

Abstract
Background
Glioblastoma multiforme (GBM) is the most malignant brain tumor, and there is no effective treatment strategy. Patients with GBM have a median overall survival of only 14.6 months. Current treatment consists of safe and maximal surgical excision, followed by concurrent chemoradiotherapy and maintenance chemotherapy. There are several obstacles that hinder the effectiveness of this aggressive treatment. Temozolomide (TMZ) is an oral alkylating drug that acts through alkylating the O6 position of guanine in DNA that leads to cell death. However, the expression and enzymatic activity of the DNA repair protein MGMT limits the therapeutic benefit from treatment with TMZ. MGMT reduces the efficacy of alkylating drugs by removing the methyl or alkyl group from damaged O6-methylguanine. Expression levels of MGMT play an important role in the outcome of GBM patients. miRNAs are a group of small regulatory RNAs that control target gene expression by binding to mRNAs. miR-142-3p has been found to be an important factor in the development and maintenance of the oncogenic state.
Results
In this study, we sought to investigate whether miR-142-3p can regulate MGMT gene expression in GBM cells. Here, we show that miR-142-3p downregulates MGMT expression through binding to the 3′-UTR of MGMT mRNA, thus affecting protein translation. Responsiveness to TMZ was significantly enhanced after transfection with miR-142-3p. Overexpression of miR-142-3p also sensitized GBM cells to alkylating drugs.
Conclusion
Above all, our findings demonstrate that miR-142-3p plays a critical role in regulating MGMT expression, has great potential for future clinical applications, and acts as a new diagnostic marker for this intractable disease.
Keywords: glioblastoma, O6-methylguanine-DNA methyltransferase, miR-142-3p, carmustine, temozolomide
Introduction
Glioma is a common type of brain tumor originating from glial cells that support and nourish the brain neurons. Glioblastoma multiforme (GBM) is the most aggressive type of glioma and is classified by the World Health Organization as a grade IV tumor. GBM accounts for >50% of all gliomas.1 GBM is characterized by pleomorphism, rapid proliferation, high invasiveness, and the capability to infiltrate and destroy the surrounding areas of the brain. Angiogenesis also occurs in neighboring tissues, which further enhances tumor growth and invasion.2–4 The estimated median survival of GBM patients is 14.6 months after diagnosis.5 Surgery cannot prevent recurrence, because tumor cells that spread and infiltrate into the surrounding brain tissue cannot be completely removed. Furthermore, the effectiveness of chemotherapeutic drugs is largely limited by the blood–brain barrier (BBB), which excludes most chemotherapeutic drugs from entering the brain.6
Most chemotherapeutic drugs for GBM treatment are alkylating agents that cross the BBB, such as carmustine (bischloroethylnitrosourea [BCNU]) and temozolomide (TMZ). Alkylating agents methylate guanine on the oxygen atom, thus forming O6-methylguanine residues in the nuclear DNA. O6-methylguanine forms cytotoxic DNA cross-links, which lead to consequent apoptosis of tumor cells.7 TMZ undergoes rapid chemical conversion in the systemic circulation under physiological conditions to become the active compound 5-(3-methyltriazen-1-yl)-imidazole-4-carboxamide (MTIC). The cytotoxicity of MTIC is primarily due to alkylation of DNA, mainly occurring at the O6 and N7 positions of guanine residues.8 Currently, concomitant and adjuvant TMZ is applied with radiation in order to improve the survival of GBM patients.1,5 However, MGMT limits the therapeutic benefit from treatment with TMZ.5,7,9,10 Hypermethylation of the MGMT promoter is associated with improved survival of GBM patients.11 The MGMT gene is located on chromosome 10q26 and consists of five exons. MGMT is a DNA repair protein that removes cytotoxic O6-methylguanine or O4-alkyl-thiamine lesions in DNA induced by drugs or exogenous agents. After receiving and covalently binding methyl/alkyl groups, MGMT is irreversibly inactivated and degraded through the ubiquitination-dependent pathway.12
Recently, it has become increasingly clear that miRNAs can function as either tumor suppressors or oncogenes.13 The dysregulation of miRNA expression has been implicated in the formation of specific cancers. Most miRNAs decrease the expression of tumor suppressors, leading to increased proliferation, invasion, and angiogenesis, and decreased cell death, thus resulting in the development of tumors. The underlying mechanisms include the activation of promoters, elevated biosynthesis, and enhanced stability of miRNAs.14–17 miR-142-3p is generated and processed from the MIR142 gene, located on the human chromosome 17q22. Several studies indicate that miR-142-3p can be involved in cancers. For example, it has been shown that miR-142-3p can inhibit the expression of the RAC1 gene in hepatocellular carcinoma cell lines, which results in suppression of their migration and invasion capacity.18 Similarly, miR-142-3p is able to inhibit proliferation and invasion of cervical cancer cells by targeting FZD7.19 In non-small-cell lung carcinoma cells, miR-142-3p can repress TGFβ-induced growth inhibition by targeting TGFβR1, thus acting as an oncogene in this type of cancer.20 Research on GBM cells has shown that IL6 inhibits miR-142-3p expression via increased methylation of its promoter. At the same time, IL6 expression is also suppressed by miR-142-3p, thus forming negative-feedback loop-based interplay between these two factors.21 It has previously been demonstrated that acquisition of TMZ resistance in GBM cells can be controlled by miRNAs, such as miR-195, miR-445-3p, and miR-10a.22
Therefore, in this study, we aimed to investigate whether miR-142-3p can regulate MGMT gene expression and to explore the effect of this regulation on resistance to alkylating agents in GBM cells. We demonstrate that miR-142-3p and MGMT had negatively correlated the patterns of expression in GBM cell lines. Furthermore, we show that miR-142-3p suppresses translation of MGMT in a 3′ -UTR-dependent manner, but does not have any effect on MGMT mRNA levels. Importantly, overexpression of miR-142-3p sensitized GBM cells to the alkylating drugs BCNU and TMZ.
Materials and methods
Cell culture
GBM cell lines were acquired from the Neurological Institute of Veterans General Hospital under the supervision and approval of the Taipei Veterans General Hospital institutional review board. The GBM cell lines S1 and R1 were maintained in MEM (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% FBS, nonessential amino acids, sodium pyruvate, l-glutamine, and 1% penicillin–streptomycin at 37°C and 5% CO2. HEK293T/17 cells were cultured in DMEM containing 10% FBS (Thermo Fisher Scientific) and 1% penicillin–streptomycin.
Vector constructs
For overexpression of miR-142-3p, its precursor sequence was cloned into a pLKO.1-puro vector (Addgene, Cambridge, MA, USA). Lentiviral particles were produced by cotransfecting HEK293T cells (ATCC, Manassas, VA, USA) with pLKO.1-puro, pCMV-dR8.91 (Addgene), and pCMV-VSV-G (Addgene) using standard calcium phosphate transfection. The virus was harvested at 48 hours posttransfection and purified by filtering culture supernatant through a 0.45 μm filter. S1 and R1 cells were infected with lentiviral particles by incubation for 24 hours in the presence of 8 μg/mL polybrene.
For luciferase reporter assays, full-length MGMT 3′ -UTR (1–522 nucleotides) and its truncated forms 3′ -UTR-del1 (1–437 nucleotides), 3′ -UTR-del2 (1–229 nucleotides), and 3′ -UTR-del3 (1–127 nucleotides) were amplified by polymerase chain reaction (PCR) from cDNA using the SpeI-tagged forward primer GCACTAGTGTATGTGCAGTAGGATG and the HindIII-tagged reverse primer AGAAGCTTCTCTGTAAGCACTGACT to amplify full-length 3′ -UTR, ATAAGCTTATGCCATCACAGCCATGGACACAGC to amplify 3′ -UTR-del1, ATAAGCTTAATGGACAT-GAAAAATAGAGCAAGG to amplify 3′ -UTR-del2, and ATAAGCTTAGAAAGGGCAGACACGCTTGTTCCC to amplify 3′ -UTR-del3. PCR products were ligated into SpeI–HindIII restriction sites of pMIR-REPORT Luciferase vector (Thermo Fisher Scientific). The resultant constructs were transfected into HEK293T cells using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Reverse-transcriptase PCR and quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Total RNA (5 μg) was transcribed into cDNA using SuperScript III reverse transcriptase (RT; Thermo Fisher Scientific) according to the manufacturer’s instructions. The following pairs of primers were used to amplify the respective transcripts: MGMT (forward GCCCCTAGAACGCTTTGCGTCCCGA, reverse CCCT-GCTCACAACCAGACAGCTCCA), miR-142-3p (forward TGCAGGGCAGCAGAGGAGCTGCTGT, reverse ACTGAGGCTCTGGGCAGTCAGGACC), and GAPDH (forward: CTCATGACCACACTCCATGC, reverse: TTCAGCTCTGGGATGACCTT). For RT-PCR, Takara Taq polymerase was used and reaction products resolved by agarose gel electrophoresis. For quantitative real-time (qRT)-PCR, SYBR green PCR Master Mix was used and reactions run on an Applied Biosystems 7900HT fast real-time PCR system (Thermo Fisher Scientific).
Methylation-specific PCR
Genomic DNA was extracted from cell lysates using the standard phenol:chloroform:isoamyl alcohol extraction method. Genomic DNA was treated with bisulfite prior to performing PCR. Two 35-cycle rounds of PCR were performed: during the first PCR, MGMT promoter methylated-specific (forward TTTCGACGTTCGTAGGTTTTCGC, reverse GCACTCTTCCGAAAACGAAACG) and unmethylated-specific (forward TTTGTCTTTTGATGTTTGTAGGTTTTTGT, reverse AACTCCACACTCTTCCAAAAACAAAACA) primer pairs were used. During the second PCR, nested primers (forward GYGTTTYGGATATGTTGGGATAGTT, reverse AAACTCCRCACTCTTCCRAAAAC) were used to amplify the 10× diluted first PCR amplification product. PCR products were resolved in 2%–3% agarose gel and visualized under ultraviolet light after staining with ethidium bromide.
Luciferase assay
HEK293T cells grown in 12-well dishes (5×104 cells/well) were transfected with the appropriate pMIR-REPORT Luciferase constructs and pMIR-REPORT β-galactosidase reporter control vector for normalizing transfection efficiency, using the standard Lipofectamine 2000 protocol. Luciferase assays were performed at 48 hours posttransfection using a Dual-Light system (Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, cells were lysed in lysis solution containing 0.5 mM dithiothreitol and transferred into microcentrifuge tubes. The lysate was centrifuged at 13,200 rpm at 4°C for 2 minutes to remove cellular debris. Lysate (5 μL) was transferred into an assay cuvette, to which 12.5 μL buffer A and 50 μL buffer B (containing 100× diluted Galacton-Plus substrate) were added. Chemiluminescence detection of luciferase activity was performed using a luminometer. Samples were maintained at room temperature for 30–60 minutes. The activities of β-galactosidase in samples were determined by the addition of a Light Emission Accelerator-II.
Cell viability assay
S1 and R1 cells (1–2×104 cells/mL) were seeded into 24-well plates and incubated overnight. On the next day, fresh media with different concentrations of drugs (0, 15, 30, 60, 125, and 250 μM BCNU (Sigma-Aldrich, St Louis, MO, USA) and 0, 15, 30, 60, 125, 250, 500, and 1,000 μM TMZ (Sigma-Aldrich) were added to the wells. Each treatment was performed in triplicate. After 48 hours of incubation with drugs, aliquots of fresh media containing 500 μg/mL MTT (Sigma-Aldrich) were added to each well, followed by incubation at 37°C for 1–2 hours in the dark. Medium (200 μL) was transferred into 96-well plates and absorbance measured at 560 and 670 nm using an enzyme-linked immunosorbent assay reader. IC50 was determined by Excel and graphs were plotted using SigmaPlot.
Results
Expression profiles of MGMT protein and miR-142-3p in different GBM cell lines
Initially, we sought to investigate the expression patterns of MGMT protein and miR-142-3p in different GBM cell lines. We used in-house S1 and R1 cell lines derived from patients with first-diagnosed GBM and recurrent GBM, respectively, as well as popular commercially available GBM cell lines: LN18, U87MG, GBM8401, and GBM8901. The control fetal glial cell line SVG p12 was used as a representative of normal glial cells. Using Western blotting, we observed high levels of MGMT protein expression in GBM cell lines S1, R1, and LN18; however, its expression was not detected in U87MG, GBM8410, GBM8901, or SVG p12 cells (Figure 1B). In contrast, using qRT-PCR, we found that miR-142-3p was expressed at lower levels in the cell lines with higher levels of MGMT protein expression (Figure 1A). Based on these findings, further miR-142-3p gain-of-function experiments were performed using the cell lines S1 and R1, which had the lowest endogenous levels of miR-142-3p and at the same time expressed high levels of MGMT protein.
Figure 1.

Expression of MGMT and miR-142-3p in GBM cell lines.
Notes: (A) qRT-PCR analysis of expression of miR-142-3p in GBM cell lines S1, R1, LN18, U87MG, GBM8401, GBM8901, and fetal glial SVG p12 cells. Expression levels in GBM cell lines quantified relative to expression levels in the SVG p12 cell line. (B) Western blotting analysis of MGMT expression in GBM cell lines. β-Actin was detected as a loading control.
Abbreviations: GBM, glioblastoma multiforme; qRT-PCR quantitative real-time polymerase chain reaction.
Marginal effect of overexpression of miR‑142-3p on MGMT promoter methylation in GBM cell lines
Preliminary results indicated that GBM cell lines with higher MGMT protein expression exhibited lower miR-142-3p expression. Many previous studies have indicated that GBM patients with a methylated MGMT promoter were more resistant to alkylating agents, and thus, their treatment efficacy was reduced. Therefore, we tested the effect of miR-142-3p overexpression on the methylation status of the MGMT promoter. For this purpose, we used lentivirus vector to overexpress miR-142-3p in S1 (Figure 2C) and R1 cell lines (Figure 2D), using low and high multiplicities of infection. The effect of overexpressed miR-142-3p on methylation status of the endogenous MGMT promoter was analyzed using methylation-specific PCR (Figure 2A and B). Overexpression of miR-142-3p marginally enhanced methylation of the MGMT promoter compared with the control (shLuc) in both S1 (Figure 2A) and R1 (Figure 2B) cell lines.
Figure 2.

Overexpression of miR-142-3p increases MGMT promoter methylation in GBM cell lines.
Notes: Methylation-specific PCR assay showing methylation status of MGMT promoter in S1 cells (A) and R1 cells (B) infected with control (shLuc) and miR-142-3p-encoding recombinant lentivirus at low and high multiplicities of infection. Lane U represents amplification of unmethylated MGMT promoter and lane M represents methylated MGMT promoter. Total DNA extracted from PBMCs was used as control: lane U represents amplification of unmethylated PBMC DNA and lane M represents amplification of methylated PBMC DNA pretreated with SssI methyltransferase. qRT-PCR showing overexpression of miR-142-3p in S1 cells (C) and R1 cells (D) after infection with recombinant lentivirus at low (Low) and high (High) multiplicities of infection. Expression levels in miR-142-3p-overexpressing cells quantified relative to expression level in control cells infected with shLuc lentiviral construct, with ΔΔCt quantification with GAPDH amplification used as internal control.
Abbreviations: GBM, glioblastoma multiforme; qRT-PCR quantitative real-time polymerase chain reaction; PBMCs, peripheral blood mononuclear cells.
Overexpression of miR-142-3p does not affect MGMT mRNA, but decreases MGMT protein level
As the next step, we examined whether miR-142-3p overexpression affected the expression of MGMT at an mRNA or a protein level (Figure 3). S1 and R1 cells were infected with miR-142-3p-encoding recombinant lentiviruses, and levels of MGMT mRNA were analyzed by qRT-PCR. Overexpression of miR-142-3p did not change MGMT mRNA levels in Marginal effect of overexpression of miR-142-3p on MGMT promoter methylation in GBM cell lines either S1 (Figure 3A) or R1 (Figure 3B) cell lines. On the other hand, MGMT protein levels examined by Western blotting were noticeably reduced in both S1 (Figure 3C) and R1 (Figure 3D) cells, with more pronounced effect at the high multiplicity of infection.
Figure 3.

Overexpression of miR-142-3p does not affect MGMT mRNA, but decreases MGMT protein level.
Notes: qRT-PCR analysis of expression of MGMT mRNA in S1 (A) and R1 cells (B) infected with control (shLuc) and miR-142-3p-encoding recombinant lentiviruses at low and high multiplicities of infection. Expression levels of MGMT mRNA in miR-142-3p-infected cells quantified relative to control shLuc-infected cells and presented as mean from three independent experiments with SD error bars. Western blotting analysis of expression of MGMT protein in S1 (C) and R1 cells (D) after infection with shLuc control and miR-142-3p-encoding recombinant lentivirus at low and high multiplicities of infection. β-Actin and tubulin were used as loading controls.
Abbreviation: qRT-PCR quantitative real-time polymerase chain reaction.
miR-142-3p binds MGMT 3′-UTR directly to regulate MGMT gene
The aforementioned findings showed that increased expression of miR-142-3p suppressed the expression of MGMT protein, but MGMT mRNA expression was not affected. To determine whether this effect of miR-142-3p on MGMT expression was dependent on complementary binding to the 3′-UTR of MGMT mRNA, the 3′-UTR sequence of MGMT was cloned downstream of the luciferase-coding sequence in the luciferase-reporter vector pMIR-REPORT Luciferase. Easily transfectable HEK293T cells were cotransfected with the MGMT 3′-UTR-luciferase construct and different amounts (0, 50, 100, and 150 ng) of plasmid expressing miR-142-3p (pLKO-miR-142-3p). As shown in Figure 4A, miR-142-3p overexpression significantly decreased luminescence generated by MGMT 3′-UTR luciferase reporter in a dose-dependent manner. To confirm further that decreased luminescence in HEK293T cells was induced by the inhibitory action of miR-142-3p on MGMT 3′-UTR, cells were cotransfected with 0, 15, and 30 ng of an miR-142-3p-specific miRNA sponge construct. The effects of the miRNA sponge were determined by luminescence measurement in HEK293T cells cotransfected with MGMT 3′-UTR and miR-142-3p. miR-142-3p diminished luminescence in HEK293T cells in the absence of the miRNA sponge; however, in cells cotransfected with 15 and 30 ng miRNA sponge, miR-142-3p-induced decrease in luminescence was not observed (Figure 4B).
Figure 4.

Regulatory effect of miR-142-3p on MGMT 3′-UTR.
Notes: (A) Luciferase-reporter assay demonstrating the effect of miR-142-3p on MGMT 3′-UTR-mediated luciferase activity. The luciferase-reporter construct (pMIR-REPORT Luciferase) containing MGMT 3′-UTR insert was cotransfected into HEK293T cells with the indicated amounts of miR-142-3p-encoding plasmid (pLKO-miR-142-3p) and luminescence determined at 48 hours posttransfection. Readouts from three wells were normalized by β-galactosidase activity and expressed as mean with SD error bars. *P<0.05 (Student’s t-test). (B) Inhibitory effect of miR-142-3p overexpression on MGMT 3′-UTR-mediated luciferase activity reduced by coexpression of miR-142-3p-specific miRNA sponge. MGMT 3′-UTR-tagged pMIR-REPORT Luciferase construct was cotransfected into HEK293T cells with different amounts (0, 50, 100, and 150 ng) of pLKO-miR-142-3p and 0, 15, and 30 ng of miRNA sponge construct. Luminescence was determined at 48 hours posttransfection. Readouts from three wells were normalized by β-galactosidase activity and expressed as mean with SD error bars. *P<0.05 (Student’s t-test). (C) Design of luciferase reporter tagged with full-length MGMT 3′-UTR (3′-UTR-FL [1–522 nucleotides]) and three truncated MGMT 3′-UTRs (3′-UTR-del1, 3′-UTR-del2, and 3′-UTR-del3). (D) miR-142-3p-dependent suppression of luciferase activity mediated by FL MGMT 3′-UTR, but not by its truncated forms. pMIR-REPORT Luciferase reporter vectors with various lengths of MGMT 3′-UTR (FL, del1, del2, and del3) were cotransfected into HEK297T cells with different amounts of pLKO-miR-142-3p (0, 50, 100, and 150 ng). After 48 hours, luciferase activity was measured and normalized by β-galactosidase activity. Experiments were performed with three replicates, with mean and SD error bars shown. *P<0.05 (Student’s t-test).
As our findings implied that miR-142-3p mediated MGMT mRNA translation by direct interaction with its 3′-UTR, we tried to predict the binding site using the common miRNA target prediction tools TargetScan, PicTar, and miRanda. However, no specific miR-142-3p-binding site was identified, which could have been due to the design of these algorithms, which are based on 3′-UTR alignment with the region between the second and eighth nucleotides of miRNA, ignoring the possibility of base pairing with other regions. Therefore, we aimed to identify the miR-142-3p-interacting region within MGMT 3′-UTR experimentally. For this purpose, we constructed luciferase-reporter constructs with various truncated fragments of MGMT 3′-UTR, as shown in Figure 4C, and coexpressed them with different doses of miR-142-3p (Figure 4D). The results showed that miR-142-3p markedly reduced only the luciferase activity of the full-length MGMT 3′-UTR reporter construct (3′-UTR-FL) in a dose-dependent manner, but did not affect other reporter constructs with truncated 3′-UTRs (3′-UTR-del1, 3′-UTR-del2, and 3′-UTR-del3; Figure 4D).
miR-142-3p overexpression inhibits GBM-cell viability after treatment with alkylating agents
Current treatment regimens for GBM patients include alkylating agents that cross the BBB. However, outcomes are limited by the level and activity of MGMT expression. Given the fact that elevated miR-142-3p can inhibit MGMT protein expression (Figure 2), we analyzed the cytotoxic and survival effects of increased miR-142-3p expression in GBM cell lines. Two alkylating drugs – BCNU dissolved in ethanol and TMZ dissolved in dimethyl sulfoxide – were tested. The cell viabilities were assessed by MTT assay after incubation with different concentrations of alkylating drugs for 72 hours. As shown in Figure 5, the IC50 values of both BCNU and TMZ decreased in S1 and R1 cells infected with miR-142-3p-encoding recombinant lentivirus compared to the control. Moreover, this decrease in IC50 values was more pronounced in the case of BCNU treatment compared to TMZ treatment. As such, our findings demonstrate that increased miR-142-3p expression can reduce resistance to alkylating drugs in GBM cell lines.
Figure 5.

Overexpression of miR-142-3p leads to reduction of resistance to alkylating drugs in GBM cell lines.
Notes: S1 cells (A, C) and R1 cells (B, D) were infected with control (shLuc) and miR-142-3p-encoding recombinant lentiviruses and treated with the indicated concentrations of BCNU (A, B) or TMZ (C, D). Cell viability was determined by MTT colorimetric assay after 48 hours of incubation. Cell viability of drug-treated cells expressed as percentage of 560/670 nm absorbance values relative to untreated cells (100%). Three independent measurements were performed for each experimental condition.
Abbreviations: GBM, glioblastoma multiforme; BCNU, bis-chloroethylnitrosourea (carmustine); TMZ, temozolomide.
Discussion
It is commonly known that miRNAs are important regulatory molecules whose dysregulation is widely implicated in oncogenesis.23 Similarly to other miRNAs, miR-142-3p has been shown to be involved in different kinds of cancer by regulating various targets implicated in both oncogenic and tumor-suppressor pathways.24 For example, in non-small-cell lung carcinoma cells, miR-142-3p can repress TGFβ-induced growth inhibition by targeting the TGFβR1 receptor, thus acting as an oncogene.20 On the other hand, tumor suppressor functions of miR-142-3p are often associated with targeting the HMG family of proteins, such as HMGB1 in lung cancer25,26 and osteosarcoma,27 HMGA1 in osteosarcoma,28 and HMGA2 in GBM.21 In our recent study, we found that oncogenic cytokine IL6 promotes GBM propagation by suppressing expression of miR-142-3p.21 Given the fact that high expression of the DNA repair protein MGMT is one of the most crucial factors of poor prognosis in GBM patients,29 we sought to investigate a possible functional link between miR-142-3p and MGMT. Therefore, we tested the expression levels of miR-142-3p and MGMT protein in different GBM cell lines and found that they were negatively correlated. For our further experiments, we used two GBM cell lines, S1 and R1, derived from patients with first-diagnosed and recurrent GBM, respectively, in which miR-142-3p was expressed at very low levels and MGMT was highly expressed (Figure 1). In comparison, in several other tested GBM cell lines and the normal glial cell line SVG p12, the high level of expression of miR-142-3p correlated with the absence of expression of MGMT (Figure 1). Such patterns of expression made S1 and R1 cells a good model for miR-142-3p gain-of-function study to investigate whether miR-142-3p has an effect on MGMT expression.
Here, we demonstrated that overexpression of miR-142-3p suppressed MGMT protein expression; however, it did not have any effect on its mRNA levels (Figure 3), which indicated that miR-142-3p might inhibit translation of MGMT mRNA, but did not affect its stability. As is commonly known, the regulatory functions of miRNAs depend on the base-pairing region between the second and seventh nucleotides of miRNA, known as the seed region, which binds the 3′-UTR of a target gene. Currently, most analyses also follow this canonical miRNA-binding rule to predict the potential targets of miRNAs. However, we were unable to predict a binding site of miR-142-3p within the MGMT 3′-UTR using the available bioinformatic tools (TargetScan, PicTar, and miRanda). Nevertheless, using the luciferase-reporter assay, we have experimentally proven that miR-142-3p inhibits translation of MGMT mRNA via a 3′-UTR-binding mechanism (Figure 4). Moreover, by testing the ability of different truncated forms of MGMT 3′-UTR to mediate the repressive effect of miR-142-3p on translation, we showed that only the full-length 3′-UTR consisting of 522 nucleotides was capable of this. Given that the shortest tested truncated form comprised nucleotides 1–437 of the 3′-UTR, we could assume that nucleotides 438–522 of MGMT 3′-UTR contained an miR-142-3p-response element. miR-142-3p has partial complementarity with this region in at least two possible ways, albeit without a particular bias to the seed region (Figure 6A). In a number of recent studies, the roles of miRNAs involved in regulating downstream gene expression were not limited to the seed region, and binding sites were not only within the sequence of a target-gene 3′-UTR. miRNAs also show the inhibitory activities through binding to the 5′-cap region and open reading frames of target genes.30–33 The genome-wide mapping of miRNA–mRNA interaction sites in mouse brain revealed that a significant proportion of them were mediated by noncanonical seed-independent mechanisms.34,35 Based on a recently described mechanism in Caenorhabditis elegans, although noncanonical interaction may not be sufficient on its own to recruit the miRNA-induced silencing complex, it facilitates its recruitment by neighboring canonical seed-pairing interactions.36 Interestingly, two miRNAs, miR-181d-5p and miR-409-3p, have been shown cooperatively to suppress MGMT expression, augmenting each other’s effect.37 Therefore, in spite of the apparent noncanonical mode of interaction of miR-142-3p with MGMT 3′-UTR, it may exert its effect by cooperating with other miRNAs. In a recent study, it was shown that miR-603 directly regulated MGMT expression and targeted MGMT mRNA on five 3′-UTR sites.38 One of these sites is located adjacently to our predicted miR-142-3p response element (Figure 6A), which makes miR-603 a good candidate for a partner of miR-142-3p in a cooperative model of miRNA-induced silencing-complex recruitment.
Figure 6.
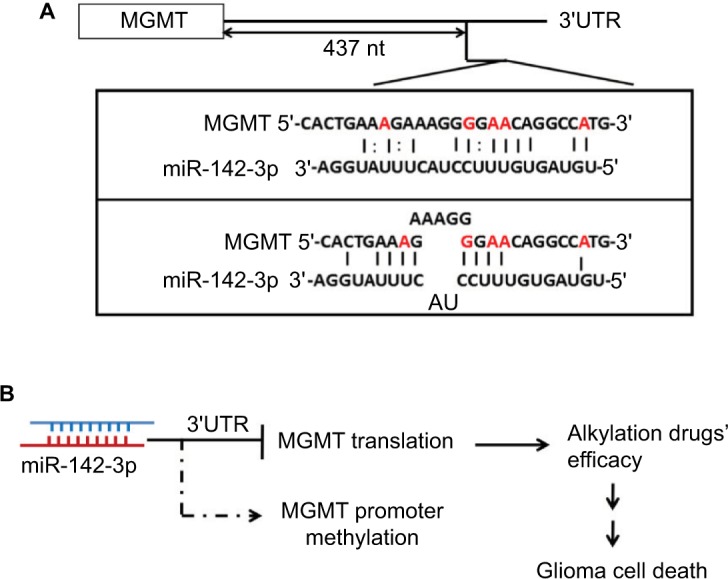
Model of interaction between miR-142-3p and MGMT.
Notes: (A) Best complementarity of miR-142-3p to 3′-UTR of MGMT mRNA within the region 438–522 nucleotides, experimentally proven to mediate the repressive effect on translation. (B) miR-142-3p inhibits MGMT mRNA translation and thus diminishes MGMT protein level by binding to MGMT 3′-UTR. Therefore, overexpression of miR-142-3p in GBM cell lines enhanced alkylating agents’ efficacy, which caused GBM cell death.
Abbreviation: GBM, glioblastoma multiforme.
Many previous studies have indicated that methylation of the MGMT promoter is associated with treatment outcomes in GBM patients.39,40 In the current study, we have shown that the ratio of methylated:unmethylated MGMT promoter was slightly increased in GBM cell lines, exogenously overexpressing miR-142-3p, whereas the MGMT mRNA level was not affected. Although this effect was very mild and did not result in a change in MGMT mRNA levels in S1 or R1 cells, it is highly possible that it could be more pronounced in a different cellular environment of other cell lines.
Currently, the best standard treatment of GBM is maximal safe surgical resection, followed by concurrent radiochemotherapy and TMZ maintenance. However, the options of chemotherapeutic drugs are limited by drug resistance. Although alkylating agents are characterized by better BBB permeability, their efficacy is decreased by the DNA-repairing capability of MGMT protein. MGMT expression is known to be negatively regulated epigenetically by methylation of its promoter. Traditionally, MGMT promoter methylation is the most commonly used predictor for alkylating agent treatment outcome.9,41 However, different studies have produced ambiguous results on the predictive power of MGMT promoter methylation, as high methylation status is not always associated with positive alkylating agent treatment outcome in GBM patients.42,43 In addition, this criterion is not applicable to 60% of GBM patients, who have unmethylated MGMT promoters, but still can be potentially responsive to alkylating agent treatment.44 Therefore, given the fact that MGMT expression is abundantly regulated at the posttranscriptional level by miRNAs, it makes this class of molecules good candidates as potential biomarkers to predict alkylating agent treatment.45 In cell viability assays, we observed that overexpression of miR-142-3p increased the resistance of S1 and R1 cells to the alkylating drugs TMZ and BCNU, commonly used in chemotherapy of GBM, which was highly consistent with the molecular effect of suppression of MGMT activity by miR-142-3p (Figure 5). We also observed that miR-142-3p overexpression was more efficient in increasing the sensitivity to BCNU compared to TMZ, and thus, it may potentially be considered to be a better prognostic factor for the former drug, a possibility that can be tested in clinical studies.
Conclusion
To summarize, in our study, we found that miR-142-3p expression was negatively correlated with MGMT protein levels in GBM cells. Overexpression of miR-142-3p in MGMT-expressing GBM cell lines S1 and R1 diminished MGMT protein levels, but not mRNA levels. Our data indicate that miR-142-3p downregulates MGMT through binding to MGMT 3′-UTR by a noncanonical seed-independent mechanism. MGMT promoter methylation status was slightly increased after miR-142-3p overexpression; however, it did not result in changes in transcription. miR-142-3p overexpression inhibited GBM cell viability after treatment with alkylating drugs, commonly used in chemotherapy for GBM, particularly to BCNU (Figure 6B). This makes miR-142-3p a good candidate as a potential biomarker and a therapeutic target in GBM.
Acknowledgments
This study was funded by the Ministry of Science and Technology (MOST; 106-2633-B-009-001, 104-0210-01-09-02, 105-0210-01-13-01, and 106-0210-01-15-02), NHRI-EX106-10621BI, Taipei Veterans General Hospital (V104B-036), and the Department of Health Cancer Center Research of Excellence (MOHW106-TDU-B-211-113001 and MOHW106-TDU-B-211-144003).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6(7):2585–2597. [PubMed] [Google Scholar]
- 2.Birnbaum T, Hildebrandt J, Nuebling G, Sostak P, Straube A. Glioblastoma-dependent differentiation and angiogenic potential of human mesenchymal stem cells in vitro. J Neurooncol. 2011;105(1):57–65. doi: 10.1007/s11060-011-0561-1. [DOI] [PubMed] [Google Scholar]
- 3.di Tomaso E, Snuderl M, Kamoun WS, et al. Glioblastoma recurrence after cediranib therapy in patients: lack of “rebound” revascularization as mode of escape. Cancer Res. 2011;71(1):19–28. doi: 10.1158/0008-5472.CAN-10-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smits M, Nilsson J, Mir SE, et al. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget. 2010;1(8):710–720. doi: 10.18632/oncotarget.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF. Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med. 2011;13:e17. doi: 10.1017/S1462399411001888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 8.Stupp R, Gander M, Leyvraz S, Newlands E. Current and future developments in the use of temozolomide for the treatment of brain tumours. Lancet Oncol. 2001;2(9):552–560. doi: 10.1016/S1470-2045(01)00489-2. [DOI] [PubMed] [Google Scholar]
- 9.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 10.Yoshino A, Ogino A, Yachi K, et al. Gene expression profiling predicts response to temozolomide in malignant gliomas. Int J Oncol. 2010;36(6):1367–1377. doi: 10.3892/ijo_00000621. [DOI] [PubMed] [Google Scholar]
- 11.Weller M, Tabatabai G, Kästner B, et al. MGMT promoter methylation is a strong prognostic biomarker for benefit from dose-intensified temozolomide rechallenge in progressive glioblastoma: the DIRECTOR trial. Clin Cancer Res. 2015;21(9):2057–2064. doi: 10.1158/1078-0432.CCR-14-2737. [DOI] [PubMed] [Google Scholar]
- 12.Srivenugopal KS, Yuan XH, Friedman HS, Ali-Osman F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-ben-zylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry. 1996;35(4):1328–1334. doi: 10.1021/bi9518205. [DOI] [PubMed] [Google Scholar]
- 13.Esquela-Kerscher A, Slack FJ. Oncomirs: microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 14.Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol Rep. 2006;16(4):845–850. [PubMed] [Google Scholar]
- 15.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65(14):6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 16.Cowland JB, Hother C, Grønbaek K. MicroRNAs and cancer. APMIS. 2007;115(10):1090–1106. doi: 10.1111/j.1600-0463.2007.apm_775.xml.x. [DOI] [PubMed] [Google Scholar]
- 17.Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65(16):7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Cai C, Wang X, Liu M, Li X, Tang H. MicroRNA-142-3p, a new regulator of RAC1, suppresses the migration and invasion of hepatocellular carcinoma cells. FEBS Lett. 2011;585(9):1322–1330. doi: 10.1016/j.febslet.2011.03.067. [DOI] [PubMed] [Google Scholar]
- 19.Deng B, Zhang Y, Zhang S, Wen F, Miao Y, Guo K. MicroRNA-142-3p inhibits cell proliferation and invasion of cervical cancer cells by targeting FZD7. Tumour Biol. 2015;36(10):8065–8073. doi: 10.1007/s13277-015-3483-2. [DOI] [PubMed] [Google Scholar]
- 20.Lei Z, Xu G, Wang L, et al. MiR-142-3p represses TGF-β-induced growth inhibition through repression of TGFβR1 in non-small cell lung cancer. FASEB J. 2014;28(6):2696–2704. doi: 10.1096/fj.13-247288. [DOI] [PubMed] [Google Scholar]
- 21.Chiou GY, Chien CS, Wang ML, et al. Epigenetic regulation of the miR-142-3p/interleukin-6 circuit in glioblastoma. Mol Cell. 2013;52(5):693–706. doi: 10.1016/j.molcel.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 22.Ujifuku K, Mitsutake N, Takakura S, et al. miR-195, miR-455-3p and miR-10a* are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010;296(2):241–248. doi: 10.1016/j.canlet.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Palanichamy JK, Rao DS. miRNA dysregulation in cancer: towards a mechanistic understanding. Front Genet. 2014;5:54. doi: 10.3389/fgene.2014.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shrestha A, Mukhametshina RT, Taghizadeh S, et al. MicroRNA-142 is a multifaceted regulator in organogenesis, homeostasis, and disease. Dev Dyn. 2017;246(4):285–290. doi: 10.1002/dvdy.24477. [DOI] [PubMed] [Google Scholar]
- 25.Xiao P, Liu WL. MiR-142-3p functions as a potential tumor suppressor directly targeting HMGB1 in non-small-cell lung carcinoma. Int J Clin Exp Pathol. 2015;8(9):10800–10807. [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Zhou X, Qiao J, Bao A. MiR-142-3p overexpression increases chemo-sensitivity of NSCLC by inhibiting HMGB1-mediated autophagy. Cell Physiol Biochem. 2017;41(4):1370–1382. doi: 10.1159/000467896. [DOI] [PubMed] [Google Scholar]
- 27.Liu K, Huang J, Ni J, et al. MALAT1 promotes osteosarcoma development by regulation of HMGB1 via miR-142-3p and miR-129-5p. Cell Cycle. 2017;16(6):578–587. doi: 10.1080/15384101.2017.1288324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu G, Wang J, Jia Y, Shen F, Han W, Kang Y. MiR-142-3p functions as a potential tumor suppressor in human osteosarcoma by targeting HMGA1. Cell Physiol Biochem. 2014;33(5):1329–1339. doi: 10.1159/000358700. [DOI] [PubMed] [Google Scholar]
- 29.Chen Y, Hu F, Zhou Y, Chen W, Shao H, Zhang Y. MGMT promoter methylation and glioblastoma prognosis: a systematic review and meta-analysis. Arch Med Res. 2013;44(4):281–290. doi: 10.1016/j.arcmed.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 30.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson AL, Burchard J, Schelter J, et al. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12(7):1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 33.Mathonnet G, Fabian MR, Svitkin YV, et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science. 2007;317(5845):1764–1767. doi: 10.1126/science.1146067. [DOI] [PubMed] [Google Scholar]
- 34.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460(7254):479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seok H, Ham J, Jang ES, Chi SW. MicroRNA target recognition: insights from transcriptome-wide non-canonical interactions. Mol Cells. 2016;39(5):375–381. doi: 10.14348/molcells.2016.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flamand MN, Gan HH, Mayya VK, Gunsalus KC, Duchaine TF. A non-canonical site reveals the cooperative mechanisms of microRNA-mediated silencing. Nucleic Acids Res. 2017;45(12):7212–7225. doi: 10.1093/nar/gkx340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khalil S, Fabbri E, Santangelo A, et al. miRNA array screening reveals cooperative MGMT-regulation between miR-181d-5p and miR-409-3p in glioblastoma. Oncotarget. 2016;7(19):28195–28206. doi: 10.18632/oncotarget.8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kushwaha D, Ramakrishnan V, Ng K, et al. A genome-wide miRNA screen revealed miR-603 as a MGMT-regulating miRNA in glioblastomas. Oncotarget. 2014;(12):4026–4039. doi: 10.18632/oncotarget.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anda T, Shabani HK, Tsunoda K, et al. Relationship between expression of O6-methylguanine-DNA methyltransferase, glutathione-S-transferase pi in glioblastoma and the survival of the patients treated with nimustine hydrochloride: an immunohistochemical analysis. Neurol Res. 2003;25(3):241–248. doi: 10.1179/016164103101201445. [DOI] [PubMed] [Google Scholar]
- 40.Brell M, Tortosa A, Verger E, et al. Prognostic significance of O6-methylguanine-DNA methyltransferase determined by promoter hypermethylation and immunohistochemical expression in anaplastic gliomas. Clin Cancer Res. 2005;11(14):5167–5174. doi: 10.1158/1078-0432.CCR-05-0230. [DOI] [PubMed] [Google Scholar]
- 41.Gusyatiner O, Hegi ME. Glioma epigenetics: from subclassification to novel treatment options. Semin Cancer Biol. 2017 Nov 21; doi: 10.1016/j.semcancer.2017.11.010. Epub. [DOI] [PubMed] [Google Scholar]
- 42.Blanc JL, Wager M, Guilhot J, et al. Correlation of clinical features and methylation status of MGMT gene promoter in glioblastomas. J Neurooncol. 2004;68(3):275–283. doi: 10.1023/b:neon.0000033385.37098.85. [DOI] [PubMed] [Google Scholar]
- 43.Sadones J, Michotte A, Veld P, et al. MGMT promoter hypermethylation correlates with a survival benefit from temozolomide in patients with recurrent anaplastic astrocytoma but not glioblastoma. Eur J Cancer. 2009;45(1):146–153. doi: 10.1016/j.ejca.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Taylor JW, Schiff D. Treatment considerations for MGMT-unmethylated glioblastoma. Curr Neurol Neurosci Rep. 2015;15(1):507. doi: 10.1007/s11910-014-0507-z. [DOI] [PubMed] [Google Scholar]
- 45.Low SY, Ho YK, Too HP, Yap CT, Ng WH. MicroRNA as potential modulators in chemoresistant high-grade gliomas. J Clin Neurosci. 2014;21(3):395–400. doi: 10.1016/j.jocn.2013.07.033. [DOI] [PubMed] [Google Scholar]