

Abstract
Background & Aims
The organic solute transporter α-β (OSTα-OSTβ) mainly facilitates transport of bile acids across the basolateral membrane of ileal enterocytes. Therefore, inhibition of OSTα-OSTβ might have similar beneficial metabolic effects as intestine-specific agonists of the major nuclear receptor for bile acids, the farnesoid X receptor (FXR). However, no OSTα-OSTβ inhibitors have yet been identified.
Methods
Here, we developed a screen to identify specific inhibitors of OSTα-OSTβ using a genetically encoded Förster Resonance Energy Transfer (FRET)–bile acid sensor that enables rapid visualization of bile acid efflux in living cells.
Results
As proof of concept, we screened 1280 Food and Drug Administration–approved drugs of the Prestwick chemical library. Clofazimine was the most specific hit for OSTα-OSTβ and reduced transcellular transport of taurocholate across Madin–Darby canine kidney epithelial cell monolayers expressing apical sodium bile acid transporter and OSTα-OSTβ in a dose-dependent manner. Moreover, pharmacologic inhibition of OSTα-OSTβ also moderately increased intracellular taurocholate levels and increased activation of intestinal FXR target genes. Oral administration of clofazimine in mice (transiently) increased intestinal FXR target gene expression, confirming OSTα-OSTβ inhibition in vivo.
Conclusions
This study identifies clofazimine as an inhibitor of OSTα-OSTβ in vitro and in vivo, validates OSTα-OSTβ as a drug target to enhance intestinal bile acid signaling, and confirmed the applicability of the Förster Resonance Energy Transfer–bile acid sensor to screen for inhibitors of bile acid efflux pathways.
Keywords: Fluorescence Resonance Energy Transfer (FRET), FXR, OSTα-OSTβ, Bile Acids
Abbreviations used in this paper: ASBT, apical sodium-dependent bile acid transporter; BAS, bile acid sensor; FACS, fluorescence-activated cell sorting; FDA, Food and Drug Administration; FGF15/19, fibroblast growth factor 15/19; FRET, fluorescent resonance energy transfer; FXR, farnesoid X receptor; MDCKII, Madin–Darby canine kidney epithelial cells; mRNA, messenger RNA; nucleoBAS, nucleus-localized bile acid sensor; OSTα-OSTβ, organic solute transporter α-β; TCDCA, taurochenodeoxycholic acid; TICE, transintestinal cholesterol excretion; U2OS, human bone osteosarcoma epithelial cells
Graphical abstract

Summary.
A fluorescent resonance energy transfer–based, high-throughput screen was established to identify inhibitors of organic solute transporter (OST)α-OSTβ–mediated bile acid efflux. We identified clofazimine as an inhibitor of OSTα-OSTβ and showed that such inhibition enhanced intestinal farnesoid X receptor activation.
Bile acids are released in the duodenum during a meal, where they act as digestive detergents crucial for intestinal absorption of lipids, fat-soluble vitamins, and other lipophilic nutrients.1 After the meal, most bile acids are reabsorbed from the ileum and return to the liver via the hepatic portal vein. In the hepatocyte, the bile acid pool is replenished via de novo bile acid synthesis from cholesterol.2 Subsequently, bile acids are conjugated and stored in the gallbladder until the next meal. The meal-regulated dynamics renders bile acids as potent signaling molecules that modulate triglyceride, lipid, glucose, and energy homeostasis, making bile acid signaling an interesting target for metabolic diseases.3 Activation of the farnesoid X receptor (FXR), a major nuclear receptor involved in bile acid signaling, has been suggested to be beneficial in many metabolic disorders. In particular, selective intestinal activation of FXR led to metabolic improvements such as enhanced glucose tolerance, reduced diet-induced weight gain, and reduced inflammation,4 but also enhanced transintestinal cholesterol excretion (TICE).5
The organic solute transporter α-β (OSTα-OSTβ) mainly facilitates transport of bile acids across the basolateral membrane of ileal enterocytes.6, 7 It consists of 2 proteins forming a heterodimer, OSTα (encoded by SLC51A) and its subunit OSTβ (encoded by SLC51B), which are both required for normal trafficking and function of OSTα-OSTβ.
OSTα knockout mice show a reduction in the bile acid pool and serum levels combined with increased FXR activation in ileal enterocytes.8 In addition, deficiency of OSTα is protective for liver injury during obstructive cholestasis, and leads to decreased body fat and lipid accumulation and improved insulin sensitivity.9, 10, 11 Furthermore, OSTα knockout mice show increased elimination of cholesterol in the feces and decreased levels of cholesterol and triglyceride in serum.8, 12 These data point to OSTα-OSTβ as a novel target to treat diabetes and obesity, but also lipid and cholesterol disorders, for instance, by inducing the TICE pathway via intestinal FXR activation.13 However, no inhibitors for OSTα-OSTβ have yet been identified and techniques available for measuring bile acid efflux are limited. Therefore, we developed a novel assay for cell-based high-throughput screening to identify specific inhibitors for OSTα-OSTβ, making use of a fluorescent resonance energy transfer (FRET)-based bile acid sensor that enables rapid visualization of bile acid efflux in living cells.14 This screen specifically measures bile acid efflux, which simultaneously provides a readout based on increased FXR activation as a consequence of OSTα-OSTβ inhibition. We screened 1280 Food and Drug Administration (FDA)-approved drugs of the Prestwick chemical library and confirmed several positive hits. Here, we show that inhibition of OSTα-OSTβ leads to intestinal FXR activation.
Materials and Methods
Reagents
The Prestwick chemical library (1280 FDA approved compounds in 96-well plates) was purchased from Prestwick Chemical (Illkirch, France). Bifonazole, bromhexine HCl, clofazimine, lovastatin, meclozine 2HCL, and simvastatin were purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). The fibroblast growth factor (FGF)19 human enzyme-linked immunosorbent assay kit was purchased from BioVendor R&D (Brno, Czech Republic). [3H]-taurocholate and [14C]-inulin were purchased from Perkin Elmer (Groningen, The Netherlands).
Cell Culture
Human bone osteosarcoma epithelial cells (U2OS) wild-type cells (HTB-96; American Type Culture Collection, Wesel, Germany) were cultured in Dulbecco's modified Eagle medium (high glucose) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% L-glutamine. NucleoBAS (Nuclear-localized Bile Acid Sensor),14 NA+-taurocholate co-transporting polypeptide, and OSTα-OSTβ–expressing U2OS cells were engineered by transfecting cells using polyethylenimine. Stable cell lines were generated by colony picking using cloning rings over well-separated colonies. Apical sodium-dependent bile acid transporter (ASBT) and mouse OSTα-OSTβ co-expressing Madin–Darby canine kidney epithelial cells (MDCKII) cells were a gift from Paul Dawson (Emory University School of Medicine, Atlanta, GA).15 All cells were cultured at 5% CO2 at 37°C.
Fluorescence-Activated Cell Sorting
Two days before the experiments, wild-type and transfected U2OS cells were cultured in 5% charcoal-treated fetal bovine serum to prevent bile acid overload of the sensor. The adherent cell layer was trypsinized by 5 mmol/L EDTA to create a suspension of single cells for fluorescence-activated cell sorting (FACS) analysis. Cells were harvested by centrifugation and the resulting pellet was suspended in FACS uptake buffer (0.3 mmol/L EDTA, 0.5% bovine serum albumin, 0.01% NaN3, and 10 mmol/L D-glucose), plated in 96-wells and subsequently incubated with 10 μmol/L of 1 of 1280 compounds of the Prestwick Chemical library. After 5 minutes, 3 μmol/L TCDCA was added and incubated for 30 minutes at room temperature while shaking. Citrine and cerulean intensity was measured by a violet 405-nm laser. Spectral range for emission detections were as follows: cerulean, 450/40 nm; and citrine, 525/20 nm.
Taurocholate Uptake Assay in MDCKII–ASBT Cells
Cells were cultured in a 24-well plate format at 50%–60% confluency. The next day, cells were washed with uptake buffer and incubated with compounds at 37°C for 30 minutes. Uptake buffer was aspirated and a mix of trace amounts of [3H]-taurocholate and 20 μmol/L nonradiolabeled Taurocholic Acid was administered. After 2 minutes of incubation at 37°C, the cells were washed using ice-cold phosphate-buffered saline and lysed in 0.05% sodium dodecyl sulfate in distilled water. Tritium signal of each sample was measured in 3 mL of scintillation liquid.
Transcellular Transport Assay Across MDCKII Monolayers
MDCKII cells stably expressing ASBT/OSTα-OSTβ were cultured on 6.5-mm Transwell filter inserts (Costar, London, United Kingdom) in a density of 15,000 cells per well. The medium was refreshed every 2–3 days. The formation of a tight monolayer (normally between days 6 and 10) was measured with the EVOM2 (World Precision Instruments [WPI], Berlin, Germany) epithelial voltohmmeter for Trans Epithelial Electrical Resistance. A minimal resistance of 500 Ω was required before the experiment was continued. On the day of the experiment, cells were washed and incubated with the compounds for 1 hour at 37°C. A mix of [3H]-taurocholate, [14C]-inulin, and 20 μmol/L unlabeled trichloroacetic acid was added. At t = 10, 30, 60, and 120 minutes, 10 μL of the basolateral compartment was measured for radioactivity. The diffusion of [14C]-inulin was used as a measurement for leakage of the monolayer and confirmed monolayer integrity. After the last time point (2 hours), cells were lysed in 0.5% sodium dodecyl sulfate in distilled water to measure intracellular accumulation of taurocholate.
Differentiation and Treatment of Caco-2 Cells
Caco-2 cells were plated at 100% confluency on 6.5-mm Transwell filter inserts (Costar). Every 2–3 days, medium was refreshed and the tightness of the monolayers were measured using the EVOM2 epithelial voltohmmeter for Trans Epithelial Electrical Resistance. After 21 days, Caco-2 cells were treated with 10 μmol/L compounds in supplemented Dulbecco's modified Eagle medium for 48 hours.
Analysis of Gene Expression Using Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted from Caco-2 cells or whole ileal tissue (most distal segment) using TRIzol reagent (Invitrogen, Bleiswijk, The Netherlands) according to the manufacturer’s instructions. Complementary DNA synthesis was initiated from 1 μg of DNase-treated RNA using oligo deoxythymine primers and Superscript II reverse transcriptase (Invitrogen). Quantitative reverse-transcription polymerase chain reaction was performed using the SensiFAST SYBR No-ROX kit (Bioloine, London, UK) in a Roche (Woerden, Netherlands) lightcycler 480 II. Expression levels of all samples were normalized to the geometric mean of housekeeping genes. Oligonucleotide sequences are available on request.
Clofazimine Treatment In Vivo
All mice were housed and treated in accordance with National Institutes of Health guidelines and enforced by the Institutional Animal Care and Use Committee at the Animal Research Institute Academic Medical Center. Clofazimine solved in corn oil was administered orally by gavage, either as a single dose or as 5 daily doses in 8-week-old male wild-type C57BL/6 mice. Afterward, mice were injected intravenously once with cholecystokinin octapeptide (50 ng/kg) to induce gallbladder contraction and thereby ensure the presence of bile acids in the intestine. Six hours later, mice were anesthetized using a mix of ketamine (100 mg/mL) and xylazine (20 mg/mL) in a dosage of 100 μL/10 g body weight. Serum, bile, urine, liver tissue, intestinal tissue, and feces were collected. Analysis of fecal and plasma bile acid composition was performed as previously described.16 All mice were kept on a 12-hour light-dark cycle and received standard chow and water ad libitum. The study design and all protocols for animal care and handling were approved by the Institutional Animal Care and Use Committee of the University of Amsterdam.
Statistical Analysis
Data are presented as the means ± SEM . Differences between 2 groups were statistically evaluated using the Student t test. Results were considered statistically significant at a P value <.05.
All authors had access to the study data and have reviewed and approved the final manuscript.
Results
Design of the Pharmacologic FRET-Based Compound Screen for Inhibitors of OSTα-OSTβ–Mediated Bile Acid Efflux
Most existing techniques to monitor bile acid efflux require modification of bile acids, possibly affecting their transport kinetics. Here, we designed a pharmacologic screen that allows for rapid live cell imaging of bile acid efflux using a genetically encoded FRET bile acid sensor (BAS).14 The sensor is a fusion protein consisting of 2 fluorescent domains, cerulean and citrine, fused via the ligand-binding domain of FXR and linked to a peptide derived from an FXR co-activator protein. Binding of bile acids to the FXR ligand-binding domain results in rapid and reversible peptide binding and association between the fluorophores, detectable as increased FRET. Advantages of this approach are its noninvasiveness, rapid live cell imaging, and the easy readout of increased FRET intensity. For this study, a cell line was created by transfecting the nucleus-localized BAS (nucleoBAS), a bile acid uptake transporter and OSTα-OSTβ in U2OS cells (Figure 1A, left). As a positive control for 100% inhibition of OSTα-OSTβ, NucleoBAS-positive cells were created that only contained the bile acid uptake transport protein and lacked OSTα-OSTβ protein expression (Figure 1A, right). The feasibility of this sensor and the accuracy of quantitative results during measurements with flow cytometry was tested in these cell lines. As expected, treatment with both 30 μmol/L taurochenodeoxycholic acid (TCDCA) and 5 μmol/L GW4064 resulted in an increased FRET ratio in the cell population compared with untreated cells (Figure 1D and E), although this effect of TCDCA was not observed in cells only expressing the sensor but no bile acid transporters (Figure 1B and C).
Figure 1.

(A) U2OS cells expressing nucleoBAS, the bile acid uptake transporter NA+-taurocholate co-transporting polypeptide, and OSTα-OSTβ (left) were used to analyze the inhibitory effect of 1280 compounds on OSTα-OSTβ–mediated transport. U2OS cells expressing nucleoBAS and NA+-taurocholate co-transporting polypeptide but no OSTα-OSTβ served as a positive control (right). (B) FACS plots of U2OS cells transfected with nucleoBAS were treated with dimethyl sulfoxide (DMSO) (left), TCDCA (middle), or GW4064 (right). (C) Percentage of cells in gate P4, plotted in a bar graph. (D) FACS plots of U2OS nucleoBAS cells co-transfected with a bile acid uptake transporter (NA+-taurocholate co-transporting polypeptide) and OSTα-OSTβ also were treated with DMSO (left), TCDCA (middle), or GW4064 (right). (E) Percentage of cells in gate P4, plotted in a bar graph. Data are represented as means ± SD. ***P < .001 (1-way analysis of variance; post hoc: Dunnett multiple comparison).
Ideally, in cells expressing both OSTα-OSTβ and a bile acid uptake transporter (Figure 1A), inhibition of OSTα-OSTβ will result in increased intracellular levels of bile acids. Indeed, the bile acid sensor in U2OS cells that expressed a bile acid uptake transporter but not OSTα-OSTβ, was activated at much lower TCDCA concentrations compared with cells that expressed both an uptake transporter and OSTα-OSTβ (Figure 2A). This implies that there is an intracellular accumulation of bile acids in cells with an uptake transporter but without OSTα-OSTβ. The most potent effect between the 2 cells lines was observed at a 3- to 5-μmol/L TCDCA concentration, and was therefore the selected dose for further research. The experimental strategy of the FRET-based screen is shown in Figure 2B. By using this assay, we were able to identify novel FDA-approved compounds (Prestwick Chemical library) that specifically inhibit OSTα-OSTβ–mediated bile acid efflux.
Figure 2.

Experimental strategy and validation of the FRET-based compound screen. (A) Response of nucleoBAS sensor-expressing cells to different TCDCA concentrations. White bars: cells only expressing the bile acid uptake transporter NA+-taurocholate co-transporting polypeptide (positive control). Blue bars: cells expressing NA+-taurocholate co-transporting polypeptide and OSTα-OSTβ (n = 4). (B) Experimental strategy used to determine which compounds inhibit OSTα-OSTβ. High FRET signal intensities represent compounds inhibiting OSTα-OSTβ–mediated transport activity. (C) Distribution of drug classes for inhibition of OSTα-OSTβ based on the top 25 hits. (D) Screen FACS plots of controls (dimethyl sulfoxide [DMSO], and 10 μmol/L GW4064), followed by FACS plots of the 6 selected compounds (10 μmol/L), bifonazole, bromhexine HCl, clofazimine, lovastatin, meclizine HCl, and simvastatin (n = 1, data are from a representative experiment replicated 2 times). (A) Data are presented as means ± SD. *P ≤ .05 (Student t test, 2-tailed).
High-Throughput Pharmacologic Screening Detects Inhibitors of OSTα-OSTβ Transport in a Large Compound Library
The initial 55 hits that tested positive for OSTα-OSTβ inhibition in the primary screen were confirmed by a subsequent secondary screen using the same assay to verify the accuracy of the hits. In addition, false positives were eliminated by testing all compounds for direct activation of the sensor in cells that expressed nucleoBAS but no bile acid transporters. At a 10 μmol/L compound concentration, 25 drugs showed more than 20% activation of the sensor compared with dimethyl sulfoxide, implying inhibition of OSTα-OSTβ in the primary screen. Figure 2C shows the distribution of drug classes among the 25 primary hits. The steroid drug class is highly represented (30%). This is probably owing to competition for the binding site of OSTα-OSTβ because this transporter is known to transport steroids as well.17, 18 Therefore, compounds of this drug class were eliminated from further research. Other significant groups in the top 25 compounds were azoles, benzenoids, and statins. From each major group, 1 or 2 compounds were selected for follow-up experiments, in which the administration route and drug safety in human beings was decisive. In the end, we selected 6 hits for further research: bifonazole, simvastatin, bromhexine hydrochloride, lovastatin, clofazimine, and meclozine dihydrochloride. FACS plots of the 6 selected compounds and controls (dimethyl sulfoxide and GW4064) are shown in Figure 2D. Table 1 provides an overview of the current applications and structure of these compounds.
Table 1.
Hits Obtained From the Screening
| Chemical name | Therapeutic effect | Structure | Structure formula | Inhibition (10 μmol/L), % |
|---|---|---|---|---|
| Bifonazole | Antifungal | 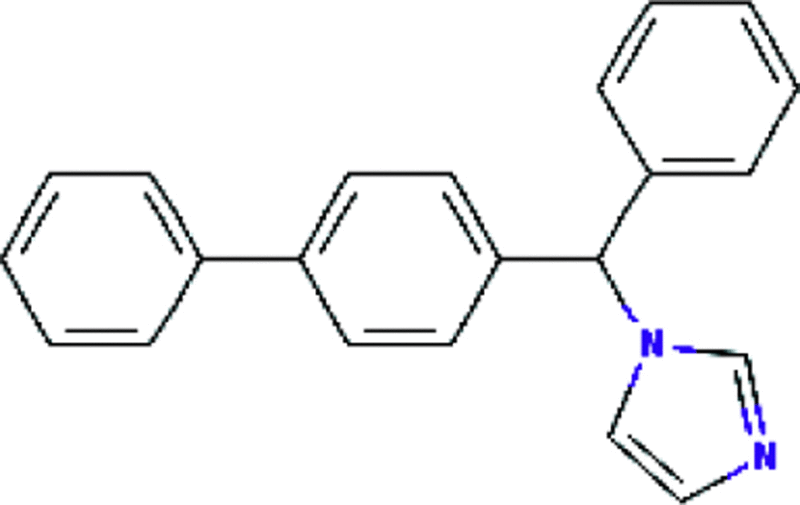 |
C22H18N2 | 98 |
| Bromhexine hydrochloride | Expectorant | 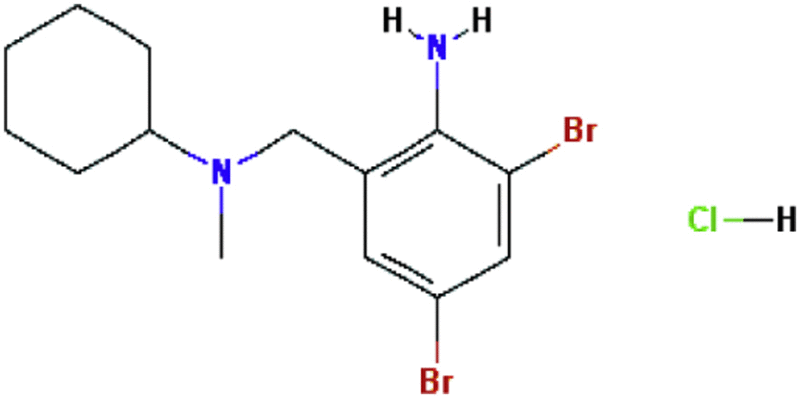 |
C14H21Br2ClN2 | 57 |
| Clofazimine | Antibacterial | 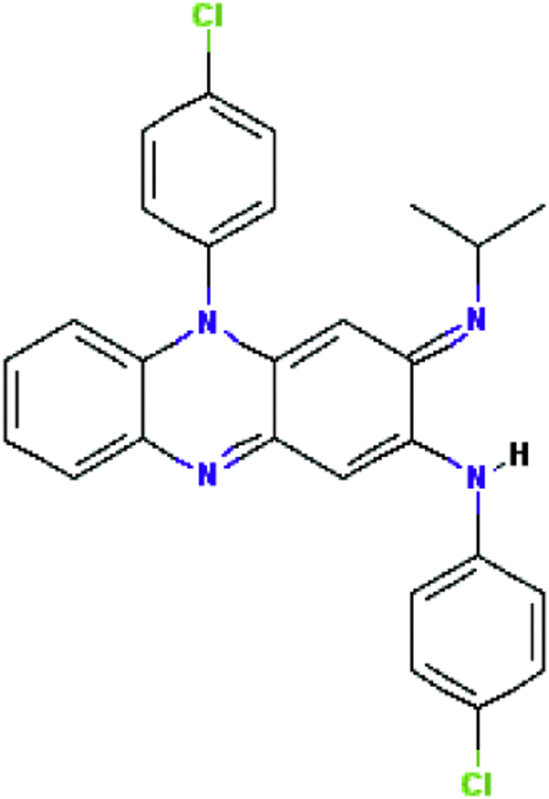 |
C27H22Cl2N4 | 47 |
| Lovastatin | Hypocholesterolemic | 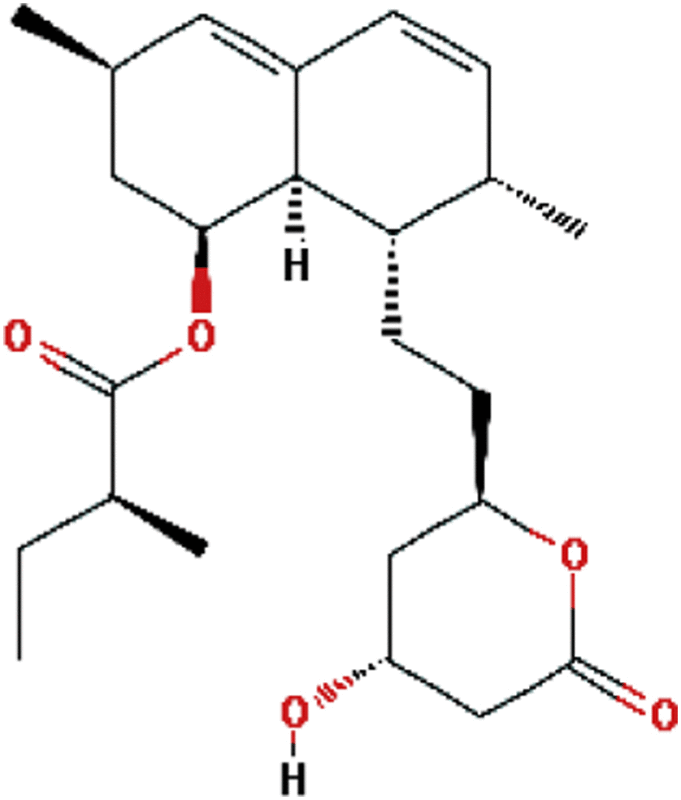 |
C24H36O5 | 51 |
| Meclozine dihydrochloride | Antiemetic | 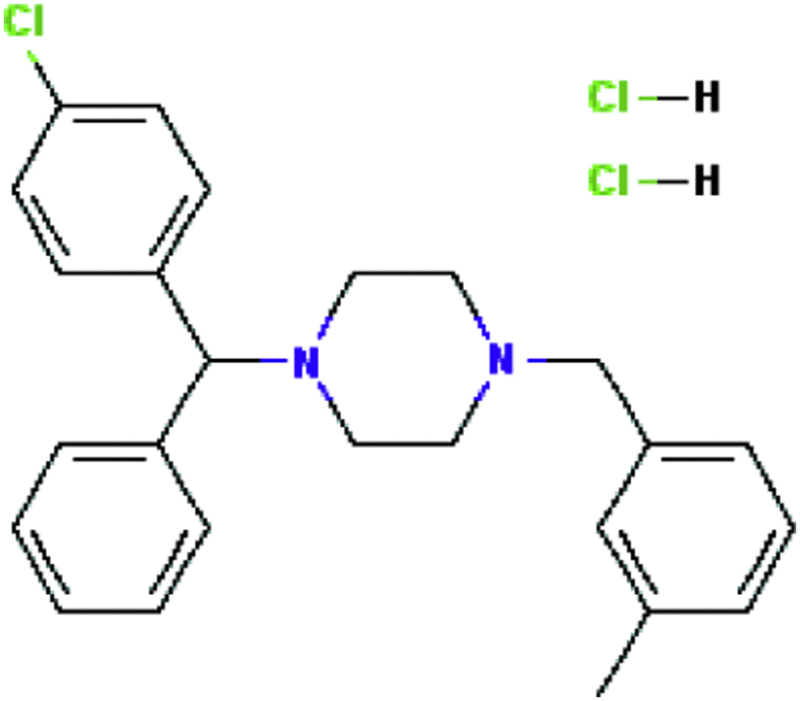 |
C25H29Cl3N2 | 53 |
| Simvastatin | Antilipemic | 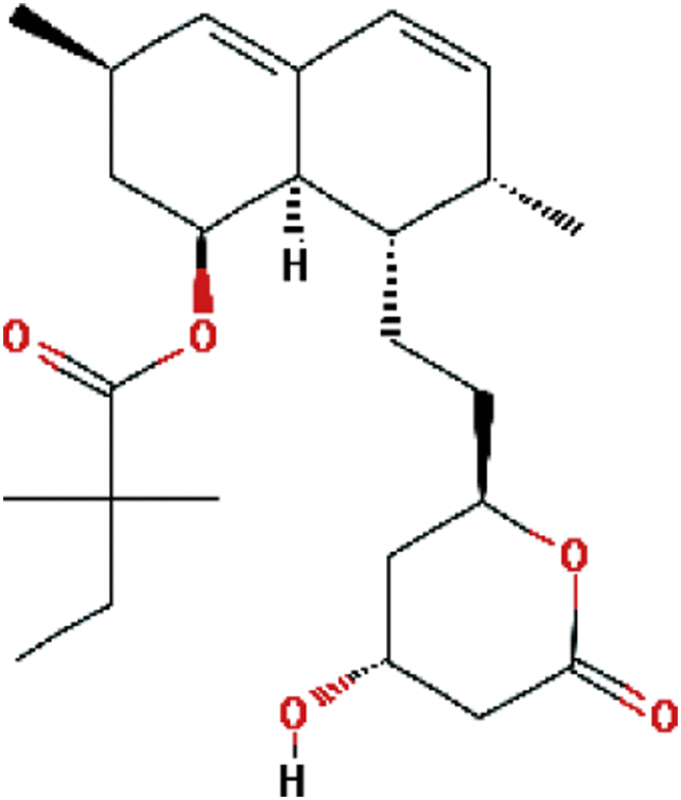 |
C25H38O5 | 77 |
Clofazimine Is a Specific Inhibitor for OSTα-OSTβ–Mediated Bile Acid Transport
The specificity for inhibition of OSTα-OSTβ–mediated bile acid transport of the 6 selected compounds was tested using ASBT as another bile acid transporter that also is expressed in the enterocytes of the ileum. MDCKII cells transfected with ASBT were used to analyze the influx of [3H]-taurocholate. Uptake values were determined by measuring the intracellular radioactivity after a short 2-minute incubation with [3H]-taurocholate. Lovastatin and simvastatin inhibited ASBT significantly in a dose-dependent manner from a concentration of ≥10 μmol/L, and were therefore excluded for further analysis (Figure 3D and F). Bifonazole, bromhexine hydrochloride, and meclozine dihydrochloride also were able to inhibit ASBT significantly, but only when administered in a high concentration of 100 μmol/L (Figure 3A, B, and E). At lower concentrations, uptake values were not decreased significantly, indicating that ASBT was not inhibited. Therefore, clofazimine is the most selective inhibitor of OSTα-OSTβ because this compound did not seem to inhibit ASBT in all tested concentrations (Figure 3C).
Figure 3.

Inhibition of taurocholate uptake by ASBT after treatment with compounds. (A–F) Intracellular taurocholic acid values in MDCKII cells expressing ASBT after a 2-minute uptake after treatment with the following: (A) bifonazole, (B) bromhexine hydrochloride, (C) clofazimine, (D) lovastatin, (E) meclozine dihydrochloride, and (F) simvastatin. Data are normalized as a percentage from control (dimethyl sulfoxide [DMSO], 100% is control) and are represented as means ± SD. Data are from a representative experiment replicated 2 times (n = 3). *P < .05, **P < .01, and ***P < .001 (1-way analysis of variance; post hoc: Dunnett multiple comparison).
Clofazimine Inhibits OSTα-OSTβ In Vitro in a Concentration-Dependent Manner
The 4 compounds that did not inhibit ASBT at a concentration of 10 μmol/L were validated for OSTα-OSTβ inhibition using another method than the primary screen. Transcellular transport of [3H]-taurocholate across MDCKII monolayers expressing ASBT and OSTα-OSTβ was measured. As shown in Figure 4A, apical-to-basal flux was decreased in all compounds tested compared with control cells. To determine whether this effect was really caused by OSTα-OSTβ inhibition, intracellular concentrations of the cells in monolayer culture were measured as well. Indeed, increased accumulation of taurocholate was observed after 2 hours treatment with 10 μmol/L bromhexine hydrochloride, clofazimine, and meclozine dihydrochloride, indicating that the decreased efflux observed is owing to OSTα-OSTβ inhibition (Figure 4B). Bifonazole decreased the intracellular amount of taurocholate and was excluded from follow-up analysis. The low counts of [14C]-inulin transport during the assay that was similar to control cells excludes transport by nonspecific diffusion (leakage <2.5%) and confirms the integrity of the monolayer (Figure 4C). In conclusion, all 4 compounds that underwent further evaluation were confirmed to be OSTα-OSTβ inhibitors. Among the tested compounds, clofazimine showed the highest accumulation of bile acids while not able to inhibit ASBT, and is therefore probably the most specific inhibitor for OSTα-OSTβ and the most potent for increasing bile acid levels in enterocytes. Therefore, subsequent experiments were performed with clofazimine only.
Figure 4.

Transcellular transport of taurocholic acid across MDCKII monolayers. (A) Time profile for taurocholic acid transport across MDCKII monolayers expressing ASBT and OSTα-OSTβ. Cells were treated with 10 μmol/L bromhexine HCl, clofazimine, meclozine 2HCl, or bifonazole for 1 hour before the experiment and during the 2 hours of the experiment when taurocholic acid was added and measured. (B) Intracellular levels of taurocholic acid 2 hours after addition of apical taurocholic acid when treated with dimethyl sulfoxide (DMSO) or 10 μmol/L bromhexine HCl, clofazimine, meclozine 2HCl, and bifonazole. (C) Disintegrations per minute of [14C]-inulin after 30, 60, 90, and 120 minutes of transport in MDCKII cells administered with bromhexine, clofazimine, meclizine, bifonazole, or DMSO (0.01%). (D) Time profile for transcellular transport of taurocholate from the apical to basolateral compartment after treatment with 1, 10, or 30 μmol/L clofazimine. (E) Increased intracellular levels of taurocholate in MDCKII cells after 2 hours uptake in cells treated with 1, 10, or 30 μmol/L clofazimine. (F) Disintegrations per minute of [14C]-inulin after 30, 60, 90, and 120 minutes of transport in MDCKII cells administered with 1, 10, and 3 μmol/L clofazimine or DMSO (0.03%). (G) Apical to basolateral [3H]-taurocholic acid transport in MDCKII parental cells vs MDCKII cells expressing OSTα-OSTβ. (H) [14C]-inulin transport in MDCKII parental cells vs MDCKII-OSTαβ cells. Data are represented as means ± SD. Data are from a representative experiment replicated 2 times (n = 3). *P < .05, **P < .01, and ***P < .001. (A–F) One-way analysis of variance; post hoc: Dunnett multiple comparison; (G and H) Student t test, 2-tailed.
Clofazimine is an iminophenazine drug that originally was developed to treat tuberculosis, but was later used as a key drug in the medication for leprosy.19, 20 We performed a dose-response with clofazimine and found that the inhibiting effect of clofazimine is already present at a concentration of 1 μmol/L. An increase in the administered clofazimine concentration resulted in a modestly increased inhibition of OSTα-OSTβ (Figure 4D). Concentrations of 100 μmol/L and greater led to increased inulin diffusion, suggestive of impaired monolayer integrity, and therefore could not be analyzed (data not shown). In addition, intracellular amounts of taurocholate were increased significantly after 2 hours of treatment of 10 and 30 μmol/L clofazimine (Figure 4E), showing that clofazimine is able to specifically inhibit OSTα-OSTβ in vitro. No paracellular flux of [14C]-inulin was measured in the various concentrations of clofazimine (Figure 4F), and passive diffusion of bile acids through diffusion was not possible (Figure 4G and H).
OSTα-OSTβ Inhibition in Caco-2 Cells Leads to Intracellular Accumulation of Bile Acids
Next, monolayers of a well-differentiated human intestinal epithelial cell line (Caco-2) were used as a model to determine whether inhibition of OSTα-OSTβ results in increased FXR activation. After 21 days of differentiation on Transwell filters, Caco-2 cells endogenously express OSTα and OSTβ, which is strongly up-regulated after 24 hours of treatment with GW4064, indicating functional FXR signaling as well (Figure 5A and B). Quantitative real-time polymerase chain reaction analysis was used on differentiated Caco-2 cells to asses messenger RNA (mRNA) expression of intestinal FXR target genes after 6 hours of treatment of 10 μmol/L clofazimine combined with 3 μmol/L TCDCA. Small heterodimer partner, ileal bile acid binding protein, and OSTα mRNA expression was increased significantly after 6 hours of clofazimine treatment (Figure 5C-F). FGF19 mRNA expression showed the same trend but did not reach significance (data not shown). ASBT mRNA expression was not increased or decreased significantly in Caco-2 cells. Together, these results suggest that clofazimine treatment in differentiated Caco-2 cells leads to increased FXR activation as a result of increased intracellular bile acid concentrations resulting from OSTα-OSTβ inhibition.
Figure 5.

Clofazimine-mediated inhibition of OSTα-OSTβ results in increased FXR target gene expression. (A) Gene expression levels of Ostα before and after 24 hours of treatment with FXR agonist GW4064. (B) mRNA expression levels of the OSTβ gene before and after 24 hours treatment of GW4064. (C–F) mRNA levels of 21-day differentiated Caco-2 cells after treatment with dimethyl sulfoxide (DMSO) (light box plots) or clofazimine (dark box plots). Gene expression of (C) small heterodimer partner (SHP), (D) ileal bile acid binding protein (IBABP), (E) Ostα, or (F) Ostβ. Values were normalized to Cyclophilin B and 36B4 and expressed as arbitrary units. (A and B) Data are represented as means ± SD. ***P < .001 (Student t test, 2-tailed). (C–F) Data are represented as box plots (box, 25%–75%; whisker, 10%–90%; line, median) from 3 independent experiments combined (n = 4 per experiment). *P < .05 (Student t test, 2-tailed).
Clofazimine Is an OSTα-OSTβ Inhibitor In Vivo
C57BL/6 wild-type mice were used to determine whether oral clofazimine is capable of inhibiting OSTα-OSTβ in ileal enterocytes in vivo. To this end, mice were orally administered clofazimine (high dose, 250 mg/kg; low dose, 25 mg/kg) or placebo, followed by an intravenous cholecystokinin octapeptide injection to induce gallbladder contraction to ensure the presence of bile acids in the intestine. All mice were killed 6 hours after treatment. We hypothesized that OSTα-OSTβ inhibition will lead to increased, but nontoxic, intracellular bile acid levels and thereby activate FXR. Therefore, we measured gene expression levels of the FXR target genes Fgf15, Ibabp, Ostα, and Ostβ in the distal part of the ileum in mice. In the low-dose group, all FXR target genes (Ibabp, Fgf15, Ostα, and Ostβ) were increased significantly compared with the placebo-treated mice (Figure 6A–D). This effect was confirmed in the high-dose group, which showed an even more significant increase in mRNA expression of FXR target genes. These results imply that clofazimine is able to inhibit OSTα-OSTβ both in vitro and in vivo.
Figure 6.

Oral clofazimine administration results in increased intracellular bile acid concentrations in ileal enterocytes. (A–D) Eight-week old mice were orally administered placebo, 25 mg/kg clofazimine, or 250 mg/kg clofazimine solved in corn oil followed by a cholecystokinin octapeptide injection (50 ng/kg). After 6 hours, mice were killed and mRNA levels were measured in the distal part of the ileum. Gene expression of (A) Ibabp, (B) Fgf15, (C) Ostα, and (D) Ostβ were measured. Values were normalized to Hypoxanthine-guanine phosphoribosyltransferase and 36B4 and expressed as arbitrary units. Data are represented as means ± SD (n = 13). *P < .05, **P < .01, and ***P < .001 (1-way analysis of variance; post hoc: Dunnett multiple comparison).
Clofazimine-Mediated Inhibition of OSTα-OSTβ Only Transiently Increases Intestinal FXR Activation
Because clofazimine moderately inhibits OSTα-OSTβ, and intestinal FXR activation leads to up-regulation of both OSTα and OSTβ, a longer experiment was conducted to identify whether clofazimine-mediated OSTα-OSTβ inhibition is durable. Currently, clofazimine is used as a slow-responding antibiotic in the treatment of leprosy (and to a lesser extent tuberculosis). Possible bacteriocidal effects could interfere with bile acid signaling, so we limited exposure to 5 days. Mice were orally gavaged for 5 days with placebo (corn oil), a low dose of clofazimine (25 mg/kg), or a high dose of clofazimine (75 mg/kg). There were no changes in bile acid composition/conjugation in bile, plasma, feces, and urine (Figure 7A–D), suggesting that major effects on microbiota are unlikely at this treatment duration. Bile acid concentrations in plasma (Figure 7E) or urine (Figure 7F) also were unchanged. The only difference found was the reduced total fecal output in mice orally gavaged with clofazimine (Figure 7G). The absence of an increase in fecal bile acid output suggests that OSTα-OSTβ inhibition was transient and largely lost on a 5-day clofazimine exposure. In line with this is the mRNA expression levels of FXR target genes Ostβ and Fgf15 showing a significant decrease instead of increase (Figure 8A), and Ostα and Ibabp levels remained unchanged. To test whether a reduced bile acid pool was the cause of the observed reduced FXR activation in intestine, bile acid synthesis gene expression levels were measured in liver tissue. However, there was no apparent change in either Cyp7a1 or Cyp8b1 levels in clofazimine-treated mice compared with placebo-treated mice (Figure 8B). Shp and Ostβ levels were increased in livers of clofazimine-treated mice. There was no significant change in total body weight or epididymal white adipose tissue weight (Figure 8C–E). Furthermore, blood glucose levels were slightly increased in the low-dose compared with placebo-treated mice, but in the high-dose group there was no apparent change (Figure 8F).
Figure 7.

Bile acid levels and composition are unchanged after 5 days of oral clofazimine treatment. (A–D) Bile acid composition in bile, feces, urine, and plasma of mice orally gavaged with placebo, 25 mg/kg clofazimine (low dose), or 75 mg/kg clofazimine (high dose). (E–G) Bile acid concentration in (E) blood plasma and (F) urine, or (G) total fecal bile acid output of mice treated with placebo, a high dose, or a low dose of clofazimine for 5 days. Bile acid composition is expressed as a percentage of total bile acid concentration (μmol/L). Data are represented as means ± SD. *P < .05, n = 15 mice per group (1-way analysis of variance; post hoc: Dunnett multiple comparison). αMC, alpha muricholate; BA, bile acids; βMC, beta-muricholate; CA, cholic acid; DC, deoxycholate; GCDC, glycochenodeoxycholic acid; omega-muricholate (ΩMC)TαMC, tauro-alpha muricholate; TβMC, tauro-beta muricholate; TCA, taurocholic acid; TC, Taurocholate; TDC, Taurodeoxycholic acid.
Figure 8.

Relative quantitative reverse-transcription polymerase chain reaction analysis of FXR target gene expression in intestine and liver. (A) Intestinal mRNA levels of Ibabp, Fgf15, Ostα, and Ostβ after 5 days of oral gavage of placebo corn oil (white bars), 25 mg/kg clofazimine (grey bars), or 75 mg/kg clofazimine (black bars). (B) Hepatic mRNA levels of Cyp8b1, Cyp7a1, Shp, and Ostβ of mice orally gavaged with placebo or a high dose or a low dose of clofazimine for 5 days. Values were normalized to TATA-box binding protein and 36B4 and expressed as arbitrary units. (C) Relative body weight of 8-week-old male mice over a 5-day period. Relative body weight was calculated as a function of the body weight at the beginning of the experiment (day -1). (D) Body weight at the end of the experiment (day 5). (E) Weight of epididymal white adipose tissue (eWAT) per kilogram of body weight in mice treated with clofazimine for 5 days. (F) Blood glucose levels (mmol/L) on the day of killing (day 5). Data are represented as means ± SD. N = 13 mice per group. *P < .05, **P < .01, and ***P < .001 (1-way analysis of variance; post hoc: Dunnett’s multiple comparison).
Discussion
In this study we proposed OSTα-OSTβ inhibition as a novel strategy to intestine-specifically activate the nuclear bile acid receptor FXR, developed a high-throughput live cell FRET-based method that can be used to screen large libraries of natural and synthetic compounds, and showed (transient) in vivo proof of concept using clofazimine as the first identified OSTα-OSTβ inhibitor.
Before this study, no inhibitors for OSTα-OSTβ were discovered. The ability of the FRET bile acid sensor to measure bile acid transport in living cells enables its use in robust high-throughput and real-time screening with the advantage of ratiometric detection.21 This screen is capable of imaging export of bile acids instead of import without the requirement of modified bile acids or the necessity for destruction of the sample. The selected hits from the primary screen were confirmed by a secondary screen, and were tested positive for inhibition of OSTα-OSTβ–mediated bile acid transport, supporting the reliability of this screen. Counter-screening for ASBT inhibition seems advisable because most hits also inhibited this bile acid transporter at high dosages. Although the identification from such a small library of FDA-approved compounds shows proof of concept for this screening strategy, this drug is not an ideal OSTα-OSTβ inhibitor yet. The ability of clofazimine to inhibit OSTα-OSTβ–mediated bile acid transport was effective in a short-term experiment because all expression levels of FXR target genes seemed induced, but is not sustained owing to compensatory OSTα-OSTβ up-regulation. Furthermore, clofazimine can moderately inhibit the hepatic bile acid efflux machinery bile salt export pump as well at high concentrations.22 This possibly may explain the small but significant increase in expression of the hepatic FXR target gene Shp seen on 5 days of clofazimine exposure. The discovery of a more potent and specific OSTα-OSTβ inhibitor will abolish this effect.
FXR activation in the intestine is of clear importance in several (patho)physiological processes, including bile acid synthesis. Intestinal FXR activation leads to FGF15/FGF19 secretion, repression of Cyp7a1, and, consequently, a reduced and more hydrophilic bile salt pool.23, 24 This leads to protection against hepatocellular damage in cholestatic liver disorders25 and stimulated cholesterol removal via direct TICE.5 TICE is a major contributor to cholesterol removal from the body in mice and human beings and likely contributes to the prevention of cardiovascular disease.26 Similarly, OSTα-/- mice show a decreased bile acid pool, increased cholesterol excretion, decreased Cyp7a1 mRNA expression and an increased Fgf15 mRNA expression,8 and reduced hepatic damage in a severe model for cholestasis.10 Although we here identified clofazimine as the first OSTα-OSTβ inhibitor, its efficacy is insufficient to overcome the compensatory up-regulation of OSTα-OSTβ seen on FXR activation. However, short-term treatment in vitro and in vivo show that Fgf15/FGF19 is induced significantly, showing proof of concept that OSTα-OSTβ inhibitors can be identified/developed.
Inhibition of OSTα-OSTβ can activate intestinal FXR in an indirect way. Two main advantages of this approach are that FXR activation will now take place specifically in enterocytes, thereby largely eliminating unwanted FXR activation in other tissues,4 and the prolonged and enhanced postprandial FXR activation is more moderate compared with synthetic FXR agonists and better matches the circadian rhythm. Together, this is expected to reduce the risks for excessive FGF19 signaling implicated with hepatocellular carcinoma.27 The mild phenotype of OSTα knockout mice suggests that this strategy is safe. Other bile acid efflux pathways at the basolateral membrane of ileocytes prevent toxic accumulation of bile salts. The clear protective role of FXR in intestine suggests a third possible application of OSTα-OSTβ inhibitors. FXR activation dampens intestinal inflammation, improves epithelial barrier function and permeability, and inhibits proinflammatory cytokine production in the mouse colonic mucosa.28, 29 Notably, clofazimine also is incidentally used in inflammatory bowel disease and has an anti-inflammatory capacity.30, 31
Finally, OSTα-/- mice are more resistant to age-related weight gain and body fat accumulation and less prone to lipid accumulation in liver and muscle tissue. However, these effects are subtle and it is still unclear whether these positive effects are generalizable in patients. Furthermore, OSTα-deficient mice, and especially male mice, showed an improved glucose tolerance and insulin sensitivity,9 albeit less prominently on a high-fat/high-cholesterol diet.32 In contrast, we observe slightly increased glucose levels in mice receiving the low clofazimine dose. However, this effect was not observed in the high-dose group, suggesting that the effect on glucose levels is not caused by OSTα-OSTβ inhibition itself. Therefore, the concept of inhibiting OSTα-OSTβ and thereby specifically activating intestinal FXR could provide an effective therapeutic approach in diseases such as cholestasis, specific aspects of the metabolic syndrome, and perhaps also for patients suffering from low FGF19 levels as seen in primary bile acid diarrhea.
In conclusion, by using our screening strategy we have discovered a novel characteristic of an existing drug, clofazimine, that selectively inhibits OSTα-OSTβ and we have shown the utility of our high-throughput screen FRET-based assay to identify novel inhibitors for OSTα-OSTβ. This report shows that pharmacologic inhibition of OSTα-OSTβ in vitro and in vivo results in activation of FXR specifically in the intestine. Therefore, OSTα-OSTβ appears to be a potential therapeutic target for diseases in which intestinal FXR activation and the resulting increased FGF15/19 levels seem beneficial. Furthermore, this method also could have use for the identification of novel inhibitors for other bile acid transporters, and therefore has an application in elucidating candidates regulating bile acid signaling that could have a therapeutic effect in other metabolic diseases.
Acknowledgments
The authors would like to thank Dr James Boyer for conceptual discussion and Dr Paul Dawson (Emory University School of Medicine, Atlanta, GA) for generously providing constructs and the MDCK cell lines expressing ASBT, OSTα, and OSTβ.
Footnotes
Author contributions Sandra M. W. van de Wiel and Stan F. J. van de Graaf were responsible for the study concept and design; all authors acquired data, analyzed and interpreted data, and drafted and revised the manuscript; and Stan F. J. van de Graaf obtained funding and performed study supervision.
Conflicts of interest The authors disclose no conflicts.
Funding Supported by The Netherlands Organization for Scientific Research (Vidi 91713319) and European Research Council grant 337479 (S.F.J.v.d.G.).
References
- 1.Hofmann A.F. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 2.Chiang J. Regulation of bile acid synthesis. Front Bbiosci. 1998;3:D176–D193. doi: 10.2741/a273. [DOI] [PubMed] [Google Scholar]
- 3.Kuipers F., Bloks V.W., Groen A.K. Beyond intestinal soap–bile acids in metabolic control. Nat Rev Endocrinol. 2014;10:488–498. doi: 10.1038/nrendo.2014.60. [DOI] [PubMed] [Google Scholar]
- 4.Fang S., Suh J.M., Reilly S.M., Yu E., Osborn O., Lackey D., Yoshihara E., Perino A., Jacinto S., Lukasheva Y. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21:159–165. doi: 10.1038/nm.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Boer J.F., Schonewille M., Boesjes M., Wolters H., Bloks V.W., Bos T., van Dijk T.H., Jurdzinski A., Boverhof R., Wolters J.C. Intestinal farnesoid X receptor controls transintestinal cholesterol excretion in mice. Gastroenterology. 2017;152:1126–1138. doi: 10.1053/j.gastro.2016.12.037. [DOI] [PubMed] [Google Scholar]
- 6.Dawson P.A., Lan T., Rao A. Bile acid transporters. J Lipid Res. 2009;50:2340–2357. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballatori N., Christian W.V., Lee J.Y., Dawson P.A., Soroka C.J., Boyer J.L., Madejczyk M.S., Li N. OSTα-OSTβ: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 8.Rao A., Haywood J., Craddock A.L., Belinsky M.G., Kruh G.D., Dawson P.A. The organic solute transporter α-β, Ostα-Ostβ, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A. 2008;105:3891–3896. doi: 10.1073/pnas.0712328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wheeler S.G., Hammond C.L., Jornayvaz F.R., Samuel V.T., Shulman G.I., Soroka C.J., Boyer J.L., Hinkle P.M., Ballatori N. Ostα−/− mice exhibit altered expression of intestinal lipid absorption genes, resistance to age-related weight gain, and modestly improved insulin sensitivity. Am J Physiol Gastrointest Liver Physiol. 2014;306:G425–G438. doi: 10.1152/ajpgi.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soroka C.J., Mennone A., Hagey L.R., Ballatori N., Boyer J.L. Mouse organic solute transporter α deficiency enhances renal excretion of bile acids and attenuates cholestasis. Hepatology. 2010;51:181–190. doi: 10.1002/hep.23265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soroka C.J., Velazquez H., Mennone A., Ballatori N., Boyer J.L. Ostα depletion protects liver from oral bile acid load. Am J Physiol Gastrointest Liver Physiol. 2011;301:G574–G579. doi: 10.1152/ajpgi.00141.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ballatori N., Fang F., Christian W.V., Li N., Hammond C.L. Ostα-Ostβ is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am J Physiol Gastrointest Liver Physiol. 2008;295:G179–G186. doi: 10.1152/ajpgi.90319.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ballatori N., Christian W.V., Wheeler S.G., Hammond C.L. The heteromeric organic solute transporter, OSTα–OSTβ/SLC51: a transporter for steroid-derived molecules. Mol Aspect Med. 2013;34:683–692. doi: 10.1016/j.mam.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van der Velden L.M., Golynskiy M.V., Bijsmans I.T., Van Mil S.W., Klomp L.W., Merkx M., Van de Graaf S.F. Monitoring bile acid transport in single living cells using a genetically encoded Förster resonance energy transfer sensor. Hepatology. 2013;57:740–752. doi: 10.1002/hep.26012. [DOI] [PubMed] [Google Scholar]
- 15.Dawson P.A., Hubbert M., Haywood J., Craddock A.L., Zerangue N., Christian W.V., Ballatori N. The heteromeric organic solute transporter alpha-beta, ost alpha-ost beta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slijepcevic D., Roscam Abbing R.L.P., Katafuchi T., Blank A., Donkers J.M., van Hoppe S., de Waart D.R., Tolenaars D., van der Meer J.H.M., Wildenberg M., Beuers U., Oude Elferink R.P.J., Schinkel A.H., van de Graaf S.F.J. Hepatic uptake of conjugated bile acids is mediated by both NTCP and OATPs and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology. 2017;66:1631–1643. doi: 10.1002/hep.29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballatori N., Li N., Fang F., Boyer J.L., Christian W.V., Hammond C.L. OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front Biosci. 2009;14:2829. doi: 10.2741/3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang F., Christian W.V., Gorman S.G., Cui M., Huang J., Tieu K., Ballatori N. Neurosteroid transport by the organic solute transporter OSTα–OSTβ. J Neurochem. 2010;115:220–233. doi: 10.1111/j.1471-4159.2010.06920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barry V.C., Belton J., Conalty M.L., Denneny J.M., Edward D.W., O'sullivan J., Twomey D., Winder F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature. 1957;179:1013–1015. doi: 10.1038/1791013a0. [DOI] [PubMed] [Google Scholar]
- 20.Arbiser J.L., Moschella S.L. Clofazimine: a review of its medical uses and mechanisms of action. J Am Acad Dermatol. 1995;32:241–247. doi: 10.1016/0190-9622(95)90134-5. [DOI] [PubMed] [Google Scholar]
- 21.Merkx M., Golynskiy M.V., Lindenburg L.H., Vinkenborg J.L. Rational design of FRET sensor proteins based on mutually exclusive domain interactions. Biochem Soc Trans. 2013;41:1201–1205. doi: 10.1042/BST20130128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Te Brake L.H., Russel F.G., Van Den Heuvel J.J., De Knegt G.J., De Steenwinkel J.E., Burger D.M., Aarnoutse R.E., Koenderink J.B. Inhibitory potential of tuberculosis drugs on ATP-binding cassette drug transporters. Tuberculosis. 2016;96:150–157. doi: 10.1016/j.tube.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Gadaleta R.M., Cariello M., Sabbà C., Moschetta A. Tissue-specific actions of FXR in metabolism and cancer. Biochim Biophys Acta. 2015;1851:30–39. doi: 10.1016/j.bbalip.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Inagaki T., Choi M., Moschetta A., Peng L., Cummins C.L., McDonald J.G., Luo G., Jones S.A., Goodwin B., Richardson J.A. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Modica S., Petruzzelli M., Bellafante E., Murzilli S., Salvatore L., Celli N., Di Tullio G., Palasciano G., Moustafa T., Halilbasic E. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology. 2012;142:355–365. e4. doi: 10.1053/j.gastro.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 26.Jakulj L., van Dijk T.H., de Boer J.F., Kootte R.S., Schonewille M., Paalvast Y., Boer T., Bloks V.W., Boverhof R., Nieuwdorp M. Transintestinal cholesterol transport is active in mice and humans and controls ezetimibe-induced fecal neutral sterol excretion. Cell Metab. 2016;24:783–794. doi: 10.1016/j.cmet.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Nicholes K., Guillet S., Tomlinson E., Hillan K., Wright B., Frantz G.D., Pham T.A., Dillard-Telm L., Tsai S.P., Stephan J.-P. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol. 2002;160:2295–2307. doi: 10.1016/S0002-9440(10)61177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadaleta R.M., van Erpecum K.J., Oldenburg B., Willemsen E.C., Renooij W., Murzilli S., Klomp L.W., Siersema P.D., Schipper M.E., Danese S. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472. doi: 10.1136/gut.2010.212159. [DOI] [PubMed] [Google Scholar]
- 29.Inagaki T., Moschetta A., Lee Y.-K., Peng L., Zhao G., Downes M., Ruth T.Y., Shelton J.M., Richardson J.A., Repa J.J. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patton P.H., Parker C.E., MacDonald J.K., Chande N. Anti-tuberculous therapy for maintenance of remission in Crohn's disease. Cochrane Database Syst Rev. 2016;7:CD000299. doi: 10.1002/14651858.CD000299.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon G.S., Sud S., Keswani R.K., Baik J., Standiford T.J., Stringer K.A., Rosania G.R. Phagocytosed clofazimine biocrystals can modulate innate immune signaling by inhibiting TNFα and boosting IL-1RA secretion. Mol Pharma. 2015;12:2517–2527. doi: 10.1021/acs.molpharmaceut.5b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammond C.L., Wheeler S.G., Ballatori N., Hinkle P.M. Ostα−/− mice are not protected from western diet-induced weight gain. Physiol Rep. 2015;3:e12263. doi: 10.14814/phy2.12263. [DOI] [PMC free article] [PubMed] [Google Scholar]