zilberstein, Liberman and Corfas, 2012; Tong et al., 2015; Kaur et al., 2015). Instead, the loss of SGNs occurs over a period of months to years (Liberman and Kiang, 1978; Spoendlin et al., 1975, Kujawa and Liberman, 2009, Oesterle and Campbell, 2009; Schmitz, Johnson and Santi, 2014). Considered together, these findings underscore the complexity of noise-induced hearing loss (NIHL) and point to our present lack of knowledge regarding the cellular and molecular mechanisms that regulate the survival of cochlear afferents. Spiral ganglion neurons are the sole means by which information from the cochlea is conveyed to the brain. As such, long-term survival of SGNs is critical for the preservation of residual hearing and for the success of cochlear prosthetics or any future hair cell restoration strategies in hearing impaired patients.
We recently demonstrated that selective hair cell ablation without any accompanying cochlear pathology in huDTR-Pou4f3DTR/+ mice (Tong et al., 2015) is sufficient to recruit leukocytes into the cochlea and results in sustained elevation of macrophage numbers in the spiral ganglion. Our data further suggest that SGNs communicate with macrophages via the ligand fractalkine (CX3CL1), that is expressed on expressed by SGNs, interacting with its receptor, CX3CR1, that is expressed by cochlear macrophages. We also found that disruption of fractalkine signaling after hair cell death results in decreased macrophage recruitment into the injured cochlea as well as enhanced loss of SGNs (Kaur et al., 2015). Together, our findings have revealed a critical role for macrophages in auditory pathology and suggest that macrophages have a neuroprotective role via fractalkine signaling in the injured cochlea.
Fractalkine is a chemokine that is expressed as a membrane bound glycoprotein on neurons (Harrison et al., 1998), peripheral endothelial cells (Bazan et al., 1997; Rossi et al., 1998; Harrison et al., 2001), and epithelial cells (Lucas et al., 2001). The fractalkine receptor (CX3CR1) is present on microglia and circulating monocytes, dendritic cells and natural killer (NK) cells (Jung et al., 2000; Cook et al., 2001) and also expressed on cochlear macrophages (Hirose, Discolo, Keasler, and Ransohoff, 2005). Fractalkine occurs in two different forms: as a membrane-bound protein tethered to neuronal membranes by a mucin-like stalk, and as a soluble factor released upon cleavage of its N-terminal chemokine domain (Garton et al., 2001). The extracellular chemokine domain of fractalkine is proteolytically cleaved from the membrane-bound fraction by the lysosomal cysteine protease, cathepsin S and members of ADAM (a disintegrin and metalloproteinase) family such as ADAM-10 and ADAM-17. The soluble chemokine domain of fractalkine, when cleaved, can act as chemoattractant promoting cellular migration, whereas, membrane-tethered mucin-stalk of fractalkine has been proposed to act as an adhesion molecule for leukocytes during inflammation (Haskell, Cleary and Charo, 1999; Hermand et al., 2008). In the central nervous system, fractalkine signaling has been suggested to control microglial neurotoxicity during certain neurodegenerative and neuroinflammatory conditions (Cardona et al., 2006). In humans, two single-nucleotide polymorphisms produce four allelic receptor variants (Faure et al., 2000; McDermott et al., 2000). Most individuals carry CX3CR1V249/T280, whereas 20% population carry CX3CR1I249/M280 Biochemical studies have suggested that the polymorphic variant CX3CR1I249/M280 exhibits defective adhesive properties, therefore leading to deficits in fractalkine signaling (McDermott et al., 2003). Several studies support a role of CX3CR1 in multiple neurodegenerative and neuroinflammatory disorders however, it is still yet to be determined how these changes affect macrophage-neuronal communication and to what extent dysregulated macrophage responses in absence of fractalkine signaling contribute to neuronal pathology in the damaged cochlea.
The present study examined how the lack of CX3CR1 influences monocyte/macrophage recruitment and the survival of afferent neurons after both aminoglycoside ototoxicity and acoustic trauma. CX3CR1 wild type, heterozygous, and knockout mice, were given a combination of kanamycin and furosemide or exposed to 120 dB SPL noise. Macrophage numbers and SGN density was quantified at acute and prolonged recovery periods after these injuries. Aminoglycoside ototoxicity and acoustic trauma both led to increased macrophage numbers within the cochlea and to a prolonged increase in macrophage numbers in the spiral ganglion. Disruption of fractalkine signaling by genetic deletion of CX3CR1 did not affect macrophage density, but did lead to enhanced loss of SGN cell bodies and increased cochlear expression of IL-1β at longer recovery periods after cochlear injury. The results complement our previous findings and point to a crucial role for inflammation in promoting long-term survival of afferent neurons through CX3CL1-CX3CR1 signaling following cochlear injury.
2. Materials and Methods
Animals
Experiments utilized young adult (6-8 weeks old) CX3CR1+/+(Wild type), CX3CR1GFP/+ (Heterozygous) and CX3CR1GFP/GFP (Homozygous) mice (Dan Littman, New York University, New York) of both sexes. A targeted deletion of CX3CR1 and replacement with the gene encoding green fluorescent protein (GFP) rendered all monocytes and macrophages endogenously fluorescent (Jung et al., 2000), thereby permitting the tracking of mononuclear phagocytes after cochlear damage. The CX3CR1GFP/+ mice (also denoted as CX3CR1+/−) retained one copy of CX3CR1, while mice homozygous for GFP (CX3CR1GFP/GFP, also denoted as CX3CR1−/−) lacked fractalkine signaling. Identification of CX3CR1GFP/+ and CX3CR1GFP/GFP by PCR followed previously described methods (Jung et al., 2000). All transgenic mice were on a C57BL/6J background. Mice were housed in the animal facility at the Central Institute for the Deaf (Washington University School of Medicine) and were maintained on a 12-hr/day-night light cycle with open access to food and water. All experimental protocols involving animals were approved by the Animal Studies Committee of the Washington University School of Medicine (Saint Louis, MO).
Aminoglycoside ototoxicity model
Kanamycin sulfate was purchased from Sigma (St. Louis, MO) and prepared at 50 mg/ml in saline. Furosemide (10 mg/ml, Butler animal health supply (Dublin, OH)) was provided by Dr. Keiko Hirose (Washington University School of Medicine). Hair cell death was induced by administrating a single dose of kanamycin (1000 mg/kg, subcutaneous) followed 30-45 min later by a single injection of furosemide (250 mg/kg, intraperitoneal) (Oesterle et al., 2008, modified). Saline-injected animals served as controls. Animals were sacrificed at either two-weeks or two-months post injections.
Acoustic trauma model
Mice were exposed to octave band noise (8-16 kHz) at 120 dB sound pressure level (SPL) for 2 hr and then allowed to recover for 1, 7, 14, or 60 days. In all cases, non-exposed age-, strain- and genotype-matched animals served as controls. Briefly, noise exposures were performed in a foam-lined, single-walled soundproof room from Industrial Acoustics Company (IAC). Fully awake and unrestrained animals were placed singly or in pairs in modified cages (food, water, bedding removed) positioned up to two cages at once directly under an exponential horn. All noise was octave band (8-16 kHz), generated digitally using custom Labview routines running on a PC. The signal was output to a Tucker-Davis Technologies RZ6 signal processor, then to a Crown D-150A power amplifier that drove the speaker.
Auditory brainstem response
Auditory brainstem responses (ABRs) to tone pips were quantified prior to and at multiple time points after either KF injection or noise exposure for both experimental and control subjects. Mice were first anaesthetized via i.p. injections of ketamine (100 mg/kg) and xylazine (20 mg/kg). Subcutaneous electrodes were placed behind the right pinna (inverting) and vertex (active). A ground electrode was placed near the tail of the mouse. Stimuli were 5-ms tone pips (0.5-ms cos2 rise-fall), delivered at 40/s with alternating stimulus polarity. Recorded electrical responses were amplified (~10,000×), filtered (100 Hz to 3 kHz) and averaged, using BioSig software (Tucker-Davis Technologies, Alachua, FL, USA). The sound level was decreased in 5-dB steps from 99 dB SPL down to 15 dB SPL. At each sound level, 1024 responses were averaged, and response waveforms were discarded as artifacts if the peak-to-peak voltage exceeded 15 μV. Thresholds at 5,10, 20, 28.3, 40, and 56.6 kHz were determined by a single observer, who noted the lowest sound level at which a recognizable waveform could be obtained. Waveforms were confirmed as auditory-evoked responses by their increasing latency and decreasing amplitude as the intensity of the stimulus was lowered. If a hearing threshold at a particular stimulus frequency could not be detected at a sound level of <99 dB, a threshold of ‘105 dB’ was assigned for that frequency. These threshold values (actual or assigned) were then used to calculate the mean ABR thresholds at each stimulus frequency.
Histological Preparation
Mice were deeply anesthetized with Fatal Plus and perfused with phosphate-buffered 4% paraformaldehyde (PFA) (Electron Microscopy Sciences). Temporal bones were removed and post fixed for 40 minutes in 4% PFA, rinsed in PBS, and placed in 0.1 M EDTA, to allow decalcification for whole-mount dissections and frozen mid-modiolar sectioning.
Immunohistochemistry
Decalcified cochleae were processed for immunohistochemical labeling of proteins in both cochlear whole mounts (surface preparations) and frozen mid-modiolar (radial) sections using standard immunofluorescence methods. Briefly, tissue was rinsed with PBS (3X) and incubated at room temperature for 2 hr in blocking solution (5% normal horse serum in 0.2% Triton X-100 in PBS). Cochleae were incubated overnight at room temperature with combinations of following primary antibodies: Hair cells were labeled with antibody against anti-Myosin VIIa (catalogue # 25-6790, Proteus Biosciences, 1:500). Neuronal peripheral processes and ganglion cell bodies were labeled using a combination of anti-Neurofilament (NF) (mouse monoclonal, clone # 2H3, Developmental Studies Hybridoma Bank, University of Iowa, 1:100) and anti-β-III Tubulin antibodies (mouse monoclonal, catalogue # MMS-435P, Covance, 1:500). Visualization of GFP-expressing macrophages was enhanced using rabbit anti-GFP (catalogue # A11122, Life technologies, 1:500). Cytotoxic cytokine IL-1β was immunolabeled using an antibody directed against mouse IL-1β (catalogue# AF-401-NA, R&D systems, 5ug/ml). Following incubation in primaries, specimens were rinsed 5X in PBS and treated for 2 h in secondary antibodies, conjugated to either Alexa Flour 488, 546, 555 or 647 (1:500; Invitrogen or Jackson Immuno Research). The secondary antibody solutions also contained DAPI (catalogue # D9542, Sigma-Aldrich, 1μg/ml), in order to label cell nuclei. All specimens were coverslipped in glycerol: PBS (9:1) before microscopic imaging.
Cellular imaging and analyses
Fluorescence imaging was performed using a Zeiss LSM 700 confocal microscope. For both cochlear whole mount or frozen section preparations, Z-series images were obtained using either 10×, 20× or 63× objectives. Image processing and quantitative analysis was performed using Volocity 3D image analysis software (Ver 6.1.1, PerkinElmer).
Macrophage counts
To assess macrophages per 100μm of sensory epithelium, GFP-labeled macrophages were counted from 20× images taken from the middle region of cochlear whole mounts. To assess macrophages in the spiral ganglia, GFP-labeled macrophages were counted from 20× images taken from the lower basal (base), upper basal (middle) and apical portions of the Rosenthal’s canal of sectioned specimens. Macrophages in spiral ganglia were counted from at least 4-5 sections per cochlea, and normalized to the cross-sectional area of the Rosenthal’s canal of the respective cochlear turn and averaged and reported as number per 1000 µm2.
Spiral ganglion neuron counts
Serial floating radial sections (30μm) were immunolabeled using standard immunofluorescence methods. Briefly, tissue was rinsed with PBS (3X) and incubated at room temperature for 2 h in blocking solution (5% normal horse serum in 0.2% Triton X-100 in PBS). Sections were then incubated overnight at room temperature and neurons were labeled with a combination of anti-Neurofilament and anti-β-III Tubulin (as described above). Specimens were then rinsed 5X in PBS and incubated for 2 h in secondary antibody (Alexa Flour 546 anti-mouse IgG; Invitrogen; 1:500). Specimens were coverslipped in glycerol: PBS (9:1) and fluorescent imaging was carried-out using a Zeiss LSM 700 confocal microscope. Z-series images at 1μm intervals were obtained using a 20× objective. Image processing and quantitative analysis was performed using Volocity 3D image analysis software (Ver 6.1.1, PerkinElmer). To assess the numbers of spiral ganglion cell bodies, Neurofilament and β-III Tubulin labeled-somata within Rosenthal’s canal were counted from the maximum intensity projections of each section. Cell bodies counted from 4-5 sections per cochlea were normalized to the cross-sectional area of Rosenthal’s canal per cochlear turn and averaged and reported as spiral ganglion neuron density per 1000 µm2.
IL-1β Immununofluroscence intensity measurements
Fluroscent intensity was measured using Fiji/ImajeJ software (ver 1.47b) for Mac from National Institute for Health (NIH). Briefly, a region of interest (ROI) i.e., spiral ganglion (SG) was drawn and its area, integrated density and mean gray value were measured from 4-5 sections per cochlea. Similary, a region next to SG was selected that had no fluroscence to get background area, integrated density and mean gray values. Following formula was used to compute corrected total fluorescence (CTF): CTF= Integrated density - (Area of selected ROI × Mean fluorescence of background readings). CTF was averaged and reported as IL-1β fluorescent intensity in spiral ganglion (arbitrary units).
Statistical analysis
All the data analysis and statistics were carried out using GraphPad Prism version 7.0a. Data are presented as mean±SD. Analyses of variance (ANOVA) was applied as appropriate and significance main effects or interactions were followed with the use of Bonferroni’s or Tukey’s posthoc tests. Results were considered statistically significant when p < 0.05.
3. Results
Exposure to systemic aminoglycosides or loud noise results profound hair cell death and hearing loss
In order to test whether fractalkine signaling influences macrophage numbers and neuronal survival in biologically-relevant models of acquired sensorineural hearing loss, we employed two previously-established murine models of cochlear injury and hair cell death: aminoglycoside ototoxicity and acoustic trauma (Wang, Hirose and Liberman, 2002; Hirose, Discolo, Keasler, and Ransohoff, 2005; Oesterle et al., 2008). CX3CR1 wild type, heterozygous, and knockout mice were exposed to either a single dose of kanamycin/furosemide or octave-band (8-16 kHz) noise at 120 dB SPL for 2 hours. Saline treated or unexposed age-matched littermates of either genotype served as controls. ABR thresholds in saline-injected (Fig. 1a) or unexposed control mice (Fig. 1c) were similar in all three genotypes. Kanamycin-furosemide treatment (Fig. 1a) or loud noise exposure (Fig. 1c) resulted in profound hearing loss as assessed by ABRs. ABR thresholds typically exceeded 90 dB SPL at all frequencies tested. In most cases we were unable to obtain clear ABR responses, even at highest sound intensities that could be generated by our test apparatus (indicated by arrow). The differences in ABR thresholds were statistically significant at all days and frequencies tested, when compared to respective controls (p<0.0001). Differences in ABR thresholds were insignificant among the three genotypes at all days and frequencies tested after ototoxic- or noise-injury (p>0.05). Based on Myosin 7a immunolabeling (from 4-6 temporal bones at each time point from CX3CR1+/+ mice), both inner and outer hair cells were intact in saline treated (Fig. 1b) or unexposed control mice (Fig. 1d). A single KF treatment resulted in nearly-complete ablation of outer hair cells (OHC) along the entire length of the cochlea by 14 days post injection (dpi). Inner hair cells (IHC) were completely missing in the base region of these cochleae and a partial loss of IHCs was detected in the middle and apical region of the cochlea at 60 days post KF injection (Fig 1b). Hair cells were intact at one day following 2 hr exposure to 8-16 kHz noise (120 dB) but, by 7 days post-noise exposure (DPNE), a nearly-complete loss of OHCs was observed in the base and middle region of the cochlea (Fig. 1d). There was also profound loss of IHCs in the base and middle region of the cochlea at the same recovery time points post exposure. By 60 DPNE there were very few intact hair cells in the organ of Corti. Furthermore, there was no difference in the degree of hair cell damage among the three genotypes after ototoxic- or noise-injury (data not shown). These results demonstrate that a single exposure to aminoglycosides or noise in CX3CR1 wild type, heterozygous, and knockout mice can induce hearing loss and hair cell death.
Figure 1.
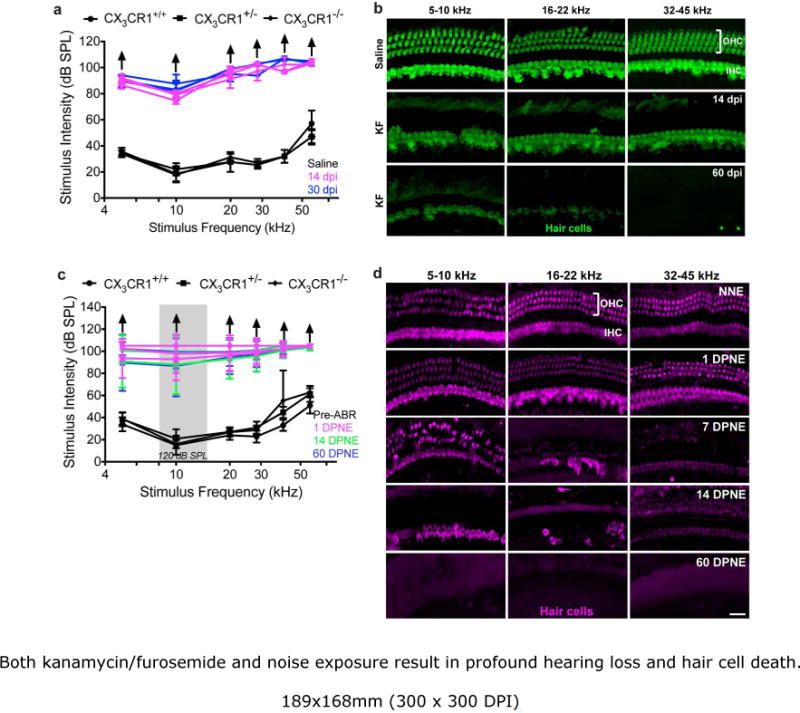
Both kanamycin/furosemide and noise exposure result in profound hearing loss and hair cell death. a, c, All three genotypes have normal ABR thresholds prior to noise exposure or saline injection (black line). ABR thresholds are permanently elevated without any evident recovery after cochlear damage induced by either aminoglycoside injection (a) or noise over exposure (c). (
 ) indicate that mice did not respond to any stimulus frequency. Data expressed as mean±SD. n=4-9 for each genotype for either injury model. Statistical significance computed using two-way ANOVA followed by Tukey’s multiple comparisons test yielded highly significant (p<0.0001) effect of days and frequency. Difference among genotypes prior noise exposure or saline injection and at several times post injury is not significant. b, d, Representative confocal images of cochlear whole mounts immunolabeled for hair cells (Myosin VIIa) from CX3CR1+/+ mice injected with kanamycin/furosemide (KF) (b) or exposed to loud noise (d) demonstrate extensive loss of both inner and outer hair cells in all the frequency regions of the cochlea. dB SPL, decibel sound pressure level; kHz, kilohertz; OHC, outer hair cell; IHC, inner hair cell; dpi, days post injection; DPNE, days post noise exposure. Scale=55μm
) indicate that mice did not respond to any stimulus frequency. Data expressed as mean±SD. n=4-9 for each genotype for either injury model. Statistical significance computed using two-way ANOVA followed by Tukey’s multiple comparisons test yielded highly significant (p<0.0001) effect of days and frequency. Difference among genotypes prior noise exposure or saline injection and at several times post injury is not significant. b, d, Representative confocal images of cochlear whole mounts immunolabeled for hair cells (Myosin VIIa) from CX3CR1+/+ mice injected with kanamycin/furosemide (KF) (b) or exposed to loud noise (d) demonstrate extensive loss of both inner and outer hair cells in all the frequency regions of the cochlea. dB SPL, decibel sound pressure level; kHz, kilohertz; OHC, outer hair cell; IHC, inner hair cell; dpi, days post injection; DPNE, days post noise exposure. Scale=55μm
Leukocytes infiltrate and accumulate in the damaged cochlea
Consistent with previous studies (Fredelius and Rask-Andersen, 1990; Hirose, Discolo, Keasler, and Ransohoff, 2005; Sato, Shick, Ransohoff and Hirose, 2010), systemic KF exposure or acoustic trauma resulted in enhanced numbers of leukocytes within the cochlea. The cochleae of saline-treated and unexposed control mice contained very few macrophages below the sensory epithelium (~1-2/100μm) or within the spiral ganglion (~10/1000μm2, Fig. 2a,d, h, l). Kanamicin/furosemide-induced ototoxicity was accompanied by a ~2-fold increase in macrophage numbers within the sensory epithelium and spiral ganglion (Fig. 2b,e) at two-weeks post KF injection. Macrophages numbers in the sensory epithelium then began to decline (Fig. 2c) but macrophages numbers remained elevated in the ganglion at two months after ototoxic injury (Fig. 2f). Complete quantitative data are shown in Figure 2g.
Figure 2.
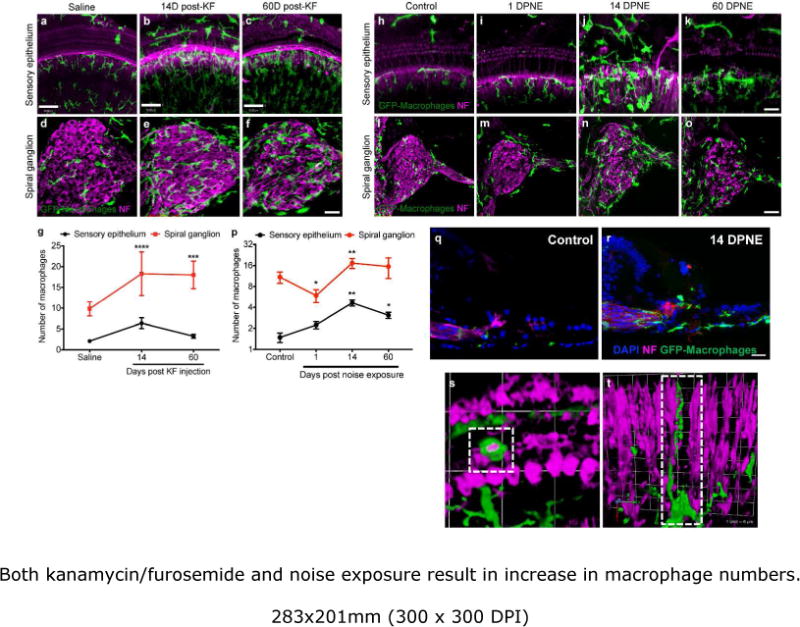
Both kanamycin/furosemide and noise exposure result in increase in macrophage numbers. Cochlear whole mounts (a-c) and mid-modiolar sections (d-f) labeled for GFP (macrophages, green) and neurofilament (neurons, magenta) show increased recruitment of macrophages into both the sensory epithelium and spiral ganglion of KF-treated mice at 14 dpi, compared to saline treated animals. The number of macrophages in the sensory epithelium subsided by 60 dpi, but remained elevated in the ganglion even at longer recovery time points. g, Macrophage density in the sensory epithelium (per 100μm) and spiral ganglion (per 1000μm2). h-k, Cochlear whole mounts showing GFP-labeled macrophages (green) and afferent nerve fibers (magenta), demonstrate increased numbers of macrophages within sensory epithelium of noise exposed mice (i-k), compared with controls (h). l-o, Mid-modiolar section from unexposed (control, l) and noise exposed mouse at various recovery times (m-o), showing GFP-expressing macrophages and neurons (magenta). p, Macrophage density in the sensory epithelium (per 100μm) and spiral ganglion (per 1000μm2) after noise overexposure. q, r, s,t, Representative 3D rendering of cochlear image stacks of noise-damaged cochlea showing macrophages (green) actively phagocytosing hair cell debris (magenta) in the organ of Corti (s) and macrophages wrapped around degenerating afferent nerve fiber (magenta) in the osseous spiral lamina (t). Data expressed as mean±SD. n= 3-7 for each time point for either injury model. Statistical significance was computed using one-way ANOVA. *p<0.05, ** p<0.01, ***p-value <0.001, ****p< 0.0001. Scale=30μm.
A similar trend was observed in the cochlear epithelium after acoustic injury (Fig. 2i-k, p). However, in the noise-damaged epithelium, some macrophages were seen within the scala media (Fig. 2r) in close proximity to the dying hair cells (Fig. 2s), and were also observed wrapped around degenerating auditory afferent nerve fibers (Fig. 2t). In the Rosenthal’s canal, there was a significant decrease (45%) in the leukocyte numbers at 1 day post noise overexposure, followed by a sustained elevation (~2-fold) at longer recovery time points (Fig. 2l-p). Complete quantitative data on macrophage numbers after acoustic overexposure are shown in Figure 2p. We also noted an apparent increase in CX3CR1 positive immune cell accumulation in the spiral ligament, spiral limbus, borders of scala tympani and scala vestibule and in the modiolus following noise exposure (data not shown). These results resemble our previous finding that leukocyte density increases in the cochlea after sterile injury with density peaking around two weeks post injury and remains elevated in the ganglion in close association with SGN cell bodies at longer recovery times.
CX3CR1 modulates macrophage numbers in the damaged cochlea in an injury dependent manner
In a previous study, we found that the chemokine CX3CL1 is expressed by mature SGNs as well as by inner hair cells and certain supporting cells in the sensory epithelium. We also reported that disruption of fractalkine signaling reduces the recruitment of mononuclear phagocytes into the cochlea after selective hair cells ablation (Kaur et al., 2015). In the present study, we sought to determine whether fractalkine signaling also regulates monocyte/macrophage numbers in the cochlea after ototoxic- or noise-injury. To resolve this issue, macrophage numbers were compared in the cochleae of CX3CR1−/− mice with those in CX3CR1+/− mice at different recovery times after ototoxic- or acoustic exposure. We observed a ~3-fold increase in macrophage numbers associated with the cochlear sensory epithelium of CX3CR1+/− mice at two weeks post KF injection (Fig. 3c,e), compared to saline-treated controls (Fig. 3a,e). However, lack of CX3CR1 resulted in significantly fewer macrophages (36%) in or near the sensory epithelium after KF injection (Fig. 3d, e). Deletion of CX3CR1 did not affect the numbers of resident macrophages in the saline treated animals (Fig. 3b,e), which were similar to those in saline-treated CX3CR1+/− mice. As described previously (Hirose, Discolo, Keasler, and Ransohoff, 2005), acoustic injury resulted in a 2-3-fold increase in macrophage numbers in the sensory epithelium. Notably, deficiency of CX3CR1 did not significantly affect those numbers in the noise-damaged epithelium (data not shown).
Figure 3.
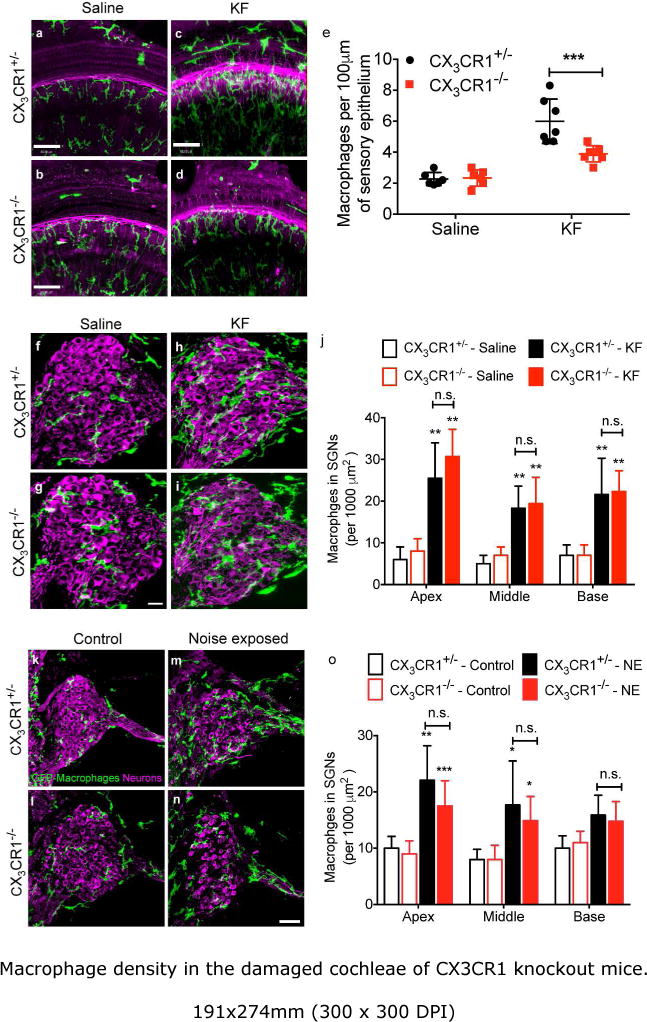
Macrophage density in the damaged cochleae of CX3CR1 knockout mice. a,b, Cochlear whole mounts labeled for GFP (macrophages, green) and neurons (magenta) show normal number of macrophages in saline injected CX3CR1+/− (a) and CX3CR1−/− (b) mice. c,d, KF-induced cochlear damage in CX3CR1+/− mice resulted in an increase in macrophage numbers in the sensory epithelium at 14 days post injection (dpi) (c) however, lack of CX3CR1 (CX3CR1−/−) resulted in fewer macrophages in the KF-damaged sensory epithelium (d). e, Number of macrophages in KF-lesioned sensory epithelia. f,g,k,l, Mid-modiolar sections processed to label macrophages and neurons in the spiral ganglia show normal macrophage density in saline treated or unexposed CX3CR1+/− (f,k) and CX3CR1−/− (g,l) mice. h,i,m,n, Genetic deletion of CX3CR1 did not affect macrophage density at 14 days post-KF treatment (i) or 60 days post noise exposure (DPNE) (n) compared to CX3CR1+/− mice (h,m). j,o, Macrophage density in the spiral ganglion. Data expressed as mean±SD. n=3-9 for each genotype and treatment group for either injury models. Statistical significance was computed using two-way ANOVA followed by Tukey’s multiple comparison test. *p<0.05, ** p<0.01, ***p-value < 0.001, n.s., not significant. Scale=30 μm (a-d), 35 μm (f-i), 63 μm (k-n)
Both ototoxic injury and noise exposure resulted in nearly 2-3 fold significant increase in the numbers of macrophages in the spiral ganglia of both CX3CR1+/− and CX3CR1−/− mice (Fig. 3h,i,j,m,n,o) compared to unexposed animals (Fig. 3f,g,j,k,l,o). However, disruption of fractalkine signaling did not affect macrophage numbers in the spiral ganglia of either ototoxic- or noise-damaged cochleae. Macrophage numbers in the SG of CX3CR1−/− mice were not significantly different from those in CX3CR1+/− mice in all the cochlear regions following ototoxic or acoustic injury (Fig. 3i,j,n,o p=0.2573 in apex; p=0.6704 in middle and p=0.9702 in base). The results suggest that fractalkine signaling may have distinct effects for different types of cochlear injuries (DT-mediated selective hair cell ablation, ototoxic or acoustic) as well as cochlear locations (epithelium or ganglion).
Deficiency of CX3CR1 led to enhanced neuronal loss in damaged cochlea
In light of our previous finding that CX3CR1 signaling can influence neuronal survival after diphtheria toxin-mediated lesion of cochlear hair cells (Kaur et al., 2015), we next compared SGN density in the cochleae of CX3CR1−/− mice and CX3CR1+/− mice at two months after ototoxicity or acoustic exposure. Cochlear mid-modiolar sections immunolabeled for neurofilament and β-III-tubulin revealed significant loss of spiral ganglion cell bodies in the CX3CR1−/− mice (Fig. 4d, i, e, j), compared with CX3CR1+/− (Fig. 4b, g, e, j) in response to both forms of cochlear damage. At 2 months after KF ototoxicity, there was a reduction in the density of SGN cell bodies in all cochlear regions of CX3CR1−/− mice (31% reduction), when compared to CX3CR1+/− littermate controls (Fig. 4e). With acoustic trauma, significant neuronal loss (29% reduction) in CX3CR1−/− mice was mainly observed in the apical and middle region of the cochleae (Fig. 4j). We also noted an approximately 10% loss of neurons in the CX3CR1+/− mice after ototoxic- or acoustic exposure, but this loss was not statistically significant (p=0.5421 (apex), p=0.5949 (middle), p=0.8796 (base)) when compared to saline treated or unexposed CX3CR1+/− animals. Finally, we observed no reduction in SGNs in the saline treated or unexposed CX3CR1−/− animals, suggesting that the loss of neurons occurred only after damage (Fig 4c, h, e, j).
Figure 4.
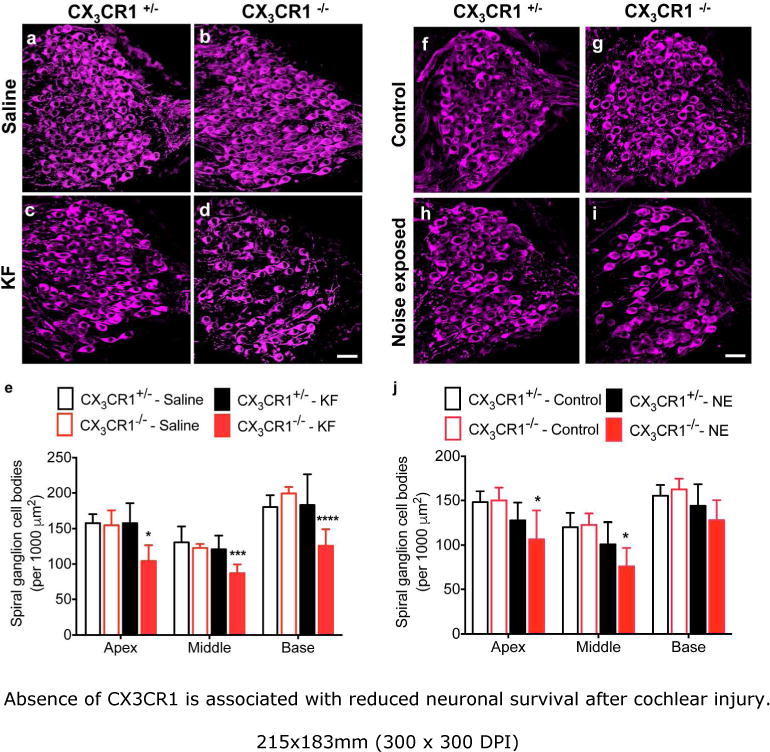
Absence of CX3CR1 is associated with reduced neuronal survival after cochlear injury. Cochlear mid-modiolar sections from the middle and basal cochlear turns immunolabeled for neurofilament (neurons, magenta) revealed significant loss of spiral ganglion cell bodies in the CX3CR1−/− mice (d, i) compared with CX3CR1+/− mice (c,h), at 60 days post KF injection or noise exposure, respectively. There was no evident loss of SGNs in saline treated or unexposed CX3CR1+/− (a,f) and CX3CR1−/− (b,g) mice. e,j, Quantitative data on SGN density (per 1,000 μm2) from all cochlear turns. Data expressed as mean±SD. n=3-9 for each genotype and treatment group for either injury models. Statistical significance was computed using two-way ANOVA followed by Tukey’s multiple comparison post hoc tests. *p<0.05, ***p <0.001, ****p <0.0001. Scale=30μm.
Enhanced production of IL-1β in CX3CR1 deficient injured cochlea
Our data show that disruption of fractalkine signaling leads to reduced SGN survival after cochlear injury. How might macrophages and/or fractalkine signaling affect the survival of cochlear afferents? Studies in the CNS have suggested that fractalkine signaling can regulate the activation status of microglia (brain resident macrophages), thereby influencing the production of several cytokines (Cardona et al., 2006; Cardona et al., 2015). Such neuronal pathology has been attributed to enhanced microglial expression of cytotoxic cytokines such as IL-1β. In order to determine whether a similar mechanism occurs in the cochlea after deletion of CX3CR1, we assessed macrophage production of IL-1β after cochlear damage. Cochlear mid-modiolar frozen sections from saline and KF-treated mice were immunolabeled for IL-1β. Saline treated (CX3CR1+/− and CX3CR1−/−) tissue staining revealed low levels of IL-1β in the spiral ganglia of the cochlea (Fig. 5c). However, following KF injury, a significant upregulation of IL-1β was detected in CX3CR1−/− mice (Fig. 5b,c). In contrast, increased IL-1β was not observed in comparably injured CX3CR1+/− mice (Fig. 5a,c). The source of IL-1β in the SG is not clear, but double immunofluorescence labeling in the spiral ligament of CX3CR1−/− mice indicate that macrophages may contribute to IL-1β production (Fig. 5d). However, other cells types such as glia and neurons might also produce IL-1β (Cardona et al., 2015). It should also be noted that IL-1β upregulation was not observed in the noise-damaged cochlea of CX3CR1−/− mice (data not shown) at any post recovery time point. The lack of IL-1β in the noise exposed CX3CR1−/− mice does rule out the possibility that IL-1β may have been upregulated earlier than 24 hours post noise exposure or that other cytokines could be responsible for SGN loss.
Figure 5.
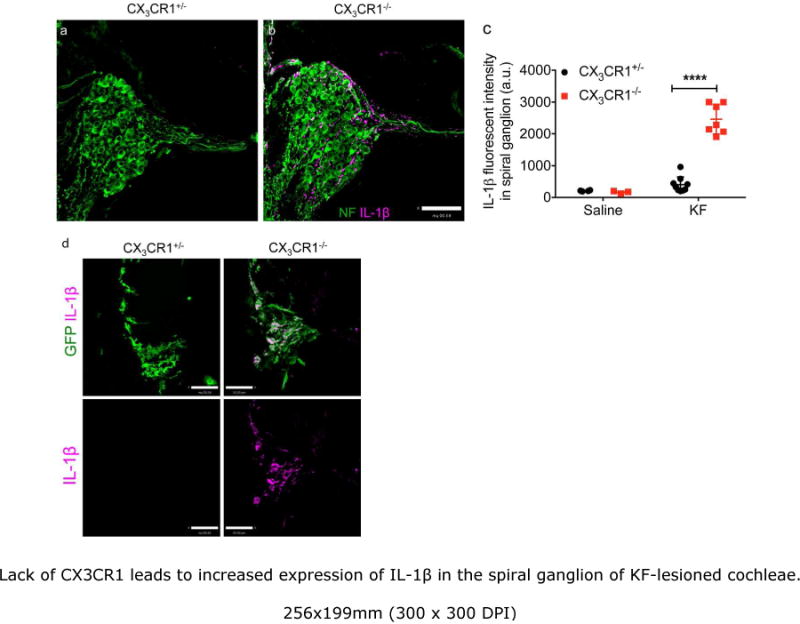
Lack of CX3CR1 leads to increased expression of IL-1β in the spiral ganglion of KF-lesioned cochleae. a,b, Representative mid-modiolar sections labeled for neurons (green) and IL-1β (magenta) from CX3CR1+/− (a) and CX3CR1−/− (b) cochleae at two months post-KF treatment. c, IL-1β fluorescent intensity in spiral ganglion. d, Mid-modiolar frozen sections from CX3CR1+/− and CX3CR1−/− cochleae stained with anti GFP (green) and IL-1β (magenta) antibodies. IL-1β immunoreactivity was detected in GFP-macrophages of CX3CR1−/− mice in the spiral ligament after two months post KF treatment, whereas no immunoreactivity was detected in the macrophages of injured CX3CR1+/− mice. Data expressed as mean±SD n=3-9,****p-value <0.0001. Scale=63 μm (a, b), 33μm (d).
4. Discussion
Our previous study demonstrated that genetic deletion of CX3CR1 in mice led to decreased macrophage numbers and enhanced neuronal loss in the cochlea after selective hair cell ablation (Kaur et al., 2015). We have now extended these results to more biologically relevant scenarios, and examined whether CX3CR1 deletion also resulted in enhanced SGN loss after kanamycin/furosemide ototoxicity or acoustic trauma. Our data show enhanced neuronal loss in both ototoxic- and noise-damaged cochleae that lack fractalkine signaling. These results point to a key role of cochlear inflammation in regulating the long-term survival of target-deprived afferent neurons.
Although the inner ear was previously thought to be immune-privileged, cochlear injury has been shown to cause recruitment of immune cells to lesion sites (Fredelius and Rask-Andersen, 1990). These immune cells have subsequently been identified as monocytes and macrophages based on the expression of CD45, F4/80, Iba-1, CD11b, and CX3CR1 (Hirose, Discolo, Keasler, and Ransohoff, 2005, Tornabene et al., 2006, Sautter et al., 2006; Ladrech et al., 2007; Sato, Shick, Ransohoff, and Hirose, 2010; Yang et al., 2015). It has been further proposed that circulating monocytes and macrophages migrate into the cochlea through the vasculature of the lateral wall, modiolus and ganglion (Hirose, Discolo, Keasler, and Ransohoff, 2005). These cells then accumulate in both sensory and non-sensory regions of the injured cochlea. The present study primarily focused on CX3CR1-postive immune cells in the organ of Corti and spiral ganglion at acute (1 day, 14 days) and delayed (60 days) recovery time periods after ototoxic drug or noise exposure. Consistent with previous findings, macrophage numbers peaked at two weeks post exposure and but then subsided in the sensory epithelium by two months. Although macrophage phagocytosis of dead hair cells was rarely observed after DT–mediated hair cell ablation in Pou4f3DTR mice (Kaur et al., 2015), we frequently noted macrophages in the scala media engulfing hair cell debris and surrounding the tectorial membrane immediately after noise overexposure. This may be attributed to the breach in the reticular lamina at 120 dB noise level (Wang, Hirose, and Liberman, 2002). Such breach can cause intermixing of cochlear fluids thereby diluting the high potassium ion concentration in the endolymph that has been proposed to be incompatible with macrophage survival (Sato, Shick, Ransohoff, and Hirose, 2010). It is also possible that macrophages in the scala media helps promote repair of the ruptured reticular lamina by clearing debris in the organ of Corti and/or by secreting unknown factors necessary for repair. In this light, it is notable that the endocochlear potential never recovers in young C57BL/6J animals exposed to 120 dB SPL noise level (Ohlemiller, Kaur, Withnell and Warchol, unpublished observations).
Macrophage numbers in the SG were significantly decreased at 24 hours after noise exposure. This observation suggests that resident macrophages of the spiral ganglion may migrate towards the osseous spiral lamina (OSL) and epithelium in response to noise exposure. There was evidence for hair cell death in the organ of Corti and fragmentation of afferent nerve fibers in the OSL at 1 day post noise exposure (Figure 2s,t). What causes macrophages to leave their SG niche and migrate towards the sensory epithelium after acoustic trauma? One possibility is that excessive release of glutamate from the inner hair cells (following prolonged depolarization) may attract macrophages. Other classical ‘find-me’ signals such as nucleotides (ATP, UTP) or fractalkine (CX3CL1) can also establish a gradient for resident macrophage/microglia attraction (Elliott et al., 2009, Ravichandran, 2010; Davalos et al., 2005; Auffray et al., 2007; Truman et al., 2008). We have previously demonstrated fractalkine expression by the inner hair cells and SGNs (Kaur et al., 2015). However, we found no difference in the macrophages numbers in the SG between CX3CR1 heterozygous, and knockout mice after 24 hours of noise exposure (data not shown) suggesting that fractalkine is unlikely to be the signal that attracts resident macrophages from the SG towards the epithelium. Macrophages express purinergic (P2RX and P2RY) receptors (Elliott et al., 2009) and ATP is released from the cochlear sensory epithelium after hair cell injury (Lahne and Gale, 2008). Future studies will focus on determining if nucleotide/P2R signaling is the ‘find-me’ signal. The spiral ganglion contained higher numbers of CX3CR1-GFP positive monocytes/macrophages at one week after noise exposure. Also, in contrast to the cochlear epithelium, monocyte/macrophage numbers in the ganglion remained elevated at longer recovery periods. It is possible that the lack of spontaneous activity and/or sensory input following hair cell loss causes macrophage recruitment in the ganglion, but the signal that triggers this response is unclear. Also, the precise nature/phenotype of recruited immune cells in the ganglion of damaged cochlea remains to be characterized. Based on studies of injured spinal cord (Kigerl et al., 2009; Shechter et al., 2009), it is possible that the resident macrophages are phagocytic and that the recruited macrophages are resolving/protective in nature. Future studies will be necessary to understand the heterogeneity of immune cells and their differential functions in damaged cochlea.
Our previous study suggested fractalkine signaling as a chemotactic factor and reported an overall reduction in macrophage numbers in the CX3CR1-deficient mice after selective hair cell ablation (Kaur et al., 2015). However, this was not the case with KF- or noise-induced hearing loss. Consistent with Sato et al., 2010, there was an increased accumulation or activation of leukocytes in the lateral wall of CX3CR1−/− mice after KF ototoxicity (data not shown). This response resembles the increased microglial activation in CX3CR1-deficient mice that has been reported in multiple mouse models of neurodegenerative and neuroinflammatory disorders such as Parkinson’s disease, Alzheimer disease, amyotrophic lateral sclerosis or diabetic retinopathy (Cardona et al., 2006; Cardona et al., 2015). Also, lack of fractalkine signaling did not significantly affect macrophage numbers in the ganglion of KF- or noise-damaged cochlea. These findings suggest that regulation of macrophage numbers by fractalkine signaling in injured cochlea is complex and that fractalkine-chemotactic properties cannot completely account for macrophage recruitment that occurs following different forms of cochlear injury. It has been proposed that CX3CR1 is an important signal that limits the migration of monocytes/macrophages from the circulation into the damaged tissue, where the blood-brain barrier is intact (Cardona et al., 2006). When the blood-brain barrier is disrupted, the roles of CX3CL1/CX3CR1 may be quite different. The damage induced by KF or loud noise exposure is often non-selective and alters the cochlear blood-labyrinth barrier (Wang, Hirose and Liberman, 2002; Tornabene et al., 2006; Hirose, Li, Ohlemiller, and Ransohoff, 2014; Schmitz, Johnson, and Santi, 2014), and we speculate that it is the integrity of cochlear vasculature that might influence the chemotactic properties of fractalkine in different cochlear injury models. Because fractalkine is readily degraded by metalloproteases (Garton et al., 2001; Wong et al., 2014), it is possible that fractalkine may have a shorter half-life and is unlikely to serve as long-range ‘find-me’ signal to leukocytes in circulation. Instead, fractalkine may primarily attract tissue resident macrophages.
Despite the lack of significant change in macrophage density, our data extend the findings of neuronal reduction in the damaged cochlea, and provide novel insights into the inflammatory reaction in the context of hearing loss using ototoxic or acoustic trauma model and dysregulated immune response in absence of CX3CR1 signaling. Previous studies of the cochlea have demonstrated extensive hair cell loss and hearing loss in CX3CR1-knockout mice (compared to CX3CR1-expressing animals) after kanamycin exposure (Sato, Shick, Ransohoff, and Hirose, 2010). Similarly, studies in the CNS have reported enhanced neurotoxicity and inflammation in mice lacking CX3CR1 in models of low-endotoxemia, Parkinson’s disease, Amyotrophic lateral sclerosis and Diabetic retinopathy (Cardona et al., 2006; Cardona et al., 2015). Such pathology has been attributed to enhanced microglial expression of inflammatory cytokines like IL-β, TNF-α and IL-6. In a search for differentially regulated inflammatory cytokines in the damaged cochlea, we observed that IL-1β was predominantly expressed in the SG, likely by macrophages, of damaged CX3CR1−/− mice. Several studies have reported IL-1β expression in the damaged cochlea (Fujioka et al., 2006; Tornabene et al., 2006; Tan, Thorne, and Vlajkovic, 2016) but absence of CX3CR1 exaggerates this response and our data suggests a correlation between IL-1β production and neuronal loss in CX3CR1 knockout mice. IL-1β, the first discovered interleukin, may play dual roles during inflammatory disorders. In most conditions, elevated concentration of IL-1 is toxic to neurons and has been associated with multiple neurodegenerative disorders. However, neuroprotective properties of IL-1β has also been suggested in deafened guinea pigs (Komeda, Roessler and Raphael, 1999). Further studies will be necessary to demonstrate the neurotoxic properties of IL-1β in damaged cochlea and the effects of IL-1β blockade on neuronal survival. Finally, the lack of IL-1β upregulation in noise-damaged cochleae of CX3CR1 knockout mice further suggests that there may be other cytotoxic cytokines or factors that can cause SGN loss.
Human populations possess two single-nucleotide polymorphisms (SNPs) in CX3CR1: V249I and T280M (Faure et al., 2000). Most individuals carry CX3CR1V249/T280, whereas 20-30% carry CX3CR1I249/M280. Biochemical studies have suggested that the polymorphic variant CX3CR1I249/M280 exhibits defective adhesive properties, therefore leading to deficits in fractalkine signaling (Faure et al., 2000; McDermott et al., 2003). These variants have been associated with multiple neurodegenerative disorders such as age-related macular degeneration (Chan et al., 2005; Schaumberg et al., 2014), Alzheimer’s disease (Lopez-Lopez et al., 2017), multiple sclerosis (Arli et al., 2013). Based on the evidence of enhanced neuronal damage in the cochlea of CX3CR1-deficient mice, it would be of clinical relevance to dissect the effects of human reference CX3CR1V249/T280 receptor and its polymorphic variant CX3CR1I249/M280 in deaf ears. Such studies may merit evaluation as risk factors for susceptibility to sensorineural hearing loss or auditory neuropathy.
In summary, our findings suggest that impaired CX3CR1 function in the cochlea may worsen the degeneration of spiral ganglion neurons after hair cell injury and that macrophages appear to play a neuroprotective role in the damaged cochlea. Since auditory neuronal loss is irreversible, targeting IL-1β and enhancing fractalkine signaling may lead to therapies for the preservation of neurons and for the success of cochlear prosthetics in hearing impaired patients.
Acknowledgments
This work was supported by the National Institutes of Health Grants R03DC015320 (TK), R01DC006283 (MEW) and P30DC004665.
References
- Arli B, Irkec C, Menevse S, Yilmaz A, Alp E. Fractalkine gene receptor polymorphism in patients with multiple sclerosis. Int J Neurosci. 2013;123:31–37. doi: 10.3109/00207454.2012.723079. [DOI] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- Bohne BA, Harding GW. Degeneration in the cochlea after noise damage: primary versus secondary events. Am J Otol. 2000;21:505–509. [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Cardona SM, Mendiola AS, Yang YC, Adkins SL, Torres V, Cardona AE. Disruption of Fractalkine Signaling Leads to Microglial Activation and Neuronal Damage in the Diabetic Retina. ASN Neuro. 2015;7 doi: 10.1177/1759091415608204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Tuo J, Bojanowski CM, Csaky KG, Green WR. Detection of CX3CR1 single nucleotide polymorphism and expression on archived eyes with age-related macular degeneration. Histol Histopathol. 2005;20:857–863. doi: 10.14670/hh-20.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, Vassileva G, Lira SA. Generation and analysis of mice lacking the chemokine fractalkine. Mol Cell Biol. 2001;21:3159–3165. doi: 10.1128/MCB.21.9.3159-3165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure S, Meyer L, Costagliola D, Vaneensberghe C, Genin E, Autran B, Delfraissy JF, McDermott DH, Murphy PM, Debré P, Théodorou I, Combadière C. Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science. 2000;287:2274–2277. doi: 10.1126/science.287.5461.2274. [DOI] [PubMed] [Google Scholar]
- Fredelius L, Rask-Andersen H. The role of macrophages in the disposal of degeneration products within the organ of Corti after acoustic overstimulation. Acta Otolaryngol. 1990;109:76–82. doi: 10.3109/00016489009107417. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Kanzaki S, Okano HJ, Masuda M, Ogawa K, Okano H. Proinflammatory cytokines expression in noise-induced damaged cochlea. J Neurosci Res. 2006;83:575–583. doi: 10.1002/jnr.20764. [DOI] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- Golub JS, Tong L, Ngyuen TB, Hume CR, Palmiter RD, Rubel EW, Stone JS. Hair cell replacement in adult mouse utricles after targeted ablation of hair cells with diphtheria toxin. J Neurosci. 2012;32:15093–15105. doi: 10.1523/JNEUROSCI.1709-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Fong AM, Swain PA, Chen S, Yu YR, Salafranca MN, Greenleaf WB, Imai T, Patel DD. Mutational analysis of the fractalkine chemokine domain. Basic amino acid residues differentially contribute to CX3CR1 binding, signaling, and cell adhesion. J Biol Chem. 2001;276:21632–21641. doi: 10.1074/jbc.M010261200. [DOI] [PubMed] [Google Scholar]
- Haskell CA, Cleary MD, Charo IF. Molecular uncoupling of fractalkine-mediated cell adhesion and signal transduction. Rapid flow arrest of CX3CR1-expressing cells is independent of G-protein activation. J Biol Chem. 1999;274:10053–10058. doi: 10.1074/jbc.274.15.10053. [DOI] [PubMed] [Google Scholar]
- Hermand P, Pincet F, Carvalho S, Ansanay H, Trinquet E, Daoudi M, Combadière C, Deterre P. Functional adhesiveness of the CX3CL1 chemokine requires its aggregation. Role of the transmembrane domain. J Biol Chem. 2001;283:30225–30234. doi: 10.1074/jbc.M802638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose K, Discolo CM, Keasler JR, Ransohoff R. Mononuclear phagocytes migrate into the murine cochlea after acoustic trauma. J Comp Neurol. 2005;489:180–194. doi: 10.1002/cne.20619. [DOI] [PubMed] [Google Scholar]
- Hirose K, Li SZ, Ohlemiller KK, Ransohoff RM. Systemic lipopolysaccharide induces cochlear inflammation and exacerbates the synergistic ototoxicity of kanamycin and furosemide. J Assoc Res Otolaryngol. 2014;15:555–570. doi: 10.1007/s10162-014-0458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson LG. Sequence of degeneration of Corti’s organ and its first-order neurons. Ann Otol Rhinol Laryngol. 1974;83:294–303. doi: 10.1177/000348947408300303. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur T, Zamani D, Tong L, Rubel EW, Ohlemiller KK, Hirose K, Warchol ME. Fractalkine Signaling Regulates Macrophage Recruitment into the Cochlea and Promotes the Survival of Spiral Ganglion Neurons after Selective Hair Cell Lesion. J Neurosci. 2015;35:15050–15061. doi: 10.1523/JNEUROSCI.2325-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeda M, Roessler BJ, Raphael Y. The influence of interleukin-1 receptor antagonist transgene on spiral ganglion neurons. Hear Res. 1999;131:1–10. doi: 10.1016/s0378-5955(99)00006-4. [DOI] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC. Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci. 2009;29:14077–14085. doi: 10.1523/JNEUROSCI.2845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladrech S, Wang J, Simonneau L, Puel JL, Lenoir M. Macrophage contribution to the response of the rat organ of Corti to amikacin. J Neurosci Res. 2007;85:1970–1979. doi: 10.1002/jnr.21335. [DOI] [PubMed] [Google Scholar]
- Lahne M, Gale JE. Damage-induced activation of ERK1/2 in cochlear supporting cells is a hair cell death-promoting signal that depends on extracellular ATP and calcium. J Neurosci. 2008;28:4918–4928. doi: 10.1523/JNEUROSCI.4914-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman MC, Kiang NY. Acoustic trauma in cats. Cochlear pathology and auditory-nerve activity. Acta Otolaryngol. 1978;358(Suppl):1–63. [PubMed] [Google Scholar]
- López-López A, Gelpi E, Lopategui DM, Vidal-Taboada JM. Association of the CX3CR1-V249I Variant with Neurofibrillary Pathology Progression in Late-Onset Alzheimer’s Disease. Mol Neurobiol. 2017 doi: 10.1007/s12035-017-0489-3. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Lucas AD, Chadwick N, Warren BF, Jewell DP, Gordon S, Powrie F, Greaves DR. The transmembrane form of the CX3CL1 chemokine fractalkine is expressed predominantly by epithelial cells in vivo. Am J Pathol. 2001;158:855–866. doi: 10.1016/S0002-9440(10)64034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DH, Halcox JP, Schenke WH, Waclawiw MA, Merrell MN, Epstein N, Quyyumi AA, Murphy PM. Association between polymorphism in the chemokine receptor CX3CR1 and coronary vascular endothelial dysfunction and atherosclerosis. Circ Res. 2001;89:401–407. doi: 10.1161/hh1701.095642. [DOI] [PubMed] [Google Scholar]
- McDermott DH, Colla JS, Kleeberger CA, Plankey M, Rosenberg PS, Smith ED, Zimmerman PA, Combadière C, Leitman SF, Kaslow RA, Goedert JJ, Berger EA, O’Brien TR, Murphy PM. Genetic polymorphism in CX3CR1 and risk of HIV disease. Science. 2003;290:2031. doi: 10.1126/science.290.5499.2031a. [DOI] [PubMed] [Google Scholar]
- Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR. Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol. 2008;9:65–89. doi: 10.1007/s10162-007-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterle EC, Campbell S. Supporting cell characteristics in long-deafened aged mouse ears. J Assoc Res Otolaryngol. 2009;10:525–544. doi: 10.1007/s10162-009-0183-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DL, Hardiman G, Copeland NG, Gilbert DJ, Jenkins N, Zlotnik A, Bazan JF. Cloning and characterization of a new type of mouse chemokine. Genomics. 1998;47:163–170. doi: 10.1006/geno.1997.5058. [DOI] [PubMed] [Google Scholar]
- Sato E, Shick HE, Ransohoff RM, Hirose K. Expression of fractalkine receptor CX3CR1 on cochlear macrophages influences survival of hair cells following ototoxic injury. J Assoc Res Otolaryngol. 2010;11:223–234. doi: 10.1007/s10162-009-0198-3. d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautter NB, Shick EH, Ransohoff RM, Charo IF, Hirose K. CC chemokine receptor 2 is protective against noise-induced hair cell death: studies in CX3CR1(+/GFP) mice. J Assoc Res Otolaryngol. 2006;7:361–372. doi: 10.1007/s10162-006-0051-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz HM, Johnson SB, Santi PA. Kanamycin-furosemide ototoxicity in the mouse cochlea: a 3-dimensional analysis. Otolaryngol Head Neck Surg. 2014;150:666–672. doi: 10.1177/0194599813519071. [DOI] [PubMed] [Google Scholar]
- Schaumberg DA, Rose L, DeAngelis MM, Semba RD, Hageman GS, Chasman DI. Prospective study of common variants in CX3CR1 and risk of macular degeneration: pooled analysis from 5 long-term studies. JAMA Ophthalmol. 2014;132:84–95. doi: 10.1001/jamaophthalmol.2013.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoendlin H. Retrograde degeneration of the cochlear nerve. Acta Otolaryngol. 1975;79:266–275. doi: 10.3109/00016487509124683. [DOI] [PubMed] [Google Scholar]
- Tan WJ, Thorne PR, Vlajkovic SM. Characterisation of cochlear inflammation in mice following acute and chronic noise exposure. Histochem Cell Biol. 2016;146:219–230. doi: 10.1007/s00418-016-1436-5. [DOI] [PubMed] [Google Scholar]
- Tong L, Strong MK, Kaur T, Juiz JM, Oesterle EC, Hume C, Warchol ME, Palmiter RD, Rubel EW. Selective deletion of cochlear hair cells causes rapid age-dependent changes in spiral ganglion and cochlear nucleus neurons. J Neurosci. 2015;35:7878–7891. doi: 10.1523/JNEUROSCI.2179-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornabene SV, Sato K, Pham L, Billings P, Keithley EM. Immune cell recruitment following acoustic trauma. Hear Res. 2006;222:115–124. doi: 10.1016/j.heares.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–5036. doi: 10.1182/blood-2008-06-162404. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J Assoc Res Otolaryngol. 2002;3:248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HS, Jaumouillé V, Heit B, Doodnauth SA, Patel S, Huang YW, Grinstein S, Robinson LA. Cytoskeletal confinement of CX3CL1 limits its susceptibility to proteolytic cleavage by ADAM10. Mol Biol Cell. 2014;25:3884–3899. doi: 10.1091/mbc.E13-11-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Vethanayagam RR, Dong Y, Cai Q, Hu BH. Activation of the antigen presentation function of mononuclear phagocyte populations associated with the basilar membrane of the cochlea after acoustic overstimulation. Neuroscience. 2015;303:1–15. doi: 10.1016/j.neuroscience.2015.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberstein Y, Liberman MC, Corfas G. Inner hair cells are not required for survival of spiral ganglion neurons in the adult cochlea. J Neurosci. 2012;32:405–410. doi: 10.1523/JNEUROSCI.4678-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]