

Abstract
Epilepsy is a complex neurological syndrome characterized by neuronal hyperexcitability and sudden, synchronized electrical discharges that can manifest as seizures. It is now increasingly recognized that impaired astrocyte function and energy homeostasis play key roles in the pathogenesis of epilepsy. Excessive neuronal discharges can only happen, if adequate energy sources are made available to neurons. Conversely, energy depletion during seizures is an endogenous mechanism of seizure termination. Astrocytes control neuronal energy homeostasis through neurometabolic coupling. In this review we will discuss how astrocyte dysfunction in epilepsy leads to distortion of key metabolic and biochemical mechanisms. Dysfunctional glutamate metabolism in astrocytes can directly contribute to neuronal hyperexcitability. Closure of astrocyte intercellular gap junction coupling as observed early during epileptogenesis limits activity-dependent trafficking of energy metabolites, but also impairs clearance of the extracellular space from accumulation of K+ and glutamate. Dysfunctional astrocytes also increase the metabolism of adenosine, a metabolic product of ATP degradation that broadly inhibits energy-consuming processes as an evolutionary adaptation to conserve energy. Due to the critical role of astroglial energy homeostasis in the control of neuronal excitability, metabolic therapeutic approaches that prevent the utilization of glucose might represent a potent antiepileptic strategy. In particular, high fat low carbohydrate ‘ketogenic diets’ as well as inhibitors of glycolysis and lactate metabolism are of growing interest for the therapy of epilepsy.
Keywords: Gap junction coupling, neuron-glia interaction, adenosine, lactate, ketogenic diet
Introduction
Temporal lobe epilepsy (TLE) is one of the most common and most difficult to treat forms of epilepsy in the adult populations. It is characterized by hippocampal sclerosis (Malmgren & Thom, 2012), and surgically resected epileptogenic brain tissue has shown a correlation between gliotic scarring and the onset zones for chronic temporal lobe-derived and post-traumatic seizures. Astrocytes may influence the pathogenesis and pathophysiology of epilepsy by the homeostatic control of synaptic transmission via release of gliotransmitters such as glutamate, ATP, and D-serine (Haydon & Carmignoto, 2006; Clasadonte, Dong, Hines, & Haydon, 2013). Disruption of the glial component of the blood brain barrier (BBB), as well as glia-induced inflammation have been implicated not only in seizure generation (ictogenesis) but most importantly also in the pathophysiological processes that lead to the development of epilepsy (epileptogenesis). For example, inflammation-induced disturbance of astrocyte gap junction coupling and K+ clearance are key events in the etiology of TLE (Bedner et al., 2015). Astrocytes play an important ‘upstream’ homeostatic role in controlling the uptake, degradation and recycling of neurotransmitters. Impaired reuptake of neurotransmitters such as glutamate (Coulter & Eid, 2012), or dysregulated metabolism of neuromodulators such as adenosine (Boison, 2012) influence the development of epileptiform activity. Astrocytes form large intercellular networks and perturbations of glial metabolism can therefore affect the entire neuronal circuitry. Those network effects of astrocytes might indeed be a reason why neuronal networks in epilepsy synchronize; similarly, fluctuations in metabolic functions of glia might explain why seizures are sporadic. Whereas the role of astrocytes in epilepsy has been covered in more detail in various recent review articles (Clasadonte & Haydon, 2012; Coulter & Steinhäuser, 2015; De Lanerolle, Lee, & Spencer, 2010; Dulla, 2015; Eid, Williamson, Lee, Petroff, & De Lanerolle, 2008; Steinhäuser, Grunnet, & Carmignoto, 2016), we will focus here on astroglial energy metabolism and its role for the pathogenesis and pathophysiology of TLE.
Energy homeostasis in the epileptic brain
The epileptic brain is characterized by profound alterations in energy homeostasis, which can be cause or consequence of the epileptic state. Under baseline conditions glucose is the primary metabolic substrate of the brain and required to maintain the transmembrane potential of neurons. The astrocyte-to-neuron lactate shuttle plays a major role for the energy supply of neurons (Pellerin & Magistretti, 2012). According to this metabolic mechanism, glutamate stimulates glycolysis and the release of the glycolytic product lactate from astrocytes. Lactate dehydrogenase (LDH) is a metabolic enzyme essential for the lactate shuttle. Lactate is then taken up by neurons and metabolized via the tricarboxylic acid cycle (TCA) (Fig. 1).
Fig. 1.
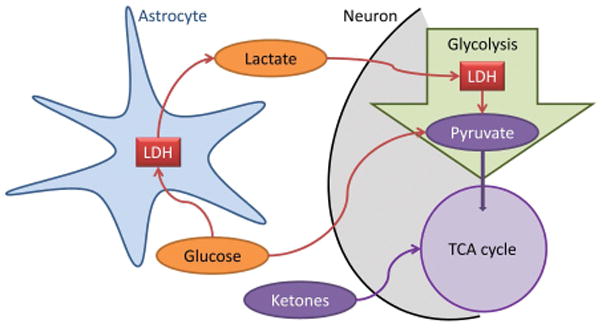
Astrocyte-neuron lactate shuttle. Glucose can directly be transported into neurons and then converted into pyruvate via glycolysis. Alternatively, glucose can be transported into astrocytes, or astrocytes can mobilize stored glycogen as an endogenous source for glucose. Astrocytes convert glucose into lactate by lactate dehydrogenase (LDH). Lactate is then released into the extracellular space and taken up into neurons, which convert lactate into pyruvate. This alternative pathway constitutes the astrocyte-neuron lactate shuttle. Ketogenic diet therapy decreases glucose and increases ketones, which are transported into neurons and bypass glycolysis and thereby directly enter the neuronal TCA cycle.
Epileptic seizures are associated with an increase in extracellular glutamate (Fig 2). Increased interictal glutamate levels and stronger seizure-induced glutamate transients were found in epileptogenic hippocampi from MTLE patients (Cavus et al., 2005; During & Spencer, 1993) and those from patients with hippocampal sclerosis (HS) displayed higher interictal extracellular glutamate levels (Petroff, Errante, Kim, & Spencer, 2003; Coulter & Eid, 2012). However, as discussed by Coulter & Eid (2012), the source of glutamate in HS is obscure, since one of the hallmarks of this pathology is loss of glutamatergic neurons in the hippocampal CA1 region. Uptake of glutamate into astrocytes (Fig. 2) triggers a sustained stimulation of astrocytic glycolysis (Bittner et al., 2011). Thereby excessive synaptic activity causes a rapid drop of glucose and a corresponding rise in lactate. During the excessive energy demands of seizures, astrocyte-derived lactate becomes an essential energy source for neurons. In line with those mechanisms patients with TLE are characterized by increased glucose uptake and metabolism during seizures, whereas the interictal periods are characterized by reduced glucose uptake and hypometabolism (Engel, Jr., Kuhl, Phelps, Rausch, & Nuwer, 1983; Engel, Jr., Kuhl, & Phelps, 1983).
Fig. 2.

Astroglial dysfunction in epilepsy. (1) Seizure activity leads to an increase in [K+]o. Downregulation of astrocyte Kir4.1 channels and mutations in the KCNJ10 gene occur in human and experimental epilepsy, resulting in decreased Kir currents. (2) Gap junctions mediate spatial redistribution of K+. Genetic ablation of gap junctions entails impaired K+ buffering and hyperactivity. (3) Dislocation of water channels contributes to impaired K+ buffering. (4) Ambient adenosine is controlled by ATP release, endonucleotidases (EN) and adenosine kinase (ADK). In epilepsy, seizures result in upregulation of ADK and decreased ambient adenosine concentration, while genetic knockdown of ADK prevents seizure activity. (5) Astrocytes are primarily responsible for glutamate uptake. Reduction of EAAT1 and EAAT2 protein is observed in human epilepsy. Elevated extracellular glutamate decreases the threshold for seizure induction. (6) Glutamate is converted into glutamine through GS. Loss of GS results in elevated extracellular glutamate levels, compromised glutamine availability, reduced GABA release from interneurons and impaired inhibition. (7) Activation mGluRs or GABABRs leads to increases in astrocytic intracellular Ca2+. Enhanced mGluR levels are observed in epileptic tissue and might amplify glutamate release from astrocytes. Modified from: Steinhäuser, Grunnet, & Carmignoto (2016), with permission.
Because astrocytes can store glycogen, they can potentially provide a significant supply of energy via the lactate shuttle (Fig. 1) to sustain the high energy demands of epileptic neuronal networks. Indeed, increased levels of glycogen have been found in the hippocampus of TLE patients (Dalsgaard, Madsen, Secher, Laursen, & Quistorff, 2007). Experimentally, in mice subjected to the convulsant methionine sulfoximine (MSO), glycogen was rapidly metabolized during seizures, but returned to high levels during the interictal phase (Bernard-Helary, Lapouble, Ardourel, Hevor, & Cloix, 2000). As an inhibitor of glutamine synthetase (GS) (Ronzio & Meister, 1968), MSO decreases the activity in the glutamate-glutamine-GABA shunt resulting in reduced glutamate clearance and increased neuronal excitability (Eid et al., 2008). Alternatively, blockade of GS may cause loss of inhibition by neurons due to impaired vesicular release of GABA (Liang, Carlson, & Coulter, 2006). Therefore, impaired function of the glutamate–glutamine cycle may not only increase extracellular glutamate but also reduce vesicular release of GABA (Fig. 2). Thus, astrocytes play a major role in coordinating glutamate homeostasis with glucose metabolism in the epileptic brain.
Astrocyte uncoupling in epilepsy – consequences for neurometabolic coupling
Astrocytes fulfill key functions in the brain including supply of nutrients to neurons, control of the extracellular ion homeostasis, modulation of the BBB permeability, coupling of neuronal activity to local blood supply, and storage and release of glycogen (Attwell et al., 2010; Pellerin et al., 2007; Abbott, Ronnback, & Hansson, 2006; Kofuji & Newman, 2004). Importantly, they modulate synaptic transmission by release, uptake, degradation and recycling of gliotransmitters. To accomplish this, the fine processes of individual astrocytes ensheath thousands of synapses (Perea, Navarrete, & Araque, 2009). Indeed, in the hippocampus about 60% of all synapses are enwrapped by astrocytes (Witcher, Kirov, & Harris, 2007). The perisynaptic astrocyte membranes contain various ion channels and transporters, which control the concentration of ions and transmitters in the synaptic cleft, regulate pH and reactive oxygen species homeostasis, and provide metabolic support to neurons (see chapters 6, 8). Despite these insights, the detailed molecular and subcellular mechanisms mediating activity-dependent astrocyte-neuron energy delivery are unclear.
Part of the ignorance mentioned above is due to the still prevailing view that astrocytes interact as individual cellular units with neurons and the vasculature (Escartin & Rouach, 2013). However, astrocytes form huge networks of electrically and metabolically coupled cells (Giaume, Koulakoff, Roux, Holcman, & Rouach, 2010), a property that considerably impedes the analysis of its functional properties (Seifert et al., 2009; Ma et al., 2015). Astrocytes in the adult brain are connected via gap junction channels composed of connexin43 (Cx43) and Cx30 (Nagy & Rash, 2000), allowing intercellular exchange of ions, second messengers, nutritional metabolites, nucleotides, amino acids, small peptides, and RNA (Harris, 2007) (Fig. 2). The relative expression levels of the two connexin isoforms vary considerably between brain regions and during development. Astrocyte coupling in the hippocampus predominately depends on Cx43 (Gosejacob et al., 2011), while thalamic astrocytes mainly express Cx30 and often lack Cx43 (Griemsmann et al., 2015). In some brain areas, astrocytes even couple abundantly with oligodendrocytes to form panglial networks (Griemsmann et al., 2015; Maglione et al., 2010; Wasseff & Scherer, 2011). Cx43, but not Cx30, is highly regulated by phosphorylation via several kinases, including protein kinase A, protein kinase C, and MAPKs (Solan & Lampe, 2009).
Formation of coupled networks is a key prerequisite for the cells’ ability to exert the functions mentioned in the beginning, e.g. clearance of the extracellular space from excess K+ and glutamate and delivery of energetic metabolites to neurons. Under physiological conditions, astrocytes take up glucose from the blood through GLUT1 transporters in their endfeet, convert it to lactate which is then shuttled through monocarboxylate transporters (MCT1, MCT4) to neurons (carrying MCT2) (Belanger, Allaman, & Magistretti, 2011). In human and experimental TLE, MCT1 is upregulated in hippocampal astrocytes (Lauritzen et al., 2012). Perivascular endfeet show an enrichment of connexins and are directly coupled with each other (Langer et al., 2017; Simard, Arcuino, Takano, Liu, & Nedergaard, 2003; Nagy, Patel, Ochalski, & Stelmack, 1999). The functional impact of perivascular astrocyte coupling on neuronal activity has been demonstrated in experiments where epileptiform activity was acutely provoked. Under those conditions, the degree of glucose trafficking through the coupled astrocytic network was enhanced to match the enhanced energy need of the hyperactive neuronal circuitry (Rouach, Koulakoff, Abudara, Willecke, & Giaume, 2008). In turn, extracellular glucose deprivation-dependent loss of synaptic hyperactivity could be rescued when filling an astrocyte with glucose or lactate. Thus, astrocyte metabolic networks were functional under these experimental conditions and crucial to fuel epileptiform activity (Rouach, Koulakoff, Abudara, Willecke, & Giaume, 2008). Additionally, involvement of gap junction channels in the intercellular spread of Ca2+ waves also favors a proconvulsive role of the astroglial network, since alterations in astrocytic coupling would influence the propagation of Ca2+ waves and, therefore, neuronal synchronization and spread of ictal activity (Gomez-Gonzalo et al., 2010).
However, according to the spatial buffering concept, the astroglial network is expected to possess antiepileptic function, since reduction of astrocytic coupling would result in accumulation of extracellular K+ and glutamate and, consequently, to neuronal depolarization and a lowered threshold for seizure generation (Fig. 2). In line with this hypothesis are results from transgenic mice with coupling-deficient astrocytes (Cx30−/− Cx43flox/flox hGFAP-Cre mice; dko mice). In these mice, the clearance of K+ and glutamate was disturbed. Consistently, these mice displayed spontaneous epileptiform events, a reduced threshold to generate epileptic activity, increased synaptic transmission and enhanced activity-induced astrocytic swelling (Wallraff et al., 2006; Pannasch et al., 2011). Thus, astroglial gap junction networks might play a duel role in epilepsy, combining pro- and antiepileptic properties.
Recent studies have shown that interastrocytic gap junction coupling is significantly impaired in epilepsy. In hippocampal specimens from patients with intractable MTLE, tracer diffusion experiments found a complete loss of coupling in the sclerotic hippocampus of patients presenting with MTLE-HS, while coupled astrocytes were abundantly present in non-HS specimens (Fig. 2). Intriguingly, these alterations were recapitulated in a mouse model of MTLE-HS and the authors further demonstrated that astrocyte uncoupling and the consequential impairment of K+ clearance temporally precede neuronal death and the onset of spontaneous seizure activity, suggesting a causative role of dysfunctional astrocytes in the development of epilepsy (Bedner et al., 2015). Consistent with this finding, impaired glial coupling and K+ buffering was described in mouse models of tuberous sclerosis complex (Xu, Zeng, & Wong, 2009) and juvenile febrile seizures (Khan et al., 2016). Obviously, under particular pathological conditions in vivo, uncoupling of astrocytes exerts pro-epileptic effects on the neuronal network, potentially due to an impaired clearance of extracellular K+ and glutamate, which indeed characterizes the sclerotic hippocampus in human and experimental MTLE-HS (cf. above; reviewed by (Steinhäuser, Seifert, & Bedner, 2012; Coulter & Eid, 2012) (Fig. 2).
Altered adenosine metabolism in epilepsy
The purine ribonucleoside adenosine is an evolutionary ancient master regulator that adjusts energy consumption to energy supplies (Newby A.C., 1984). Being a structural component of ATP, adenosine levels rise during adverse conditions that result in a drop of ATP. Therefore, adenosine levels rise as a consequence of conditions that deprive the brain of energy, such as a stroke (Pignataro et al., 2008), or under conditions that lead to the consumption of excessive amounts of energy, such as an epileptic seizure (During & Spencer, 1992). As an endogenous distress signal, adenosine has been selected as a rheostat to broadly suppress all cellular processes that consume excessive amounts of energy (Fredholm, 2007). This inhibitory role of adenosine is mediated by Gi/o-coupled adenosine A1Rs, which inhibit adenylate cyclase activity, neurotransmitter release, and lead to hyperpolarization of postsynaptic neurons (Dunwiddie & Fredholm, 1989). Because adenosine is released as a response to excessive energy expenditure during a seizure and because adenosine dampens neuronal activity, adenosine is an endogenous anticonvulsant and seizure terminator of the brain and responsible for the postictal state of suppressed EEG activity (During & Spencer, 1992; Lado & Moshe, 2008). In line with a role as endogenous anticonvulsant agent, therapies directed at augmenting adenosine in the brain, for example by transplanting engineered adenosine releasing stem cells into close vicinity of a seizure focus, robustly suppress epileptic seizures in rodent models of temporal lobe epilepsy (Boison, 2007; Fedele et al., 2004).
In the adult brain adenosine metabolism is largely under the control of astrocytes and the astrocyte-based enzyme adenosine kinase (ADK) (Etherington et al., 2009; Studer et al., 2006) (Fig. 2). By phosphorylating adenosine to AMP, ADK removes adenosine and recycles the nucleoside back into the energy pool of the cell. Astrocytes express equilibrative nucleoside transporters, which rapidly equilibrate intra- with extracellular levels of adenosine (Boswell-Casteel & Hays, 2017). Therefore, intracellular ADK, and in particular the cytoplasmic isoform ADK-S, determines the flux of adenosine through the transporter, and thereby the tissue tone of adenosine, which in turn determines the level of adenosine receptor activation (Boison, 2013) (Fig. 2). Inflammatory processes, which play a key role in the epileptogenic events that turn a healthy brain into an epileptic brain, are associated with the development of astrogliosis, a common pathological hallmark of acquired epilepsies. Astrogliosis in turn is associated with increased levels of ADK, a finding, which has been reported for experimental epilepsies as well as human temporal lobe epilepsy (Boison & Aronica, 2015) (Fig. 2). Because both the transgenic overexpression of ADK-S in the brain, as well as adeno associated virus-based overexpression of ADK-S in astrocytes reliably trigger spontaneous recurrent seizures in mice (Li, Lytle, Lan, Sandau, & Boison, 2012; Li et al., 2008; Shen et al., 2014), astrocytes and the expression levels of ADK-S play a key role in the setting of seizure thresholds.
In addition to the control of neuronal excitability, ADK plays a key role in epileptogenesis (Fedele et al., 2005; Gouder, Scheurer, Fritschy, & Boison, 2004; Li et al., 2008; Williams-Karnesky et al., 2013). This role is likely mediated by the nuclear isoform ADK-L, which controls DNA methylation by determining the flux of methyl group transfers in the S-adenosylmethionine dependent transmethylation pathway (Williams-Karnesky et al., 2013). Increased expression of ADK thereby triggers increased DNA methylation, and increased DNA methylation has independently been linked to epileptogenesis (Henshall & Kobow, 2015; Kobow et al., 2013). Conversely, therapeutic adenosine augmentation blocks DNA methylation and thereby breaks the cycle of disease progression in epilepsy (Williams-Karnesky et al., 2013). Consequently, the next generation of ADK inhibitors is currently in development as promising antiepileptogenic therapeutic approach (Toti, Osborne, Ciancetta, Boison, & Jacobson, 2016).
Metabolic therapies
Dr. Hugh Conklin’s ‘water diet’ treatment from 1922 first suggested that metabolic intervention, and in particular fasting, can be used to control seizures in epilepsy (Conklin, 1922). In the 1920s it was subsequently found that a diet composed mostly of fats, i.e. a high-fat low-carbohydrate ‘ketogenic diet’ (KD), could replicate the effects of fasting, and those beneficial effects were ascribed to the production of ketones, such as β-hydroxybutyrate (BHB), acetoacetate, and acetone in the liver (Freeman & Kossoff, 2010). Ketones feed directly into the tricarboxylic acid cycle and thereby increase mitochondrial energy production (Fig. 3). Today, KDs and related metabolic therapies are established therapies for difficult-to-treat epilepsies in addition to a wider use in a variety of neurological disorders (Stafstrom & Rho, 2012). Core mechanisms of the diet have been covered in recent review articles (Boison, 2017; Masino & Rho, 2012; Rho, 2017; Yellen, 2008). Here we will focus on those mechanisms directly related to energy homeostasis in the epileptic brain.
Fig. 3.
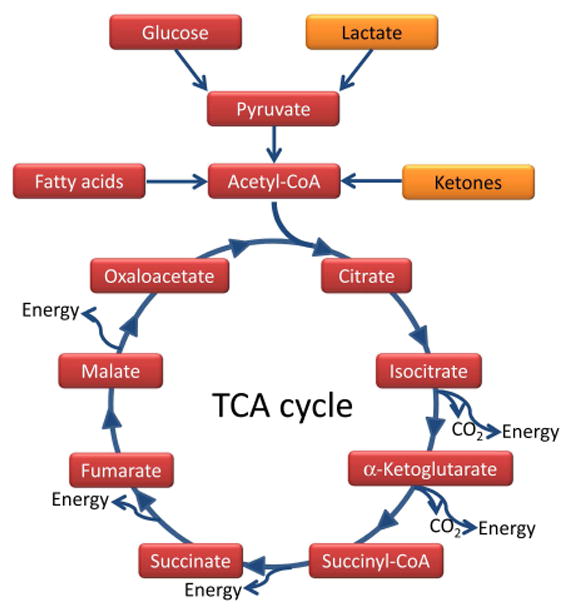
Simplified version of the TCA cycle. Conventional pathways of energy utilization (shown in red) lead from glucose via glycolysis to pyruvate and acetyl-CoA, whereas fatty acid oxidation directly forms acetyl-CoA. Acetyl-CoA feeds into the TCA cycle, where it is metabolized into CO2 and leads to the production of energy equivalents at several molecular oxidation steps. Therefore, maintaining the supply of acetyl-CoA is vital for efficient mitochondrial function and energy production. Alternative metabolites (shown in orange), such as lactate can substitute for glucose and produce pyruvate, whereas ketone bodies can be metabolized into acetyl-CoA, thereby bypassing glycolysis and boosting mitochondrial TCA cycle-based bioenergetics.
Because KD therapy improves mitochondrial function it is a rational therapeutic approach not only for the treatment of metabolic seizure disorders, but also for TLE in adulthood. The metabolic mechanisms underlying the therapeutic effects of KD therapy have best been characterized in metabolic epilepsy syndromes in the pediatric population. Thus, glucose and mitochondrial hypometabolism plays a major pathogenic role in Dravet Syndrome (DS), a catastrophic form of childhood epilepsy caused by a mutation in the voltage-sensitive Na+ channel SCN1A. In line with the metabolic benefits of KD treatment an overall positive response rate of 60 to 70% was found in a recent clinical study of 3 and 12 month old Dravet patients. A contributing molecular mechanism through which a KD in general, and ketone bodies in particular, improve mitochondrial function has been identified in a study that investigated the effects of ketone bodies on isolated brain mitochondria from Kcna1 knockout mice (Kim et al., 2015b). Ketone bodies suppressed seizures in spontaneously epileptic Kcna1 null mice, restored impaired hippocampal long-term potentiation, and raised the threshold for calcium-induced mitochondrial permeability transition (mPT). Targeted deletion of the cyclophilin D subunit of the mPT complex uncoupled the effects of ketone bodies on mPT, while mPT was directly related to the anti-seizure effects of ketone bodies (Kim et al., 2015b).
While initially considered mostly for pediatric epilepsies, KD therapy has now shown to be highly effective in adult epilepsies, including TLE (Klein, Tyrlikova, & Mathews, 2014; Kossoff, Rowley, Sinha, & Vining, 2008). The efficacy of KD therapy in TLE can best be explained by the direct correction of metabolic defects in TLE. Mechanistic studies have shown that KD therapy affects hippocampal function through ATP-sensitive K+ (KATP) channels, vesicular glutamate transporter (VGLUT), pannexin channels, and adenosine receptors (Kawamura, Ruskin, & Masino, 2016).
Because a KD has the unique ability to feed into the tricarboxylic acid cycle by bypassing glycolysis, strategies are underway to replace the rigid ketogenic dietary regimen with biochemical interventions that inhibit glycolysis or interfere with lactate formation. Thus, based on promising efficacy studies performed in acute seizure models and in the rat kindling model of TLE (Stafstrom et al., 2009; Stafstrom, Roopra, & Sutula, 2008), the glycolytic inhibitor 2-deoxy-D-glucose is currently under evaluation of antiepileptic therapy (Ockuly et al., 2012). In a seminal landmark study Tsuyoshi Inoue’s group demonstrated that the inhibition of LDH hyperpolarizes neurons and suppresses seizures in the kainate model of TLE (Sada, Lee, Katsu, Otsuki, & Inoue, 2015). Remarkably, this enzyme was also found to be a molecular target of stiripentol, a clinically-used antiepileptic drug for Dravet syndrome (Sada, Lee, Katsu, Otsuki, & Inoue, 2015). These findings are remarkable and suggest that inhibition of this metabolic pathway can mimic the effects of KD therapy, and might lead to the development of a ‘KD in a pill’.
The energy state of a cell is determined by the ATP/ADP/AMP/adenosine ratio. Importantly, a ketogenic diet was found to suppress epileptic seizures in rodent models of TLE through reducing ADK expression and augmenting adenosine signaling (Masino et al., 2011). When energy levels are low, the KATP channel, a sensor for the energy state of the cell, acts as a feedback system to restrict neuronal firing (Fig. 4). KATP activity in turn is regulated by Bcl-2-associated death promoter (BAD) protein, which plays a role in apoptosis and glucose metabolism. Genetic manipulation of BAD to reduce glucose metabolism increased the activity of neuronal KATP channels and increased seizure thresholds in vivo (Gimenez-Cassina et al., 2012). Consequently, pharmacological inhibition or genetic manipulation of KATP function abrogated or attenuated ketone-induced neuroprotection and seizure resistance (Gimenez-Cassina et al., 2012; Kim et al., 2015a). These findings support a tight mechanistic link between metabolism, BAD, KATP channel function and the control of neuronal excitation.
Fig. 4.

Proposed model for KATP-mediated anticonvulsant effects of the KD therapy. Ketones reduce glycolysis and glycolytic ATP synthesis and thereby lead to a reduction in ATP levels near the plasma membrane. This reduction of ATP can disinhibit KATP channels and thereby reduce seizure activity. Conversely, seizure activity increases ATP consumption near the plasma membrane. This constitutes a negative feedback mechanism regulating neuronal activity through KATP channels. The equilibrium at which this protective negative feedback mechanism becomes active is determined by the degree of glycolytic ATP synthesis.
Recent data suggests that KD therapy exerts additional disease modifying effects both in genetic models of metabolic epilepsy as well as in rodent models of TLE through epigenetic mechanisms. Thus, KD therapy postponed disease progression, delayed the onset of severe seizures and increased the lifespan of Kcna1 null mice, a model of progressive epilepsy (Simeone, Matthews, Rho, & Simeone, 2016). A disease modifying epigenetic mechanism of KD therapy is supported by findings that KD therapy attenuated seizure progression and ameliorated DNA methylation-mediated changes in gene expression (Kobow et al., 2013). Subsequently, it was shown that a transient KD therapy restored normal adenosine levels and global DNA methylation levels in the rat pilocarpine model of TLE, whereas epileptic control animals were adenosine deficient and hypermethylated; importantly, transient KD therapy reduced seizure activity long-term, even after reversal to a control diet (Lusardi et al., 2015). Because KD therapy increases adenosine (Lusardi et al., 2015; Masino et al., 2011) and because adenosine blocks DNA methylation (Williams-Karnesky et al., 2013) KD therapy likely exerts its disease modifying effects through an adenosine-dependent epigenetic mechanism.
Conclusions
The examples and mechanisms outlined above strongly implicate perturbations in brain energy mechanism as a major contributing factor not only for seizure generation and maintenance of the epileptic state but also for fundamental processes involved in epileptogenesis. Therefore a disturbed astrocyte-neuron metabolic crosstalk emerges as a new frontier in our understanding of this complex neurological disorder (Boison, 2016). This is an important insight because current pharmacological, target-centric therapeutic strategies fail to treat over one third of all epilepsies and fail to affect epileptogenesis. New therapies that tackle biochemical mechanisms, such as dietary interventions or more targeted biochemical manipulations, have thus the potential to improve energy metabolism and thereby to restore the equilibrium of biochemical neuron-glia interactions that are disturbed in epilepsy. Deeper understanding of the underlying biochemical mechanisms of epilepsy will lead to innovative therapeutic approaches, and – hopefully – to a cure for at least some types of epilepsy syndromes.
Main points.
Perturbations in astroglial metabolism play a major role in pathogenesis and pathophysiology of epilepsy.
Metabolic therapies, such as ketogenic diet therapy, restore glial metabolism as rational approach for seizure control.
Acknowledgments
The work of the authors quoted in this review is currently supported by grants NS084920 and NS088024 from the National Institutes of Health (DB), by generous support through the Good Samaritan Hospital Foundations (DB), and by grants (ERA-NET NEURON project BrIE; ITN project EU-GliaPhD) from the European Commission (CS).
Footnotes
Conflict of interest:
Detlev Boison has served as a consultant to UCB Biopharma and Hoffmann-La Roche.
Reference List
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature Reviews Neuroscience. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, MacVicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468(7321):232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, Deshpande T, Schramm J, Häussler U, Haas CA, Henneberger C, Theis M, Steinhäuser C. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain. 2015;138(Pt 5):1208–1222. doi: 10.1093/brain/awv067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metabolism. 2011;14(6):724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Bernard-Helary K, Lapouble E, Ardourel M, Hevor T, Cloix JF. Correlation between brain glycogen and convulsive state in mice submitted to methionine sulfoximine. Life Sci. 2000;67(14):1773–1781. doi: 10.1016/s0024-3205(00)00756-6. [DOI] [PubMed] [Google Scholar]
- Bittner CX, Valdebenito R, Ruminot I, Loaiza A, Larenas V, Sotelo-Hitschfeld T, Moldenhauer H, San MA, Gutierrez R, Zambrano M, Barros LF. Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. Journal of Neuroscience. 2011;31(12):4709–4713. doi: 10.1523/JNEUROSCI.5311-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine-based cell therapy approaches for pharmacoresistant epilepsies. Neurodegenerative Diseases. 2007;4(1):28–33. doi: 10.1159/000100356. [DOI] [PubMed] [Google Scholar]
- Boison D. Adenosine dysfunction in epilepsy. Glia. 2012;60(8):1234–1243. doi: 10.1002/glia.22285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacological Reviews. 2013;65(3):906–943. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. The Biochemistry and Epigenetics of Epilepsy: Focus on Adenosine and Glycine. Frontiers in Molecular Neuroscience. 2016;9:26. doi: 10.3389/fnmol.2016.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. New insights into the mechanisms of the ketogenic diet. Current Opinion in Neurology. 2017;30(2):187–192. doi: 10.1097/WCO.0000000000000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Aronica E. Comorbidities in Neurology: Is adenosine the common link? Neuropharmacology. 2015;97:18–34. doi: 10.1016/j.neuropharm.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell-Casteel RC, Hays FA. Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids. 2017;36(1):7–30. doi: 10.1080/15257770.2016.1210805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, Krystal JH, Spencer DD, bi-Saab WM. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Annals of Neurology. 2005;57(2):226–235. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Clasadonte J, Dong J, Hines DJ, Haydon PG. Astrocyte control of synaptic NMDA receptors contributes to the progressive development of temporal lobe epilepsy. Proceedings of the National Academy of Sciences of the US A. 2013;110(43):17540–17545. doi: 10.1073/pnas.1311967110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clasadonte J, Haydon PG. Astrocytes and Epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- Conklin HW. Cause and treatment of epilepsy. The Journal of the American Osteopathic Association. 1922;(26):11–18. [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60(8):1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter DA, Steinhäuser C. Role of astrocytes in epilepsy. Cold Spring Harbor Perspectives in Medicine. 2015;5(3):a022434. doi: 10.1101/cshperspect.a022434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard MK, Madsen FF, Secher NH, Laursen H, Quistorff B. High glycogen levels in the hippocampus of patients with epilepsy. Journal of Cerebral Blood Flow & Metabolism. 2007;27(6):1137–1141. doi: 10.1038/sj.jcbfm.9600426. [DOI] [PubMed] [Google Scholar]
- De Lanerolle NC, Lee TS, Spencer DD. Astrocytes and epilepsy. Neurotherapeutics. 2010;7(4):424–438. doi: 10.1016/j.nurt.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulla CG. Losing Touch With Your Astrocytes Can Cause Epilepsy. Epilepsy Currents. 2015;15(6):349–350. doi: 10.5698/1535-7511-15.6.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Fredholm BB. Adenosine A1 receptors inhibit adenylate cyclase activity and neurotransmitter release and hyperpolarize pyramidal neurons in rat hippocampus. Journal of Pharmacology and Experimental Therapeutics. 1989;249(1):31–37. [PubMed] [Google Scholar]
- During MJ, Spencer DD. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Annals in Neurology. 1992;32(5):618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341(8861):1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, De Lanerolle NC. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain. 2008;131(Pt 8):2061–2070. doi: 10.1093/brain/awn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid T, Williamson A, Lee TS, Petroff OA, De Lanerolle NC. Glutamate and astrocytes - key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49(Suppl 2):42–52. doi: 10.1111/j.1528-1167.2008.01492.x. [DOI] [PubMed] [Google Scholar]
- Engel J, Jr, Kuhl DE, Phelps ME. Regional brain metabolism during seizures in humans. Advances in Neurology. 1983;34:141–148. [PubMed] [Google Scholar]
- Engel J, Jr, Kuhl DE, Phelps ME, Rausch R, Nuwer M. Local cerebral metabolism during partial seizures. Neurology. 1983;33(4):400–413. doi: 10.1212/wnl.33.4.400. [DOI] [PubMed] [Google Scholar]
- Escartin C, Rouach N. Astroglial networking contributes to neurometabolic coupling. Frontiers in Neuroenergetics. 2013;5(4) doi: 10.3389/fnene.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherington LA, Patterson GE, Meechan L, Boison D, Irving AJ, Dale N, Frenguelli BG. Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology. 2009;56(2):429–437. doi: 10.1016/j.neuropharm.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele DE, Gouder N, Guttinger M, Gabernet L, Scheurer L, Rulicke T, Crestani F, Boison D. Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain. 2005;128(Pt 10):2383–2395. doi: 10.1093/brain/awh555. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Scheurer L, Simpson EM, Mohler H, Brustle O, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neuroscience Letters. 2004;370(2–3):160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death &Differentiation. 2007;14(7):1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- Freeman JM, Kossoff EH. Ketosis and the ketogenic diet, 2010: advances in treating epilepsy and other disorders. Advances in Pediatrics. 2010;57(1):315–329. doi: 10.1016/j.yapd.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nature Reviews Neuroscience. 2010;11(2):87–99. doi: 10.1038/nrn2757. [DOI] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Martinez-Francois JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012;74(4):719–730. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, De Curtis M, Ratto GM, Carmignoto G. An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biology. 2010;8(4):e1000352. doi: 10.1371/journal.pbio.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosejacob D, Dublin P, Bedner P, Hüttmann K, Zhang J, Tress O, Willecke K, Pfrieger F, Steinhäuser C, Theis M. Role of astroglial connexin30 in hippocampal gap junction coupling. Glia. 2011;59(3):511–519. doi: 10.1002/glia.21120. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. Journal of Neuroscience. 2004;24(3):692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griemsmann S, Hoft SP, Bedner P, Zhang J, von SE, Beinhauer A, Degen J, Dublin P, Cope DW, Richter N, Crunelli V, Jabs R, Willecke K, Theis M, Seifert G, Kettenmann H, Steinhäuser C. Characterization of Panglial Gap Junction Networks in the Thalamus, Neocortex, and Hippocampus Reveals a Unique Population of Glial Cells. Cerebral Cortex. 2015;25(10):3420–3433. doi: 10.1093/cercor/bhu157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. Connexin channel permeability to cytoplasmic molecules. Progress in Biophysics & Molecular Biology. 2007;94(1–2):120–143. doi: 10.1016/j.pbiomolbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews. 2006;86(3):1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Kobow K. Epigenetics and Epilepsy. Cold Spring Harbor Perspectives in Medicine. 2015;5(12) doi: 10.1101/cshperspect.a022731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura MJ, Ruskin DN, Masino SA. Metabolic Therapy for Temporal Lobe Epilepsy in a Dish: Investigating Mechanisms of Ketogenic Diet using Electrophysiological Recordings in Hippocampal Slices. Frontiers in MolecularN euroscience. 2016;9:112. doi: 10.3389/fnmol.2016.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan D, Dupper A, Deshpande T, Graan PN, Steinhauser C, Bedner P. Experimental febrile seizures impair interastrocytic gap junction coupling in juvenile mice. Journal of Neuroscience Research. 2016;94(9):804–813. doi: 10.1002/jnr.23726. [DOI] [PubMed] [Google Scholar]
- Kim DY, Abdelwahab MG, Lee SH, O’Neill D, Thompson RJ, Duff HJ, Sullivan PG, Rho JM. Ketones prevent oxidative impairment of hippocampal synaptic integrity through KATP channels. PLoS ONE. 2015a;10(4):e0119316. doi: 10.1371/journal.pone.0119316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Simeone KA, Simeone TA, Pandya JD, Wilke JC, Ahn Y, Geddes JW, Sullivan PG, Rho JM. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Annals of Neurology. 2015b;78(1):77–87. doi: 10.1002/ana.24424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein P, Tyrlikova I, Mathews GC. Dietary treatment in adults with refractory epilepsy: a review. Neurology. 2014;83(21):1978–1985. doi: 10.1212/WNL.0000000000001004. [DOI] [PubMed] [Google Scholar]
- Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, Fritzsche I, Hauke J, Hahnen E, Coras R, Muhlebner A, El-Osta A, Blumcke I. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathologica. 2013;126(5):741–756. doi: 10.1007/s00401-013-1168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129(4):1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Rowley H, Sinha SR, Vining EP. A prospective study of the modified Atkins diet for intractable epilepsy in adults. Epilepsia. 2008;49(2):316–319. doi: 10.1111/j.1528-1167.2007.01256.x. [DOI] [PubMed] [Google Scholar]
- Lado FA, Moshe SL. How do seizures stop? Epilepsia. 2008;49(10):1651–1664. doi: 10.1111/j.1528-1167.2008.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer J, Gerkau NJ, Derouiche A, Kleinhans C, Moshrefi-Ravasdjani B, Fredrich M, Kafitz KW, Seifert G, Steinhauser C, Rose CR. Rapid sodium signaling couples glutamate uptake to breakdown of ATP in perivascular astrocyte endfeet. Glia. 2017;65(2):293–308. doi: 10.1002/glia.23092. [DOI] [PubMed] [Google Scholar]
- Lauritzen F, Perez EL, Melillo ER, Roh JM, Zaveri HP, Lee TS, Wang Y, Bergersen LH, Eid T. Altered expression of brain monocarboxylate transporter 1 in models of temporal lobe epilepsy. Neurobiology of Disease. 2012;45(1):165–176. doi: 10.1016/j.nbd.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Lytle N, Lan JQ, Sandau US, Boison D. Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia. 2012;60(1):83–95. doi: 10.1002/glia.21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. Journal of Clinical Investigation. 2008;118(2):571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SL, Carlson GC, Coulter DA. Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. Journal of Neuroscience. 2006;26(33):8537–8548. doi: 10.1523/JNEUROSCI.0329-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusardi TA, Akula KK, Coffman SQ, Ruskin DN, Masino SA, Boison D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology. 2015;99:500–509. doi: 10.1016/j.neuropharm.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Buckalew R, Du Y, Kiyoshi CM, Alford CC, Wang W, McTigue DM, Enyeart JJ, Terman D, Zhou M. Gap junction coupling confers isopotentiality on astrocyte syncytium. Glia. 2015;64(2):214–226. doi: 10.1002/glia.22924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglione M, Tress O, Haas B, Karram K, Trotter J, Willecke K, Kettenmann H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia. 2010;58(9):1104–1117. doi: 10.1002/glia.20991. [DOI] [PubMed] [Google Scholar]
- Malmgren K, Thom M. Hippocampal sclerosis--origins and imaging. Epilepsia. 2012;53(Suppl 4):19–33. doi: 10.1111/j.1528-1167.2012.03610.x. [DOI] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. Journal of Clinical Investigation. 2011;121(7):2679–2683. doi: 10.1172/JCI57813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Rho JM. Mechanisms of Ketogenic Diet Action. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- Nagy JI, Patel D, Ochalski PAY, Stelmack GL. Connexin30 in rodent, cat and human brain: Selective expression in gray matter astrocytes, co-localization with connexin43 at gap junctions and late developmental appearance. Neuroscience. 1999;88(2):447–468. doi: 10.1016/s0306-4522(98)00191-2. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Rash JE. Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Research Reviews. 2000;32(1):29–44. doi: 10.1016/s0165-0173(99)00066-1. [DOI] [PubMed] [Google Scholar]
- Newby AC. Adenosine and the concept of ‘retaliatory metabolites. Trends in Biochemical Sciences. 1984;(9):42–44. [Google Scholar]
- Ockuly JC, Gielissen JM, Levenick CV, Zeal C, Groble K, Munsey K, Sutula TP, Stafstrom CE. Behavioral, cognitive, and safety profile of 2-deoxy-2-glucose (2DG) in adult rats. Epilepsy Research. 2012;101(3):246–252. doi: 10.1016/j.eplepsyres.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N. Astroglial networks scale synaptic activity and plasticity. Proceedings of the National Academy of Sciences of the US A. 2011;108(20):8467–8472. doi: 10.1073/pnas.1016650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55(12):1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. Journal of Cerebral Blood Flow & Metabolism. 2012;32(7):1152–1166. doi: 10.1038/jcbfm.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A. Tripartite synapses: astrocytes process and control synaptic information. Trends in Neurosciences. 2009;32(8):421–431. doi: 10.1016/j.tins.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Petroff OA, Errante LD, Kim JH, Spencer DD. N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology. 2003;60(10):1646–1651. doi: 10.1212/01.wnl.0000068020.85450.8b. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Maysami S, Studer FE, Wilz A, Simon RP, Boison D. Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. Journal of Cerebral Blood Flow & Metabolism. 2008;28(1):17–23. doi: 10.1038/sj.jcbfm.9600499. [DOI] [PubMed] [Google Scholar]
- Rho JM. How does the ketogenic diet induce anti-seizure effects? Neuroscience Letters. 2017;637:4–10. doi: 10.1016/j.neulet.2015.07.034. [DOI] [PubMed] [Google Scholar]
- Ronzio RA, Meister A. Phosphorylation of methionine sulfoximine by glutamine synthetase. Proceedings of the National Academy of Sciences of the US A. 1968;59(1):164–170. doi: 10.1073/pnas.59.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322(5907):1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Sada N, Lee S, Katsu T, Otsuki T, Inoue T. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science. 2015;347(6228):1362–1367. doi: 10.1126/science.aaa1299. [DOI] [PubMed] [Google Scholar]
- Seifert G, Hüttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhäuser C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. Journal of Neuroscience. 2009;29(23):7474–7488. doi: 10.1523/JNEUROSCI.3790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Sun H, Hanthorn MM, Zhi Z, Lan JQ, Poulsen DJ, Wang RK, Boison D. Overexpression of adenosine kinase in cortical astrocytes and focal neocortical epilepsy in mice. Journal of Neurosurgery. 2014;120(3):628–638. doi: 10.3171/2013.10.JNS13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. Journal of Neuroscience. 2003;23(27):9254–9262. doi: 10.1523/JNEUROSCI.23-27-09254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone KA, Matthews SA, Rho JM, Simeone TA. Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia. 2016;57(8):e178–e182. doi: 10.1111/epi.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochemical Journal. 2009;419(2):261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, Sutula TP. Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Annals of Neurology. 2009;65(4):435–447. doi: 10.1002/ana.21603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Rho JM. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Frontiers in Pharmacology. 2012;3:59. doi: 10.3389/fphar.2012.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Roopra A, Sutula TP. Seizure suppression via glycolysis inhibition with 2-deoxy-D-glucose (2DG) Epilepsia. 2008;49(Suppl 8):97–100. doi: 10.1111/j.1528-1167.2008.01848.x. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Grunnet M, Carmignoto G. Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience. 2016;323:157–169. doi: 10.1016/j.neuroscience.2014.12.047. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60(8):1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience. 2006;142(1):125–137. doi: 10.1016/j.neuroscience.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Toti KS, Osborne D, Ciancetta A, Boison D, Jacobson KA. South (S)- and North (N)-Methanocarba-7-Deazaadenosine Analogues as Inhibitors of Human Adenosine Kinase. Journal of Medicinal Chemistry. 2016;59(14):6860–6877. doi: 10.1021/acs.jmedchem.6b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. Journal of Neuroscience. 2006;26(20):5438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasseff SK, Scherer SS. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiology of Disease. 2011;42(3):506–513. doi: 10.1016/j.nbd.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. Journal of Clinical Investigation. 2013;123(8):3552–3563. doi: 10.1172/JCI65636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher MR, Kirov SA, Harris KM. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia. 2007;5(1):13–23. doi: 10.1002/glia.20415. [DOI] [PubMed] [Google Scholar]
- Xu L, Zeng LH, Wong M. Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiology of Disease. 2009;34(2):291–299. doi: 10.1016/j.nbd.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. Ketone bodies, glycolysis, and KATP channels in the mechanism of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):80–82. doi: 10.1111/j.1528-1167.2008.01843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]