

Abstract
Human prion diseases constitute a group of infectious and invariably fatal neurodegenerative disorders associated with misfolding of the prion protein. Variant Creutzfeldt–Jakob disease (vCJD) is a zoonotic prion disease linked to oral exposure to the infectious agent that causes bovine spongiform encephalopathy (BSE) in cattle. The most recent case of definite vCJD was heterozygous (MV) at polymorphic codon 129 of the prion protein gene PRNP while all of the previous 177 definite or probable vCJD cases who underwent genetic analysis were methionine homozygous (MM). Retrospective prevalence studies conducted on lympho‐reticular tissue suggest that the number of asymptomatic vCJD carriers in the United Kingdom might be around 1 in 2000 people. In addition, there have been four known cases of the transmission of vCJD infection via blood transfusion. For these reasons, a sensitive, reliable, and fast diagnostic test is currently needed. We describe a rapid and highly sensitive seeding conversion assay that detects disease‐associated prion protein in the brain and cerebrospinal fluid in vCJD after 48–96 h of amplification, with 100% sensitivity and specificity. This method can amplify prions from definite, probable, and possible vCJD cases from patients who are either MM or MV at PRNP‐codon 129.
Keywords: prion, Creutzfeldt–Jakob disease, diagnostic test, protein misfolding
Introduction
Human prion diseases constitute a group of fatal neurological disorders classified according to their clinical, genetic, and neuropathological features as sporadic, familial, or acquired 1. A common feature of human prion diseases is the conformational change of a normal endogenous protein, the prion protein (PrPC), to the misfolded and disease‐associated form PrPSc, which accumulates in central nervous system tissue 2. Currently, variant Creutzfeldt‐Jakob disease (vCJD) in humans is considered the only zoonotic human prion disease, with evidence suggesting that it is caused by oral exposure to the infectious agent associated with bovine spongiform encephalopathy (BSE) that affected cattle in the United Kingdom and Europe 3. Although PrPSc accumulation occurs primarily in the brain in human prion disease, a notable feature of vCJD is the detection of PrPSc in the lymphoid tissues including spleen, tonsil, and appendix 2, 4. Retrospective prevalence studies, based on the analysis of anonymized appendectomy samples for the presence of PrPSc, estimate that the number of asymptomatic carriers in the United Kingdom may be around 1 in 2000 people, which differs significantly from the number of clinically reported cases 4. This raises important concerns about the potential for further secondary transmission of vCJD and underscores the need for a sensitive, reliable, and fast diagnostic test.
With this aim, we have established an optimized assay to detect prions from suspected CJD cases in life, based on the in vitro amplification method Protein Misfolding Cyclic Amplification or PMCA.
Materials and methods
Biological samples
Cerebrospinal fluid samples
Cerebrospinal fluid (CSF) samples are sent to the laboratory on dry ice and stored at −80 °C prior to analysis. For this study CSF 14‐3‐3, tau protein and phosphorylated tau were analysed (For additional details, see supplementary material, Supplementary materials and methods). Consent for research was obtained for each sample and ethical approval was given by the MREC Scotland A (05/MRE00/67).
vCJD and sCJD brain tissues
Human brain tissue was obtained from the MRC Edinburgh Brain Bank. The specimens (frontal cortex) were sampled from a frozen half brain with the appropriate consent for research use (East of Scotland Research Ethics service REC 1, reference number 16/ES/0084).
Amplification procedure (PMCA/hsPMCA), proteolytic treatment, Western blotting, and criteria for positivity
All procedures were performed in a category 3* (*with derogation) containment laboratory with strict adherence to health and safety protocols. The preparation of normal brain homogenate, human brain derived vCJD seed, preparation of amplification mixture, and amplification procedure are given in supplementary material, Supplementary materials and methods. Details of the proteolytic treatment, western blotting, and criteria for positivity 5, 6 are also provided in supplementary material, Supplementary materials and methods.
Statistical analysis
Estimated sensitivity and specificity and 95% confidence intervals were calculated using MedCalc version 17.6.
Blinded samples
CSF samples were provided as a blinded panel. They were decoded after the amplification analysis was performed and the experimental outcome evaluated.
Results and discussion
Based on the molecular susceptibility of PrPC by the abnormal and misfolded isoform PrPSc, the in vitro conversion systems such as Protein Misfolding Cyclic Amplification (PMCA) and Real‐Time Quaking Induced Conversion (RT‐QuIC) have provided important advances in the development of diagnostic opportunities for several protein misfolding disorders including human prion diseases 7, 8, 9, 10, 11, 12, 13, 14, 15.
Prior to optimization 5, 6, our standard PMCA protocol used transgenic mouse brain homogenate expressing the human PRNP gene homozygous for methionine at codon 129, as a substrate source of normal PrPC for conversion (supplementary material, Figure S1A) 16. This substrate was seeded with vCJD prions. After proteolytic treatment (Proteinase K) and western blotting, the protease resistant core of PrPSc (PrPres) was immunodetected using the monoclonal antibody 3F4. Using a single 48 h round of standard PMCA, amplified PrPSc was detected after an initial dilution of vCJD brain seed in substrate of 1 × 10−6 – 1 × 10−7 (supplementary material, Figure S1B). This level of sensitivity for a single round of PMCA is equivalent to that reported by others leading groups 8, 10, 17.
Detection of vCJD prions in human biological fluids such as whole blood, plasma, and urine demands high sensitivity of the PMCA, achieved partially by extensive rounds of in vitro amplification, causing delay in the reporting and involving laborious manipulation. By using serial rounds of amplification, vCJD prions have recently been detected in buffy coat, complete blood and plasma from vCJD patients after 2–4 rounds of PMCA 8, 10, 17. PrPSc was also identified in urine samples of vCJD patients after extensive amplification 13. However, the potential risk of cross‐contamination of samples due to the experimental handling is a concern when clinical samples need to be examined using prolonged experimental practices.
Here we report a highly sensitive PMCA procedure (hsPMCA) to detect minute quantities of vCJD prions within a relatively short‐time period requiring minimal manipulation of the human clinical samples.
This method is able to readily detect vCJD prions diluted 100 billion‐fold (1 × 10−11) from the brain homogenate of a definite case of vCJD after a single round of amplification (supplementary material, Figure S2A). The sensitivity of hsPMCA showed a minimum of five‐fold increase when compared to standard PMCA (supplementary material, Figure S1B and S2B). The hsPMCA uses the combination of heparin (previously utilized with cellular substrates 15, 18) and transgenic substrate 5, 6, and the incorporation of beads 8, 10, 13, 17, 19 as in vitro amplification enhancers. The effect of heparin and Teflon beads on the highly efficient conversion of CJD prions, but in particular of vCJD prions, may suggest direct interactions among the seed, substrate, and these two other hsPMCA components. The glycosaminoglycan heparin for example has been shown to bind to the normal and abnormal forms of the prion protein during the amplification of mouse‐adapted strains 18. Future studies are needed to clarify the nature of the in vitro interaction between human CJD prions and the amplification promoters, and its possible pathophysiological relevance. However, it would appear from our data that it is the incorporation of Teflon beads combined with the effect of heparin that may contribute to the high sensitivity reached by hsPMCA.
In view of the sensitivity of the hsPMCA assay for the detection of vCJD prions, we carried out a small pilot study, in which blinded CSF samples from two vCJD patients (one definite and one probable case), one sporadic CJD (sCJD) patient, and one other‐neurodegenerative disease (other‐ND) case were analysed by hsPMCA. After a single round of amplification, the two vCJD samples showed PrPres. No amplified product was detected for either the sCJD or the negative control samples (Figure 1A). To estimate the optimal volume of CSF required for amplification of vCJD prions, we further analysed a range of CSF volumes from these samples. Using the hsPMCA method, PrPres was detected from a 2.5 µl volume of CSF from the definite case of vCJD. The probable case of vCJD required a higher volume (around 7 µl) of CSF (seed) to propagate prion in vitro after one round of amplification (Figure 1B). In reactions seeded with 10 and 16.8 µl CSF from the case of definite vCJD, no amplified PrPres was detected, suggesting that higher concentrations of CSF components may have inhibited amplification (Figure 1B).
Figure 1.
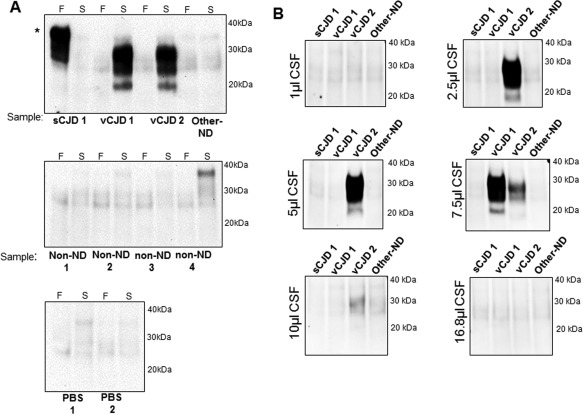
Detection of vCJD prions in cerebrospinal fluid samples by hsPMCA. (A) CSF samples from one probable and one definite vCJD (vCJD1 and vCJD2, respectively), one sporadic CJD MM1 (sCJD1), one non‐CJD neurodegenerative disease case (Other‐ND), one non‐neurodegenerative case (non‐ND), and two reactions incubated with PBS were evaluated by hsPMCA. Reactions were seeded with a final volume of 8.4 µl of sample. Non‐amplified frozen (‘F’) and amplified (‘S’) samples were analysed. (B) A range of CSF sample volumes were considered for amplification using the same CSF panel. The samples were mixed with an equal volume of substrate (83.2 µl) and normalized to a final 100 µl reaction volume. The samples were subjected to a single round of amplification. Samples were treated with Proteinase K and evaluated by western blotting using 3F4 mAb. Reference molecular mass of electrophoretic markers is shown. (*) Incomplete proteolytic digestion of PrP. [N = 5; 2 vCJD (definite and probable), 1 sCJD, 1 non‐ND, 1 Other‐ND].
The failure of the sCJD MM1 CSF, and the low amplification of MM1 and MV2K brain samples to seed detectable PrPres after hsPMCA, suggests that our methodology is not yet optimized for the detection of sCJD prions (supplementary material, Figures S2B and S2C). However, other in vitro amplification assays possess the levels of sensitivity and specificity to detect sCJD prions in CSF and in other clinical specimens 11, 12, 20.
Following on from the pilot study, we analysed a larger blinded panel of CSF samples comprising 10 definite vCJD cases, 6 sCJD MM1 cases, and 10 CSF samples from other neurodegenerative diseases, non‐neurodegenerative disease (non‐ND) and non‐CJD controls (Figure 2A, and Table 1). An additional group of 23 CSF samples from other‐ND/non‐ND/non‐CJD controls were also tested (Figure 3). In this analysis, 7.5 µl of CSF sample were included directly in the amplification mixture without any pre‐treatment of the CSF sample. After a single 48 h round of PMCA, six of the ten vCJD samples showed PrPres by western blotting. The remaining four vCJD CSF samples showed robust amplification after a second round. In contrast, none of the sCJD MM1 or the negative controls showed any evidence of PrPres formation after a single or second round of amplification (Figure 2A).
Figure 2.
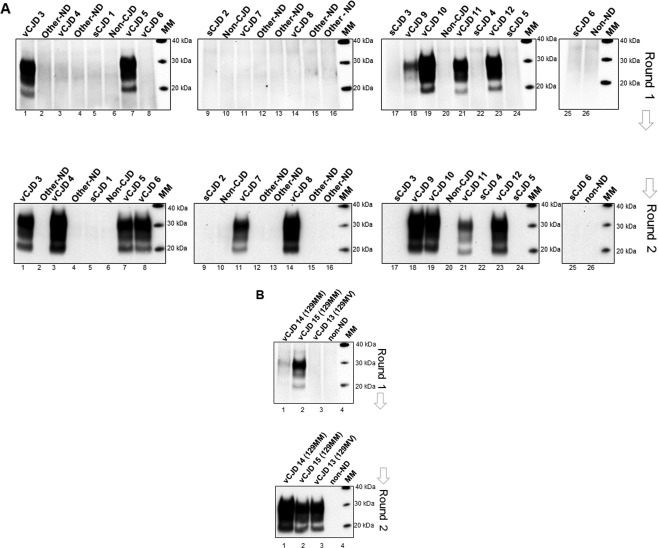
Detection of vCJD prions in CSF samples in a larger blinded panel of PRNP‐codon 129MM and MV cases by hsPMCA. (A) A second blinded panel of CSF samples were evaluated including 10 definite vCJD codon 129MM (vCJD 3–12), 6 sCJD MM1 (sCJD 1–6), and 10 CSF controls from other neurodegenerative diseases (Other–ND), non‐CJD (non‐CJD), and non‐neurodegenerative diseases (non‐ND). A volume of 7.5 μl of CSF (for each sample) was mixed with 92.5 μl volumes of substrate and subjected to amplification. (B) Analysis of two vCJD PRNP‐codon 129MM cases, one possible (vCJD 14) and one probable (vCJD 15), a definite vCJD codon 129MV case reported recently in the United Kingdom (vCJD 13), and one non‐neurodegenerative (non‐ND) control were evaluated by hsPMCA. Two amplification rounds were completed (Round 1 and 2). Samples were treated with Proteinase K and evaluated by Western blotting using 3F4 mAb. The molecular mass of electrophoretic markers (MM) is shown. [N = 30; 13 vCJD (definite, possible and probable), 6 sCJD, 5 non‐ND, 6 Other‐ND].
Table 1.
CJD and non‐CJD cases evaluated for vCJD prions by hsPMCA
| Diagnosis | Detection of vCJD prions by hsPMCA | |
|---|---|---|
| vCJD | ||
| Definite | 11 (codon 129MM) | 11/11 |
| 1 (codon 129MV) | 1/1 | |
| Possible | 1 (codon 129MM) | 1/1 |
| Probable | 2 (codon 129MM) | 2/2 |
| 15/15 | ||
| sCJD | ||
| Definite | 5 (codon 129MM) | 0/5 |
| Probable | 1 (codon 129MM) | 0/1 |
| 0/6 | ||
| Other neurodegenerative diseasesa | 21 | 0/21 |
| Non‐neurodegenerative diseases and improved cases † | 14 | 0/14 |
The ‘other neurodegenerative diseases’ group comprised cases of: Alzheimer's disease, Lewy body disease, Frontotemporal dementia, Cortical/strial/nigral degenerative disorder, Epinocerebellar ataxia, Paraneoplastic syndrome, Voltage gated K channelopathy, and Neuroaxonal dystrophy.
†The ‘other non‐neurodegenerative diseases and improved cases’ group comprise cases of: Diffuse large B cell lymphoma, Carcinomatosis of leptomeninges and brain, Progressive multifocal leukoencephalopathy, Angiotrophic lymphoma, Cerebrovascular disease, Small brain‐stem haemorrhages, Normal pressure hydrocephalus, and Intravascular B cell lymphoma.
Figure 3.
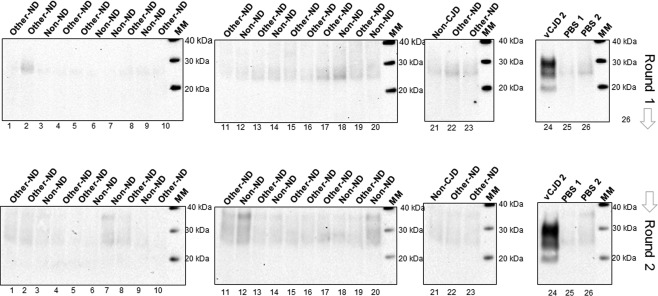
Analysis of vCJD prions in CSF samples versus non‐CJD controls by hsPMCA. Twenty three CSF samples including cases of other neurodegenerative diseases (Other–ND), non‐neurodegenerative diseases (Non‐ND), and non‐CJD controls (non‐CJD) were evaluated by hsPMCA for detection of vCJD prions. CSF from a definite vCJD case (vCJD2) and two unseeded reactions (PBS1‐PBS2) were included. Two rounds of amplification were performed (Round 1 and 2). The reactions were analysed for PrPres after PK treatment and western blotting using 3F4 mAb. The molecular mass of electrophoretic markers (MM) is shown. [N = 24; 1 vCJD, 8 non‐ND, 14 Other‐ND, 1 non‐CJD].
As a further test of the hsPMCA CSF assay, the second CSF blinded panel, also contained one possible and one probable vCJD codon 129MM case, and a third sample from the first definite codon 129MV vCJD case reported in the United Kingdom 21. The possible and probable codon 129MM cases were able to seed the hsPMCA producing detectable levels of PrPres after one round of amplification. Moreover, the CSF sample derived from the vCJD codon 129MV case showed readily detectable levels of PrPres after the second round of amplification (Figure 2B).
This study demonstrates for the first time that CSF samples from possible, probable, and definite cases of vCJD are able to propagate in vitro. The hsPMCA was able to support conversion of vCJD prions from codon 129MM and MV patients. Considering the number of samples analysed, we report an estimated sensitivity of 100% (95% CI, 69.15–100.00) and a specificity of 100% (95% CI, 89.42–100.00) of the hsPMCA assay within a 48–96 h period and with minimal manipulation of the clinical specimens. Currently, 178 cases of vCJD have been reported in the United Kingdom. It is also estimated that 1 in 2000 people are unidentified asymptomatic carriers. This study evaluates for the first time the presence of the vCJD prions in CSF samples of 15 symptomatic vCJD cases who are either MM or MV at PRNP‐codon 129, 6 sCJD, and 35 controls (supplementary material, Table S1). The relatively small absolute number of vCJD cases included in the study represents around 8.4% of the total number cases reported in the United Kingdom, not all of which have available CSF. Further analysis needs to be considered to confirm and extend the sensitivity and specificity values reported here, in a larger cohort of symptomatic, but also asymptomatic individuals. Direct comparison of our hsPMCA results with other previously published studies of CJD prions in human body fluid and tissues is challenging and complex. The use of different amplification protocols, types and substrate origin, amplification enhancers, sample treatment, and the use of human blood, urine, and CSF samples analysed complicate the comparisons and merit exhaustive review elsewhere. However, crucially, the hsPMCA assay provides a significant advantage in the development of a pre‐mortem diagnostic test for vCJD. Therefore, hsPMCA may be a valuable contribution in the surveillance for vCJD and, in combination with RT‐QuIC, may facilitate the differential diagnosis of sCJD and vCJD.
Author contributions statement
MAB conceived and designed the study in consultation with MWH. AL and MAB conducted the experiments. AG and RK processed and provided CSF samples and clinical data. MAB wrote the manuscript. All authors contributed to the editing and revision of the manuscript.
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
Supplementary materials and methods
Figure S1. Evaluation of human PrP expression levels in the humanized transgenic mouse model used for substrate and the ability to support in vitro conversion by PMCA
Figure S2. Amplification of CJD prions by hsPMCA
Table S1. Details of the CJD cases evaluated by hsPMCA
Acknowledgements
We thank the patients and families for their contribution to this work. We acknowledge Dr. Abigail Diack and Professor Jean Manson (Roslin Institute, University of Edinburgh) for providing the transgenic mouse brains utilized in this study. We thank Professor Colin Smith for the provision of the human brain tissue (MRC Edinburgh Brain Bank). We also thank Alexander Peden, and Diane Richie for reviewing the manuscript (National CJD Research & Surveillance Unit, University of Edinburgh).
This report is independent research commissioned and funded by the Department of Health Policy Research Programme and the Scottish Government. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health or the Scottish Government.
No conflicts of interest were declared.
References
- 1. Head MW. Human prion diseases: molecular, cellular and population biology. Neuropathology 2013; 33: 221–236. [DOI] [PubMed] [Google Scholar]
- 2. Head MW, Ironside JW. Review: Creutzfeldt‐Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol 2012; 38: 296–310. [DOI] [PubMed] [Google Scholar]
- 3. Will RG, Ironside JW, Zeidler M, et al A new variant of Creutzfeldt‐Jakob disease in the UK. Lancet 1996; 347: 921–925. [DOI] [PubMed] [Google Scholar]
- 4. Gill ON, Spencer Y, Richard‐Loendt A, et al Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 2013; 347: f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barria MA, Balachandran A, Morita M, et al Molecular barriers to zoonotic transmission of prions. Emerg Infect Dis 2014; 20: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ritchie DL, Barria MA, Peden AH, et al UK Iatrogenic Creutzfeldt‐Jakob disease: investigating human prion transmission across genotypic barriers using human tissue‐based and molecular approaches. Acta Neuropathol 2017; 133: 579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belondrade M, Nicot S, Béringue V, et al Rapid and highly sensitive detection of variant Creutzfeldt‐Jakob disease abnormal prion protein on steel surfaces by protein misfolding cyclic amplification: Application to prion decontamination studies. PloS One 2016; 11: e0146833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Concha‐Marambio L, Pritzkow S, Moda F, et al Detection of prions in blood from patients with variant Creutzfeldt‐Jakob disease. Sci Transl Med 2016; 8: 370ra183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fairfoul G, McGuire LI, Pal S, et al Alpha‐synuclein RT‐QuIC in the CSF of patients with alpha‐synucleinopathies. Ann Clin Transl Neurol 2016; 3: 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lacroux C, Comoy E, Moudjou M, et al Preclinical detection of variant CJD and BSE prions in blood. PLoS Pathog 2014; 10: e1004202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McGuire LI, Peden AH, Orrú CD, et al Real time quaking‐induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt‐Jakob disease. Ann Neurol 2012; 72: 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGuire LI, Poleggi A, Poggiolini I, et al Cerebrospinal fluid real‐time quaking‐induced conversion is a robust and reliable test for sporadic Creutzfeldt‐Jakob disease: An international study. Ann Neurol 2016; 80: 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moda F, Gambetti P, Notari S, et al Prions in the urine of patients with variant Creutzfeldt‐Jakob disease. New Engl J Med 2014; 371: 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shahnawaz M, Tokuda T, Waragai M, et al Development of a biochemical diagnosis of Parkinson disease by detection of alpha‐synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 2017; 74: 163–172. [DOI] [PubMed] [Google Scholar]
- 15. Yokoyama T, Takeuchi A, Yamamoto M, et al Heparin enhances the cell‐protein misfolding cyclic amplification efficiency of variant Creutzfeldt‐Jakob disease. Neurosci Lett 2011; 498: 119–123. [DOI] [PubMed] [Google Scholar]
- 16. Bishop MT, Hart P, Aitchison L, et al Predicting susceptibility and incubation time of human‐to‐human transmission of vCJD. Lancet Neurol 2006; 5: 393–398. [DOI] [PubMed] [Google Scholar]
- 17. Bougard D, Brandel J‐P, Belondrade M, et al Detection of prions in the plasma of presymptomatic and symptomatic patients with variant Creutzfeldt‐Jakob disease. Sci Transl Med 2016;8: 370ra182. 370ra182. [DOI] [PubMed] [Google Scholar]
- 18. Imamura M, Tabeta N, Kato N, et al Heparan sulfate and heparin promote faithful prion replication in vitro by binding to normal and abnormal prion proteins in protein misfolding cyclic amplification. J Biol Chem 2016; 291: 26478–26486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez‐Montalban N, Makarava N, Savtchenko R, et al Relationship between conformational stability and amplification efficiency of prions. Biochemistry 2011; 50: 7933–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Orrú CD, Bongianni M, Tonoli G, et al A test for Creutzfeldt‐Jakob disease using nasal brushings. New Engl J Med 2014; 371: 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mok T, Jaunmuktane Z, Joiner S, et al Variant Creutzfeldt‐Jakob disease in a patient with heterozygosity at PRNP Codon 129. New Engl J Med 2017; 376: 292–294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Supplementary materials and methods
Figure S1. Evaluation of human PrP expression levels in the humanized transgenic mouse model used for substrate and the ability to support in vitro conversion by PMCA
Figure S2. Amplification of CJD prions by hsPMCA
Table S1. Details of the CJD cases evaluated by hsPMCA