

Abstract
Charcot-Marie-Tooth disease (CMT) is an umbrella term for inherited neuropathies affecting an estimated 1 in 2500 people. Over 120 CMT and related genes have been identified and clinical gene panels often contain more than 100 genes. Such a large genomic space will invariantly yield variants of uncertain clinical significance (VUS) in nearly any person tested. This rise in number of VUS creates major challenges for genetic counseling. Additionally, fewer individual variants in known genes are being published as the academic merit is decreasing, and most testing now happens in clinical laboratories, which typically do not correlate their variants with clinical phenotypes. For CMT, we aim to encourage and facilitate the global capture of variant data to gain a large collection of alleles in CMT genes, ideally in conjunction with phenotypic information. The Inherited Neuropathy Variant Browser provides user-friendly open access to currently reported variation in CMT genes. Geneticists, physicians, and genetic counselors can enter variants detected by clinical tests or in research studies in addition to genetic variation gathered from published literature, which are then submitted to ClinVar bi-annually. Active participation of the broader CMT community will provide an advance over existing resources for interpretation of CMT genetic variation.
INTRODUCTION
Charcot-Marie-Tooth disease (CMT) and related disorders represent inherited conditions affecting the peripheral nervous system (Reilly et al. 2011; Saporta and Shy 2013a). Initially reported in the late 19th century by physicians Jean Martin Charcot, Pierre Marie, and Howard Henry Tooth, CMT is now considered amongst the most common inherited neurological disorders and affects an estimated 1 in 2500 people (Reilly et al. 2011). Typical findings include high arches of the feet, as well as muscle weakness and atrophy, sensory loss, and deep tendon reflexes, all more pronounced in the distal extremities (Pagon et al. 1993). These findings result from the progressive, length-dependent, degeneration of motor and/or sensory axons.
CMT is clinically and genetically heterogeneous. The clinical onset, rate of progression, nerve conduction velocities, and involvement of motor and sensory axons differ among the subtypes (Reilly et al. 2011; Saporta and Shy 2013b). The motor nerve conduction velocities in the arms separate CMT in demyelinating (CMT1), axonal (CMT2), and intermediate forms (Dyck and Lambert 1968; Reilly et al. 2011; Saporta and Shy 2013b). Each major subtype is also classified based on mode of inheritance and genetic etiology. The advances in gene discovery enabled by next-generation sequencing (NGS) have revealed the extent of locus and allele heterogeneity, with over 120 CMT and related genes identified (Timmerman et al. 2014). The “traditional” yield for definitive genetic CMT diagnosis is around 65%; however, diagnostic yields ranging from 19 to 53% have been observed from next generation sequencing approaches (Walsh et al. 2017). Because ~30% of CMT families remain without a genetic diagnosis (Saporta et al. 2011; Murphy et al. 2012; Fridman et al. 2015), and recently discovered genes typically affect a small proportion of patients, there are likely additional unidentified causes.
With so many CMT genes, Variants of Unknown Significance (VUS) are frequently identified during genetic testing. Standards and guidelines have been developed to interpret pathogenicity of VUS including their frequency within a large control population, co-segregation with disease status, and informatics/experimental evidence (MacArthur et al. 2014; Richards et al. 2015). The task of categorizing individual alleles within these genes according to pathogenicity is daunting, yet highly clinically relevant. Data sharing and disease-variant databases are salient solutions to VUS interpretation. Efforts have been implemented to collect and store genetic variation from published literature and clinical laboratories, such as the Human Gene Mutation Database (HGMD), the Human Variome Project (HVP), and ClinVar (Bean and Hegde 2016). Such initiatives are necessary for and will lead to improved variant interpretation; however, currently, erroneous pathogenicity annotations in clinical diagnostic laboratories and in the published literature have introduced false pathogenicity claims into disease-variant databases (Xue et al. 2012; MacArthur et al. 2014; Manrai et al. 2016). Discovering these errors and properly annotating novel variation will require cooperative curation efforts from experts within each disease community.
The Inherited Neuropathy Consortium (INC) is an international group of academic medical centers dedicated to clinical research of CMT. Among the INC’s goals is the construction of web-based resources for clinicians and researchers. Supported by the INC, we have created a unique, community-driven, variant sharing browser for the CMT clinical and research community. The Inherited Neuropathy Variant Browser (INVB) provides simple, user-friendly access to currently reported CMT variants, including patient-level genotypic and phenotypic information. INVB will act as a complementary satellite database, where all INVB data has been submitted to ClinVar - an open-access database of clinically observed variation (Rehm et al. 2015).
Despite current variant collection efforts, the INC recognizes that a potentially large proportion of CMT genetic variation remains uncaptured. Within the academic setting, additional observations of published variation is now rarely shared after the initial gene discovery report (Bean et al. 2013). Likewise, data sharing from clinical laboratories is limited owing to lack of funding and infrastructure, limited phenotype information, or company policies (Bean et al. 2013). Another goal of the INVB is to encourage submission of all observed genetic variation as well as obtaining reports from clinical laboratories, when possible. To minimize burden on users, the INVB accepts direct submission of genetic variation, which will be synced biannually with ClinVar. The INVB’s interactive rating system and discussion platform will lead to improved interpretation of VUS across the Mendelian CMT genes.
METHODS
Data Collection, Standardization, and Curation
The CMT genetic variation data was collected from multiple sources including the Human Gene Mutation Database, the Inherited Peripheral Neuropathies Mutation Database, results published by the Inherited Neuropathies Consortium, the supplemental data published by Athena and Quest Diagnostics, and manual data entry from collaborators.(Cooper et al. 1998) All collected and submitted data conform with institutional review boards and ethical guidelines. The initial set of genes was selected based on the published literature; however, users have the ability to add new genes as they are described as causes of CMT and related disorders. For phenotypic data, the CMT neuropathy score was imported from the Inherited Neuropathies Consortium’s natural history study when available (Fridman et al. 2015). Additionally, each variant was annotated with the minor allele frequency from the Exome Aggregation Consortium (Karczewski et al. 2016).
The coding and protein sequence variant annotation follows recommendations of the Human Genome Variation Society (Dunnen et al. 2016). HUGO Gene Nomenclature Committee’s Multi-symbol checker tool was used to validate all gene symbols. Each variant’s syntax is standardized automatically with Mutalyzer Syntax Check, and the protein sequence variation was populated with Mutation Taster (Wildeman et al. 2008; Schwarz et al. 2010). The Variant Effect Predictor was used to fill in missing variant effect data (McLaren et al. 2016). All publications require a valid PubMed ID, and the title, author, and reference metadata is automatically retrieved and saved in the database.
Continuing the community-driven nature of this database, CMT researchers and clinicians have begun to adopt individual genes to curate. In the initial release, each adoptee verified the genetic variation data. For future curation, bi-annual reports of new submissions will be generated for manual review and curation.
Database Creation and User Interface
The Inherited Neuropathy Variant Browser is built using PHP scripts for the backend and a relational database (Microsoft SQL Server) to store the data. The curated data was imported into the corresponding tables within the database using SQL scripts. The database has audit capability to track user-initiated changes. The graphical user interface for the webpage was built using the AngularJS framework, Bootstrap stylesheet, and HTML.
Data Submission
The Inherited Neuropathy Variant Browser is designed for simple and fast variation upload. User registration and log-in is required for any variant submissions. The submitter must complete all required submission fields including: variant in HGVS notation, gene (drop-down selection of current CMT genes), protein notation, variant type, genotype, and NCBI mRNA reference sequence ID. For compound heterozygous variants, the submitter should select the “Add Compounded Variant” button to submit the variants together.
For data curation purposes, the data source is also required. The possible data sources are: clinical report (including clinical lab name), published paper (including Pubmed ID), or research finding (indicated by Sanger sequencing or Next-Generation Sequencing). A comment box allows for any additional information pertinent to the variant that the submitter wishes to include.
Each variant submission is assigned a randomly generated family ID to allow for genotypic data. This family ID is displayed in the public browser. The submitter has the option to input a private family ID during submission. The private ID, along with the matched public ID, will be available only to the submitter in his/her account page. The purpose of the optional private family ID is to assist submitters with data entry tracking to minimize duplicate entries.
RESULTS
Graphical User Interface
The variant browser is accessible to users through an online graphical user interface (http://hihg.med.miami.edu/neuropathybrowser). The website consists of four different main tabs: Home, Sign Up, Contact Us, and Log In (Figure 1). The Home tab is the homepage of the website and contains most of the functionality. It contains a quick search option along with three other multi-select boxes in order to filter for a gene, variant type, or data source (Figure 1A–B). For the “gene” filter, users can select a single gene, multiple genes, or all genes. The “variant type” filter is grouped into three categories: loss-of-function (LoF), missense, and others. The “LoF” category includes frameshift insertions and deletions (INDELs), stop-gained, and splice altering variants; the “missense” category includes in-frame INDEL, stop-lost, and missense variant; the “others” category includes 3’ untranslated region (UTR), 5’ UTR, intergenic, noncoding exons, intronic, upstream of gene, and downstream of gene variants. Finally, the “data source” filter includes three options: clinical report, published paper, or research finding.
Figure 1.
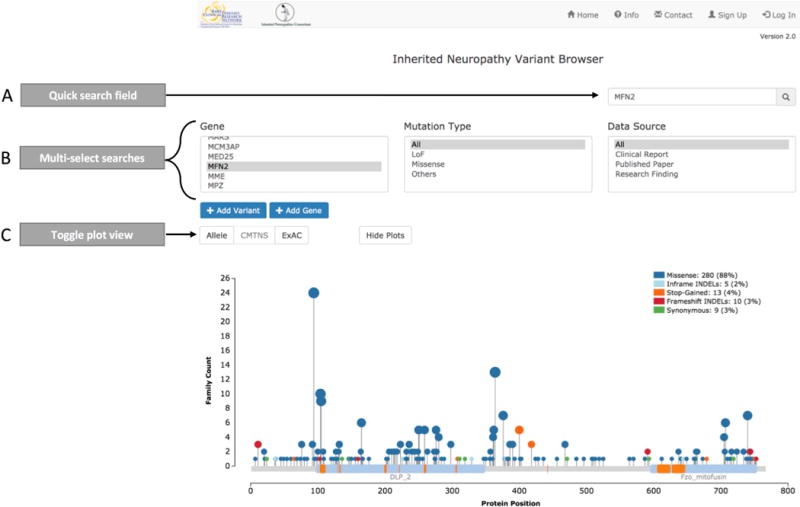
Overview of Inherited Variant Browser homepage and search options. A) The quick search field displays an autocomplete dropdown option and can be queried by: gene name, variant coding position, variant protein position, publication title, publication author, variant type, and data source. B) The multi-select searches enable multiple selections across gene, variant type, and data source. These fields allow users to conduct complex queries, such as all missense and LoF variants reported in published papers in MFN2, GJB1, and GDAP1. C) For single gene queries, variants can be displayed in three plots: allele, CMTNS, and ExAC. These plots can be toggled or hidden by selecting the buttons above each plot.
Query results are displayed in graph and table format. For each gene, the graphic result contains three viewing options: Allele, CMTNS (CMT neuropathy score) (Murphy et al. 2011), or Exome Aggregation Consortium minor allele frequency (ExAC MAF) (Karczewski et al. 2016) (Figure 1C). Each plot uses the ‘Mutations Needle Plot’ and displays the known protein sites and domains, retrieved from NCBI. The default ‘“allele” plot displays the protein location of each allele, count of observations, and variant type. The “CMTNS” and “ExAC” plots require available data for at least one variant within the gene in order to be displayed. The “CMTNS” plot shows CMTNS for each allele and the associated age at onset. The “ExAC” plot displays the ExAC MAF (release 0.3.1). Currently, not all genes have the full set of annotated information available.
The table result consists of seven sortable columns: gene, variant, protein notation, variant type, rating, links and data source (Figure 2). The data is grouped by primary variant into a gray expandable row (Figure 2A). The expanded view shows detailed information about each variant (Figure 2B). In the expanded view, each variant is grouped by the public family ID (Figure 2C) and the observed genotype and zygosity (Figure 2D). For example, the variant in Figure 2E is shown twice to indicate its homozygous zygosity (HOM). Alternatively, Figure 2F displays the variant in the compound heterozygous state (HET). For easy submission, additional families with the same genotype can be added by clicking the button next to the zygosity status (Figure 2G). Each family’s data source can be viewed, edited, or deleted in the expanded view (Figure 2H). Lastly, each individual variant can be rated through the variant rating system, described below (Figure 2I).
Figure 2.
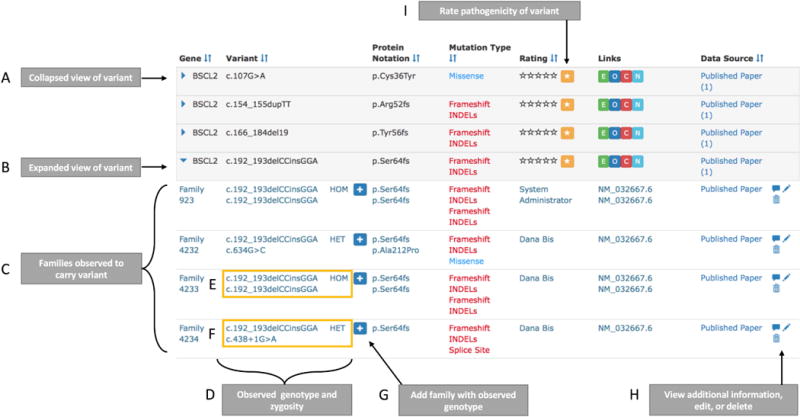
Overview of query result table. A) The collapsed view of each variant is returned by default. This view displays a high-level overview of the variant including gene symbol, cDNA sequence, protein notation, variant type, rating, link outs to ExAC (E), OMIM (O), ClinVar (C), and NCBI (N), and the data sources. B) The expanded row view shows additional information including: C) the family-level information for each variant and D) the observed variant zygosity and relevant genotype. E) Variants observed in the homozygous state is displayed twice and is marked as ‘HOM’ while (F) variants observed in the compound heterozygous state are paired with the compounded variant and marked as ‘HET.’ G) The simple-add button allows a user to quickly add another family with the same observed variant(s). H) Buttons to view additional user comments, edit variant information, or delete a variant. I) In the collapsed view, the user can view the top-level variant rating. The star button allows the user to rate the variant and display the variant rating history.
Variant Rating System
The variant pathogenicity rating system is based upon the standardized terminology of the American College of Medical Genetics and Genomics (ACMG) (Richards et al. 2015). The average rating of a variant is represented as a 5-star system in the table of query result. Each star corresponds to the ACMG terminology: benign, likely benign, uncertain significance, likely pathogenic, pathogenic (starting from 1 star rating). Registered users can rate a variant within the result table and can provide additional comments about their rating (Figure 2I). A history of all ratings and comments will be maintained for each variant. Importantly, any individual rating allows for free-text commenting. This creates a track record of evidence in support of the specific rating.
Database Summary Statistics
The Inherited Neuropathy Variant Browser currently contains 3,809 unique variants within 82 genes. The genes currently contain a median of 16 variants, an interquartile range from 4.0 to 69.5, and a maximum of 720 variants in GJB1 (Figure 3A). A total of 4,558 unrelated families exist with the following genotypes: 2,244 heterozygous, 528 homozygous, and 301 compound heterozygous (Figure 3B). Currently, 1,475 families were reported from clinical laboratories without genotypic information. All new submissions will require genotypic data. The currently available phenotypic data highlights the clinical variability of CMT with an age of onset range of 9 to 75 years and a CMT neuropathy score of 2 to 24 (Figure 3C) (Murphy et al. 2011). The majority of variants are coding (Figure 3D). However, we encourage the submission of non-coding variation to cover the full spectrum of the genetic architecture of CMT. As whole-genome sequencing usage increases we expect the amount of non-coding variation to expand. The variant rating system is becoming a popular feature with 179 ratings to date (Figure 3E).
Figure 3.
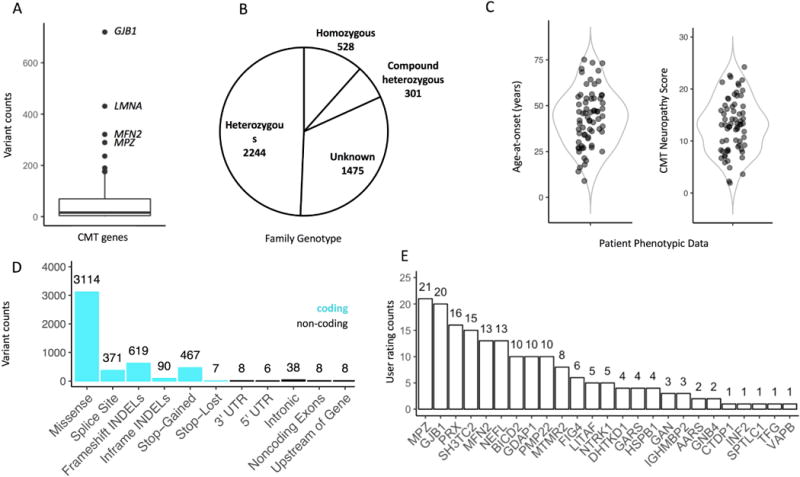
Inherited Neuropathy Browser descriptive statistics. A) Boxplot depicting the variant counts per CMT gene. B) Pie chart of variant zygosity counts. The unknown zygosity variants predominantly come from clinical lab reports that do not contain this information. C) Violin plots displaying available phenotypic data for a variant. D) Bar chart of variant functional consequences. Coding consequences are highlighted in blue. E) Bar chart of user ratings per gene.
Usage Examples
In order to illustrate use cases for the variant browser, we provide two examples that highlight current and future advantages.
Locus and allelic frequency are determinants of expected maximum tolerated population allele frequencies and directly affect pathogenicity evaluation (Whiffin et al. 2017; Wiel et al. 2017). In addition, genic sub-regions, such as protein domains and exons, have been shown valuable in determining genetic tolerance and thus pathogenicity (Gussow et al. 2016). For example, non-synonymous variants are observed across nearly all of GJB1, while such variants are clustered at specific protein domains in DNM2 (Figure 4A). By cataloging the genetic variation (and phenotypic details) observed in CMT genes, CMT-specific genetic tolerance metrics can be developed. Genetic tolerance metrics support clinical VUS interpretation as well as reveal insights into the biological mechanisms of disease genes (Samocha et al. 2017).
The Variant Rating System is a low threshold tool that allows clinicians and researchers to share and record supporting evidence for variant interpretation. Figure 4B shows a real example of a commentary from a user regarding conflicting data: the frequency of a variant in a healthy population (indicates benign) and abnormal electrophysiological data (indicates pathogenic). For quick interpretation from the table view, the variant is rated as two-star “likely benign” while the detailed view shows users’ comments. The intuitive user interface allows for simple and fast user interactions, which will greatly improve the community’s ability to interpret VUS.
Figure 4.
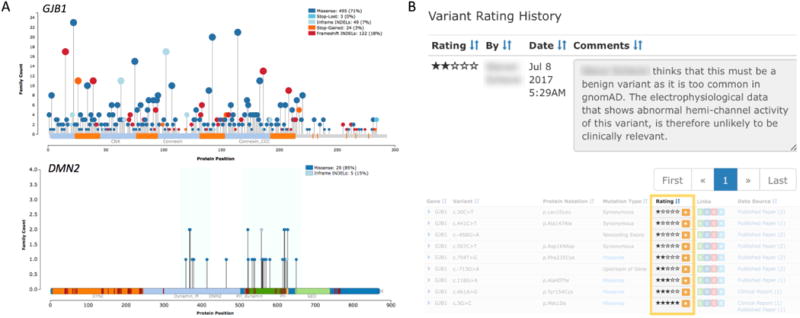
Inherited Neuropathy Browser usage examples. A) Allele plots displaying the missense and loss-of-function variants within GJB1 and DMN2. The number of families with an observed variant is shown on the y-axis and is emphasized by the size of the needle head. The protein position and domain information is displayed on the x-axis. Blue regions in DMN2 highlight the clustering of variation. B) Detailed variant rating history view displaying user comment and user ratings in collapsed table view.
DISCUSSION
At a time when clinical multi-gene testing in highly heterogeneous Mendelian diseases is becoming a standard, the burden of VUS has risen to a point where it obstructs high quality diagnosis in individual patients. Neurologists and genetic counselors working with CMT patients are confronted by this issue daily. While there are multiple potential solutions conceivable to address this problem, including future functional genomics platforms, a sensible and cost effective way forward is the comprehensive and public mapping of disease associated variation to disease-causing genes. We have collected CMT specific genetic variation, along with genotypic and phenotypic data when available, from published literature, clinical lab reports, and our own in-house data. We then created the interactive, web-based Inherited Neuropathy Variant Browser (INVB) accessible to, and relying on participation from, CMT researchers and clinicians to view the collected CMT variation. We have implemented an interactive rating system of genetic variation based on standardized terminology from the ACMG (Richards et al. 2015). The rating history for each variant, along with comments from raters, will be maintained to provide a rich perspective to users. The Inherited Neuropathy Consortium is encouraging its global membership to submit observed pathogenic variation, VUS and polymorphisms to the INVB. This platform enables a joint effort by the global CMT expert community to store, share, and discuss genetic variation to resolve VUS.
While the frequency of a DNA variant within an unaffected population helps to categorize benign variation, only repeated observation of a variant in affected families will strengthen pathogenic classification. The continued aggregation of both disease-associated and benign variation will remain essential to further resolve VUS (Bean et al. 2013). Currently, 25%-30% of variants in databases can be clearly classified as benign or pathogenic (Bean and Hegde 2016). We see an opportunity to create enthusiasm and attention in the CMT field to participate in the important task of collecting individual genetic test results from many clinics and different parts of the world. We were able to assemble an international group of experts committed to working together to classify variation and we hope the CMT community will serve as a positive example of joint enterprise within genomic medicine. The INC, the largest CMT-related consortium, has designated the interactive INVB as the CMT genetic tool of choice. The international character of the INC is attracting users from different geographic locations with diverse ancestries, such as the new Asia Oceanic Inherited Neuropathies Consortium. As minor allele frequencies can vary widely between ancestral populations, uniquely rare variants in one population might be less rare in another, thus excluding such an allele as pathogenic.
Similarly, large-scale population data, such as ExAC, a collection of over ~60,000 exomes from patients unaffected by severe pediatric diseases, has revealed previously reported pathogenic alleles as too common relative to the prevalence of CMT (Minikel et al. 2016). We therefore incorporated ExAC data into the INVB (Karczewski et al. 2016). Benign variants falsely assigned as pathogenic have been revealed and reported disease variant penetrance has been re-evaluated, such as p.Arg468His in MFN2. This variant has been implicated in variant screening studies at least four times; yet, the relatively high count of 265 heterozygous alleles out of 115,542 ExAC chromosomes excludes the variant as a causative, high penetrance allele in a dominant rare disease (Engelfried et al. 2006; Braathen et al. 2010; Casasnovas et al. 2010; McCorquodale et al. 2011).
Though variant databases excel in providing overall allele frequencies, most capture genetic variants without listing individual patients and their genotypes; however, this information may contribute to variant interpretation (Lanthaler et al. 2014). Without knowing the heterozygous, homozygous or compound heterozygous state, it may be difficult to determine causality in a recessive disorder. Furthermore, the pathogenicity of a compound heterozygous pair of alleles is directly influenced by each involved allele. For CMT specifically, genotypic information is particularly important, because several genes, for example GDAP1 and HSPB1, can cause disease in dominant and recessive inheritance modes depending on the specific variant (Houlden et al. 2008; Sivera et al. 2010; Cassereau et al. 2011). Finally, genotypic information may well guide future oligogenic models of inheritance in individual patients and influence predictable phenotypic expression. For these reasons, we require both the observed zygosity and phenotypic information with each variant submission. Beyond variant interpretation, storage of these datasets will enable future genotype-phenotype correlation analyses.
In order to benefit the wider scientific community, all collected data has been submitted to ClinVar – a public archive of the interpreted clinical significance of variants for reported conditions at the National Center for Biotechnology Information (Landrum et al. 2016). ClinVar was developed to provide access to the interpretation of human variation and is an effort critical to genomic medicine (Landrum et al. 2014). As avid supporters of the ClinVar initiative, we recommend the entire community to submit variation data directly to the database. However, we have implemented direct submission to the INVB to collect data inappropriate for ClinVar (such as variants based solely on computational predictions) or from users who have chosen not to submit to ClinVar (such as CMT researchers outside of the United States). As we do not want to add to the issue of multiple fragmented data collection efforts with yet another database, we will perform 2-way synchronization with ClinVar at least bi-annually. Users should look to ClinVar for the most up-to-date variant database and utilize the INVB as a tool to view and discuss this data. We hope the simple and easy-to-use interface of the Inherited Neuropathy Variant Browser will facilitate communication about variant pathogenicity status among CMT researchers and clinicians and provide improved diagnostic abilities.
Acknowledgments
Steven Scherer acknowledges support from the Judy Seltzer Levenson Memorial Fund for CMT Research.
Funding
Funded by the Inherited Neuropathy Consortium – Rare Disease Clinical Research Consortium (INC RDCRC - U54NS065712), NINDS/ORDR, NCATS, the Muscular Dystrophy Association (MDA), the Charcot Marie Tooth Association (CMTA) and NINDS R01NS075764.
Footnotes
Contributorship
Cima Saghira – Contributed to database concept and creation, browser design and creation, variant curation, and manuscript preparation.
Dana M Bis – Contributed to database concept, browser design, variant curation, variant annotation, and manuscript preparation.
David Stanek – Contributed to variant annotation with protein domain information.
Dr. Allene Strickland – Contributed to data collection.
Dr. David Herrmann – Contributed to the database concept, browser design, and variant curation.
Dr. Mary M Reilly – Contributed to the database concept, browser design, and variant curation.
Dr. Steven S. Scherer – Contributed to the database concept, browser design, and variant curation.
Dr. Mike Shy – Contributed to the database concept, browser design, and variant curation.
Dr. Stephan Züchner – Contributed to the database concept, browser design, and variant curation.
Competing Interests and Author Disclosures
Cima Saghira reports no disclosures.
Dana M Bis reports no disclosures.
David Stanek reports no disclosures.
Dr. Allene Strickland reports no disclosures.
Dr. David Herrmann reports no disclosures.
Dr. Mary M Reilly reports no disclosures.
Dr. Steven Scherer reports no disclosures.
Dr. Mike Shy reports no disclosures.
Dr. Stephan Zuchner reports no disclosures.
WEB RESOURCES
The Inherited Neuropathy Variant Browser (INVB): http://hihg.med.miami.edu/neuropathybrowser
Human Gene Mutation Database (HGMD): http://www.hgmd.cf.ac.uk/ac/index.php
The Human Variome Project (HVP): http://www.humanvariomeproject.org/
ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
The Inherited Peripheral Neuropathies Mutation Database: https://www.molgen.vib-ua.be/CMTMutations/
HUGO Gene Nomenclature Committee’s Multi-symbol checker tool: http://www.genenames.org/cgi-bin/symbol_checker
Mutations Needle Plot: https://www.npmjs.com/package/muts-needle-plot
References
- Bean LJH, Hegde MR. Gene Variant Databases and Sharing: Creating a Global Genomic Variant Database for Personalized Medicine. Hum Mutat. 2016;37:559–563. doi: 10.1002/humu.22982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean LJH, Tinker SW, da Silva C, Hegde MR. Free the Data: One Laboratory's Approach to Knowledge-Based Genomic Variant Classification and Preparation for EMR Integration of Genomic Data. Hum Mutat. 2013;34:1183–1188. doi: 10.1002/humu.22364. [DOI] [PubMed] [Google Scholar]
- Braathen GJ, Sand JC, Lobato A, Høyer H, Russell MB. MFN2 point mutations occur in 3.4% of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med Genet. 2010;11:48. doi: 10.1186/1471-2350-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasnovas C, Banchs I, Cassereau J, Gueguen N, Chevrollier A, Martínez-Matos JA, Bonneau D, Volpini V. Phenotypic spectrum of MFN2 mutations in the Spanish population. J Med Genet. 2010;47:249–256. doi: 10.1136/jmg.2009.072488. [DOI] [PubMed] [Google Scholar]
- Cassereau J, Chevrollier A, Gueguen N, Desquiret V, Verny C, Nicolas G, Dubas F, Amati-Bonneau P, Reynier P, Bonneau D, Procaccio V. Mitochondrial dysfunction and pathophysiology of Charcot-Marie-Tooth disease involving GDAP1 mutations. Exp Neurol. 2011;227:31–41. doi: 10.1016/j.expneurol.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998;26:285–287. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnen den JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux A-F, Smith T, Antonarakis SE, Taschner PEM, HGVS, HVP et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat. 2016;37:564–569. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch Neurol. 1968;18:619–625. doi: 10.1001/archneur.1968.00470360041003. [DOI] [PubMed] [Google Scholar]
- Engelfried K, Vorgerd M, Hagedorn M, Haas G, Gilles J, Epplen JT, Meins M. Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the mitofusin 2 gene (MFN2) BMC Med Genet. 2006;7:53. doi: 10.1186/1471-2350-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, Day J, Feely S, Finkel RS, Grider T, Kirk CA, Herrmann DN, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatr. 2015;86:873–878. doi: 10.1136/jnnp-2014-308826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussow AB, Petrovski S, Wang Q, Allen AS, Goldstein DB. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biology. 2016;17:9. doi: 10.1186/s13059-016-0869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, Laurá M, Wavrant-De Vrièze F, Blake J, Wood N, Reilly MM. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71:1660–1668. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Weisburd B, Thomas B, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha KE, Cummings BB, Birnbaum D, The Exome Aggregation Consortium, Daly MJ, et al. 2016.
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–5. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanthaler B, Wieser S, Deutschmann A, Schossig A, Fauth C, Zschocke J, Witsch-Baumgartner M. Genotype-based databases for variants causing rare diseases. Gene. 2014;550:136–140. doi: 10.1016/j.gene.2014.08.016. [DOI] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med. 2016;375:655–665. doi: 10.1056/NEJMsa1507092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorquodale DS, Montenegro G, Peguero A, Carlson N, Speziani F, Price J, Taylor SW, Melanson M, Vance JM, Zuchner S. Mutation screening of mitofusin 2 in Charcot-Marie-Tooth disease type 2. J Neurol. 2011;258:1234–1239. doi: 10.1007/s00415-011-5910-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biology. 2016;17 doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, McLean CY, Tung JY, Yu, Linda PC, Gambetti P, Blevins J, Zhang S, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9. doi: 10.1126/scitranslmed.aad5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, Pareyson D. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16:191–198. doi: 10.1111/j.1529-8027.2011.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SM, Laurá M, Fawcett K, Pandraud A, Liu Y-T, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MPT, Bull K, et al. Charcot–Marie–Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatr. 2012;83:706–710. doi: 10.1136/jnnp-2012-302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Ledbetter N, Mefford HC, Smith RJ, Stephens K. Charcot-Marie-Tooth Hereditary Neuropathy Overview. 1993 [PubMed] [Google Scholar]
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, et al. ClinGen - The Clinical Genome Resource. N Engl J Med. 2015;372:2235–2242. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly MM, Murphy SM, Laurá M. Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16:1–14. doi: 10.1111/j.1529-8027.2011.00324.x. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics. 2015 doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, Neale BM, Daly MJ. Regional missense constraint improves variant deleteriousness prediction. 2017:1–32. [Google Scholar]
- Saporta ASD, Sottile SL, Miller LJ, Feely SME, Siskind CE, Shy ME. Charcot-marie-tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011;69:22–33. doi: 10.1002/ana.22166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta MA, Shy ME. Inherited Peripheral Neuropathies. Neurol Clin. 2013a;31:597. doi: 10.1016/j.ncl.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta MA, Shy ME. Inherited Peripheral Neuropathies. Neurol Clin. 2013b;31:597–619. doi: 10.1016/j.ncl.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Meth. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Sivera R, Espinós C, Vílchez JJ, Mas F, Martínez-Rubio D, Chumillas MJ, Mayordomo F, Muelas N, Bataller L, Palau F, Sevilla T. Phenotypical features of the p.R120W mutation in the GDAP1 gene causing autosomal dominant Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2010;15:334–344. doi: 10.1111/j.1529-8027.2010.00286.x. [DOI] [PubMed] [Google Scholar]
- Timmerman V, Strickland A, Zuchner S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes. 2014;5:13–32. doi: 10.3390/genes5010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M, Bell KM, Chong B, Creed E, Brett GR, Pope K, Thorne NP, Sadedin S, Georgeson P, Phelan DG, Day T, Taylor JA, et al. Diagnostic and cost utility of whole exome sequencing in peripheral neuropathy. Ann Clin Transl Neurol. 2017;4:318–325. doi: 10.1002/acn3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, Ware JS. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158. doi: 10.1038/gim.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiel L, Venselaar H, Veltman JA, Vriend G, Gilissen C. Aggregation of population-based genetic variation over protein domain homologues and its potential use in genetic diagnostics. Hum Mutat. 2017;17:235. doi: 10.1002/humu.23313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildeman M, van Ophuizen E, Dunnen den JT, Taschner PEM. Improving sequence variant descriptions in mutation Databases and literature using the mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13. doi: 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
- Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, Tyler-Smith C, 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]