

Abstract
Cognitive behavioral therapy (CBT) and selective serotonin reuptake inhibitors (SSRIs) are both effective treatments for some patients with obsessive-compulsive disorder (OCD), yet little is known about the neurochemical changes related to these treatment modalities. Here, we used positron emission tomography and the α-[11C]methyl-l-tryptophan tracer to examine the changes in brain regional serotonin synthesis capacity in OCD patients following treatment with CBT or SSRI treatment. Sixteen medication-free OCD patients were randomly assigned to 12 weeks of either CBT or sertraline treatment. Pre-to-post treatment changes in the α-[11C]methyl-l-tryptophan brain trapping constant, K* (ml/g/min), were assessed as a function of symptom response, and correlations with symptom improvement were examined. Responders/partial responders to treatment did not show significant changes in relative regional tracer uptake; rather, in responders/partial responders, 12 weeks of treatment led to serotonin synthesis capacity increases that were brain-wide. Irrespective of treatment modality, baseline serotonin synthesis capacity in the raphe nuclei correlated positively with clinical improvement. These observations suggest that, for some patients, successful remediation of OCD symptoms might be associated with greater serotonergic tone.
Introduction
Obsessive-compulsive disorder (OCD) is a chronic mental illness involving intrusive, unwanted thoughts (obsessions) and persistent mental or behavioral rituals (compulsions) that cause significant deficits in social functioning. Cognitive behavioral therapy (CBT) and selective serotonin reuptake inhibitors (SSRIs) have, in separate multicenter trials, demonstrated efficacy and tolerability in the treatment of 40–60% of OCD patients1,2. The success of SSRIs, relative to medications targeting neurotransmitter systems other than serotonin (5-hydroxytryptamine (5-HT)), suggests that the latter may play a role in the remediation of OCD symptoms3,4. Despite the documented effectiveness of these treatments, changes in neurochemistry in vivo associated with CBT or SSRI in OCD patients, including changes in the serotonergic system, remain elusive.
Neuroimaging and neurosurgical studies have implicated the cortico-striato-thalamo-cortical (CSTC) circuit in OCD neurobiology5; indeed, effective OCD treatments with either SSRIs, clomipramine, or behavior therapy, alone or in combination, have been reported to decrease abnormally elevated CSTC circuit activity6,7. Notably, however, conflicting findings have been reported, including increased activity within CSTC circuitry following successful OCD treatment8. Positron emission tomography (PET) and single photon emission computed tomography (SPECT) studies have investigated more specific aspects of neurotransmission within CSTC circuitry, including measuring 5-HT transporter (5-HTT) and receptor binding, using tracers such as [11C]DASB, [123I]β-CIT, [11C]McN 5652, and [11C]MDL100907. Pre-treatment, baseline abnormalities in 5-HTT and 5-HT2A receptor availabilities within CSTC circuitry have been reported in OCD patients9–11, although there has been considerable variability12–14.
To date, few studies have investigated changes in the serotonergic system during OCD treatment. Early studies found changes in cerebrospinal fluid 5-HT metabolite levels and blood platelet 5-HTT levels pre–post treatment15, but these findings have not been replicated16, and peripheral 5-HT measures cannot be used to study brain regional changes in serotonergic functioning. To our knowledge, only one study has investigated within-subject brain regional changes in the serotonergic system in OCD patients before and after treatment: Zitterl et al. reported a significant reduction in 5-HTT availability in the thalamus/hypothalamus of OCD patients, using SPECT and [123I]β-CIT, following 12 weeks of clomipramine treatment17. Similar decreases during repeated exposure to SSRIs in various pathological and non-pathological conditions were also reviewed17. To our knowledge, no studies have explored the effects of CBT on the serotonergic system in OCD patients. Moreover, 5-HTT imaging has been interpreted by many to reflect density of innervation, rather than functional status per se18.
The PET tracer α-[11C]methyl-l-tryptophan (α-[11C]MTrp) is thought to reflect central 5-HT metabolism in humans in vivo19,20. α-[11C]MTrp is analogous to the 5-HT precursor, l-tryptophan, except that it is not incorporated into protein21. After crossing the blood-brain barrier, α-[11C]MTrp is taken up into serotonergic neurons, and ultimately is metabolized into α-M-5-HT. α-M-5-HT is not degraded by monoamine oxidase and cannot cross the blood–brain barrier, thereby accumulating in serotonergic neurons. The net blood-to-brain clearance of the tracer is used to calculate the α-[11C]MTrp trapping (unidirectional uptake) constant, K* (in ml/g/min). α-[11C]MTrp has been used to study 5-HT synthesis capacity, and more generally, 5-HT metabolism, in various patient populations22–24. In particular, we previously used α-[11C]MTrp to study baseline 5-HT synthesis capacity rates in OCD patients, and reported abnormally elevated α-[11C]MTrp trapping, relative to controls, in temporal, striatal, and limbic regions25.
As a follow-up to our baseline study of treatment-free OCD patients, the present study investigated the effects of drug treatment or CBT on brain regional 5-HT synthesis capacity. OCD patients were randomly assigned to either CBT or SSRI monotherapy (sertraline), and α-[11C]MTrp PET scans were repeated following 12 weeks of treatment. The goals of the present study were to (i) compare regional 5-HT synthesis capacity in OCD patients before and after treatment with CBT or sertraline, and (ii) identify brain regions where pre-treatment regional 5-HT synthesis capacity is associated with treatment outcome. Here, we expected that changes in α-[11C]MTrp uptake, particularly within CSTC circuitry, would relate to changes in obsessive-compulsive, but not mood, symptoms. However, as the first study of treatment-related changes in serotonin synthesis capacity in OCD patients, the current study was designed to be primarily exploratory in nature.
Materials and methods
Study population
Patients were referred by the OCD Clinic, Department of Psychology, McGill University Health Center (MUHC), having participated in a baseline PET study prior to beginning treatment25. Exclusion criteria included: (1) personal or family history of Tourette’s syndrome; (2) history of other Axis I disorders, except for depression secondary to OCD, as assessed using the Structured Clinical Interview for DSM-IV Axis I Disorders (SCID)26; (3) current or past substance abuse or dependence; (4) current or past use of 3,4-methylene-dioxy-methamphetamine (MDMA) or methylene-dioxy-amphetamine (MDA); and (5) history of allergy or treatment resistance to sertraline. All patients, at entry into the study, were medication-free for at least 3 weeks or more than five elimination half-lives of the drug, whichever was longer. Most patients were medication-free for considerably longer; of eight patients previously treated with antidepressants, seven were drug-free >6 months at entry into the study.
After inclusion in the study, the patients were randomly assigned (using a block randomization design with blocks of 4) to receive CBT or sertraline treatment for a period of 12 weeks. OCD symptom severity, assessed using the Yale-Brown Obsessive Compulsive Scale (Y-BOCS), and depressive symptoms, estimated with the Beck Depression Inventory (BDI), were recorded by a clinician blind to the patient’s PET data approximately every 2 weeks during treatment, beginning at baseline (week 0). Following completion of the 12-week treatment study, patients in both groups were offered further treatment as clinically indicated.
All participants provided written, informed consent. The study was carried out in accordance with the Declaration of Helsinki, and was approved by the Research Ethics Committee of the Montreal Neurological Institute (MNI) and the Institutional Review Board of McGill University.
Treatment
Patients assigned to sertraline treatment received an initial dose of 25 mg/day. Sertraline was provided in an open fashion, as 25 mg capsules ingested once daily, in the morning with food. After 1 week of treatment, unless limited by side effects, the daily dose was increased to 50 mg/day. If, after a second week, the patient’s therapeutic response did not show evidence of symptomatic improvement, this dose was increased to 100 mg/day unless limited by side effects. A third increase in dose to 150 mg/day after another 2 weeks (week 4) and a final increase to 200 mg/day (week 6) were each made if response remained unsatisfactory (<20% decrease in Y-BOCS score). The final mean ± SD daily dose of sertraline was 133 ± 52 mg/day, and all patients were prescribed a stable dose of medication during the last 2 weeks of the study.
Patients assigned to CBT received two 90-min individual sessions per week for 12 weeks. Specialized CBT was designed and administered under the close supervision of DS, an experienced OCD expert clinician and supervisor. The specialty multidimensional CBT program was individualized for each patient and included: psycho-education; cognitive therapy to collaboratively modify symptom-related appraisals and meanings of intrusive thoughts and feared situations; strategies for dysfunctional cognitive–emotional processing, intolerance of distress, and overestimation of threat; exposure and response prevention (ERP) and behavioral experiment protocols designed to optimize adaptive learning; self-directed, between-session homework with attention to treatment adherence; and interventions for relapse prevention, resilience, and self-efficacy. Therapist-assisted ERP and behavioral experiments were administered in patients’ home as needed. Interventions specifically targeted the symptom subtype characteristics for each case27.
PET and magnetic resonance imaging (MRI)
PET scans were performed before and after 12 weeks of treatment. PET and MRI procedures were carried out as per Berney et al.25. Briefly, in order to minimize variability between scans in plasma concentrations of amino acids, such as tryptophan, a low-protein diet followed by an overnight fast was required of participants before scanning days28. On PET scan days, all participants tested negative on a urine drug screen sensitive to cocaine, opiates, phencyclidine, cannabinoids, barbiturates, benzodiazepines, and amphetamines (Triage Panel for Drugs of Abuse, Biosite Diagnostics, CA, USA). Additionally, women of fertile age were scanned during their follicular phase, due to previous findings of changes in serotonergic activity in different phases of the estrous cycle in rats29 and the menstrual cycle in women30.
α-[11C]MTrp was produced as described elsewhere31. PET scanning was performed using an ECAT HR+ scanner (CTI/Siemens, Knoxville, TN; 3D mode with a resolution of 5 × 5 × 5 mm full width at half maximum (FWHM)) in the late morning/early afternoon. After a transmission scan for attenuation correction using a 68Ge/Ga source, α-[11C]MTrp was injected intravenously over 2 min (mean ± SD = 9.6 ± 0.8 mCi), and a 60-min dynamic image acquisition scan was performed. Thirteen venous blood samples were collected to compute the α-[11C]MTrp input function, as described previously32,33. Five plasma samples were used to measure free and total plasma tryptophan concentrations using high-performance liquid chromatography.
Each participant also underwent a T1-weighted MRI scan for PET-MR co-registration using a 1.5 T Philips Gyroscan scanner (Philips Medical Systems, Eindhoven, Netherlands; 3D fast-field echo scan: TR = 18 ms; TE = 10 ms; FA = 30°; 256 × 256 × 160 mm matrix; 1 mm3 isotropic resolution).
Calculation of α-[11C]MTrp trapping (K*)
The Patlak graphic approach34 was used to calculate absolute K* values (ml/g/min), using dynamic PET data collected 20–60 min after tracer injection32,33 and peripheral metabolite values. To account for any effect of global differences in α-[11C]MTrp trapping on regional values, relative regional K* values were calculated by normalizing absolute regional K* values to global K* values (defined as the mean K* value for gray matter). Given that both relative and absolute K* values were previously reported to be stable over several weeks within an individual35, we also examined within-subject changes in absolute regional K* values. Pre- and post-treatment comparisons of regional and global K* values were carried out using both Statistical Parametric Mapping (SPM) and an MRI-based region of interest (ROI) method.
Voxel-based analysis using SPM
Brain-wide voxel-wise analyses comparing K* values pre- and post-treatment were carried out using SPM12 (Wellcome Functional Imaging Laboratory). K* images were spatially normalized into MNI-305 stereotaxic space, using an algorithm described elsewhere36, and then smoothed using a 14-mm FWHM Gaussian filter to reduce the effect of anatomical variability. The t-test was applied voxel by voxel. The height threshold used to interpret the t-test in terms of probability level was set at p < 0.001, uncorrected for multiple comparisons, with an extent threshold of 100 voxels, as previously25, then at 50 voxels for exploratory analyses. The t-map threshold was T8 = 4.50 for responders/partial responders and T5 = 5.89 for non-responders.
MRI-based ROI analysis
Pre–post treatment changes in regional K* values were also analyzed using an a priori MRI-based ROI approach. Each patient’s MRI data were corrected for field inhomogeneities and spatially normalized into MNI-305 stereotaxic space. Using an automatic segmentation method37,38, ROIs were defined in the left and right caudate, hippocampus, inferior temporal gyrus, cingulate, lateral and medial prefrontal cortices, nucleus accumbens, putamen, and thalamus. ROIs were smoothed using a 7 mm FWHM Gaussian filter and resampled into PET acquisition space. Time–activity curves were then derived by applying the ROIs to dynamic native PET space.
Results
Demographics
Sixteen patients with a diagnosis of OCD as per the SCID26 were included in the study. After randomization, eight patients received CBT (6M/2F), and eight sertraline (6M/2F). Data from a post-treatment PET scan were not available for one male patient treated with CBT for technical reasons, therefore a total of 15 patients was included in all PET analyses (11M/4F; mean ± SD age = 34.4 ± 9.3 years).
The demographic and clinical characteristics of the OCD patients are summarized in Table 1 for each treatment subgroup, and in Supplementary Table 1 for each clinical response subgroup. No significant differences in age, Y-BOCS score, or BDI score were found prior to treatment between treatment subgroups, or between subgroups of “responders & partial responders” vs. “non-responders”.
Table 1.
Patient demographics
| Characteristic | CBT (n = 8) | SSRI (n = 8) | ||
|---|---|---|---|---|
| Age, y | ||||
| Mean (SD) | 33.7 (9.5) | 33.4 (8.5) | ||
| Range | 23–53 | 18–45 | ||
| Responders/partial responders | 4/8 | 6/8 | ||
| Early-onset OCD (≤10 y), No. | 5 | 5 | ||
| Predominant compulsion, No. | ||||
| Washing | 4 | 4 | ||
| Checking | 4 | 4 | ||
| Lifetime history of MDE (2° to OCD symptoms), No. | 2 | 3 | ||
| Past substance abuse, No. | 0 | 0 | ||
| Pre | Post | Pre | Post | |
| Y-BOCS score, mean (SD) | 23 (4.4) | 15.8 (7.5) | 23.6 (5.6) | 14.7 (8.2) |
| BDI score, mean (SD) | 9.8 (4.5) | 6.9 (6.0) | 14.1 (11.2) | 7.6 (9.5) |
| Plasma free tryptophan, mean (SD), nmol/La | 10.3 (2.6) | 8.4 (1.4) | 9.8 (1.7) | 9.3 (2.1) |
| Global K*, mean (SD), mL/g/mina | 5.1 (1.3) | 6.1 (1.5) | 5.8 (1.3) | 6.07 (2.0) |
| Intravenously injected, mean (SD), mCia | 9.3 (1.1) | 9.6 (0.7) | 9.6 (0.8) | 9.7 (0.4) |
Responders/partial responders demonstrated a >25% decrease in Y-BOCS score
OCD obsessive-compulsive disorder, CBT cognitive behavioral therapy, SSRI selective serotonin re-uptake inhibitor, MDE major depressive episode, Y-BOCS Yale-Brown Obsessive Compulsive Scale, BDI Beck Depression Inventory, No. number
aData not included for one patient treated with CBT
Clinical response
We observed a progressive improvement in mean Y-BOCS scores for both treatment groups, as illustrated in Fig. 1. At 12 weeks of continuous monitoring of clinical response, seven patients were deemed responders to treatment (≥35% decrease in Y-BOCS score), and three patients were deemed partial responders to treatment (≥25% but ≤35% reduction in Y-BOCS score)39; these 10 patients were combined into a group of responders/partial responders to treatment for all analyses (4 CBT/6 SSRI; mean ± SD % decrease in Y-BOCS score = 52.5 ± 20.1). Six patients were deemed non-responders to treatment (4 CBT/2 SSRI; <25% decrease in Y-BOCS score; mean ± SD % decrease = 1.9 ± 22.4). Overall, there was a significant decrease in Y-BOCS scores pre–post treatment (two-tailed paired t-test; t15 = 4.49, p < 0.001); there was no significant difference between CBT and SSRI treatment groups in the pre–post % change in Y-BOCS scores (two-tailed independent t-test, t14 = 0.72, p = 0.48). In the whole sample, BDI scores pre–post treatment decreased significantly (Wilcoxon signed-rank test, Z = −2.2, p = 0.026), though the effect was clinically minimal.
Fig. 1. OCD symptom improvement over time with CBT or sertraline treatment.
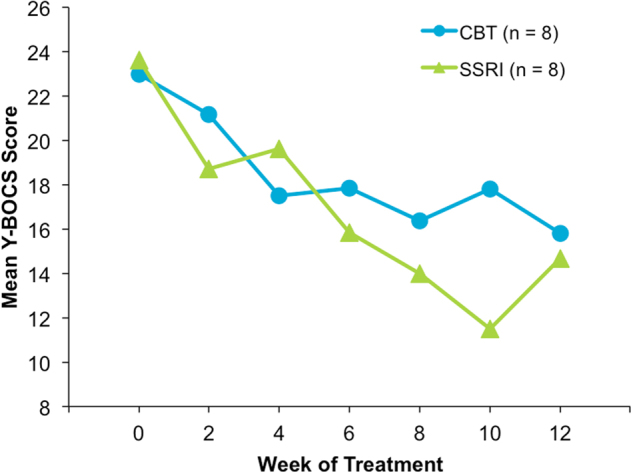
Change in mean Y-BOCS scores during 12 weeks of CBT or sertraline treatment, as measured approximately every 2 weeks. Y-BOCS Yale-Brown Obsessive Compulsive Scale, CBT cognitive behavioral therapy, SSRI selective serotonin re-uptake inhibitor
Global and regional α-[11C]MTrp trapping
Using SPM analysis, the functional images of all OCD patients from pre- and post-treatment conditions were first compared (Pre > Post and Pre < Post). No significant changes in relative (normalized) or absolute regional K* values were observed when treatment groups were combined. Accordingly, no ROIs demonstrated a significant pre–post change in relative or absolute K* values in the ROI-based analyses, and further, there was no significant pre–post change in global K* values in the whole patient sample. Similarly, when pre- and post-treatment α-[11C]MTrp trapping was compared within each treatment group separately (sertraline or CBT), no relative or absolute regional or global changes in K* values were identified.
Next, we compared pre- and post-treatment α-[11C]MTrp trapping in the sub-sample of responders/partial responders (n = 9) and non-responders (n = 6). Using SPM and ROI-based analyses, again, no significant pre–post changes in relative regional K* values were identified in treatment responders. However, responders/partial responders demonstrated a significant increase in global K* values pre–post treatment (two-tailed paired t-test, t8 = 3.05, p = 0.016; mean increase of 29.7%, Cohen’s d = 1.02), whereas non-responders showed no significant treatment-related changes in global K* values (two-tailed paired t-test, t5 = 0.63, p = 0.55; mean decrease of 6.4%). Pre-treatment values of global K* did not differ significantly between responders/partial responders and non-responders. Correspondingly, voxel-wise analyses identified brain-wide increases in absolute K* values (right » left; yet, increases in absolute K* values were observed bilaterally in the ROI analyses, see Supplementary Figure 1) in responders/partial responders pre–post treatment (Fig. 2a). By contrast, no changes in absolute K* values were observed in non-responders, pre–post treatment (Fig. 2b).
Fig. 2. Pre–post treatment increases in serotonin synthesis capacity in responders/partial responders and non-responders.

Maximum intensity projections of the t-values, showing brain regions where absolute K* values (K*Absolute) were higher post-treatment compared to pre-treatment in clinical response sub-groups. a Responders and partial responders to either CBT or SSRI treatment (n = 9) demonstrated widespread pre–post treatment increases in absolute regional K* values. b Non-responders (n = 6) did not show any significant pre–post changes in absolute regional K* values. For visualization purposes, the displayed t-map threshold was T8 = 3.4 for responders/partial responders and T5 = 4.0 for non-responders, with p = 0.005 and an extent threshold of 50 voxels
A three-way Time × ROI × Response repeated measures ANOVA yielded a significant Time × Response interaction (F1,13 = 5.67, p = 0.033) but not a three-way interaction (p = 0.61), indicating that the effects did not differ in the separate ROIs. Consistent with this, the change in global K* values pre–post treatment was significantly greater in the responders/partial responders than the non-responders (two-tailed independent t-test, t13 = 2.37, p = 0.034, Hedges’ g = 1.25).
Correlations between α-[11C]MTrp trapping and clinical scores
Using SPM analysis and ΔY-BOCS scores as a covariate, we evaluated the correlation between ΔY-BOCS scores and pre-treatment K* values in the whole patient sample. Both baseline K* and ΔY-BOCS values were normally distributed. Improvement in Y-BOCS scores correlated positively with baseline α-[11C]MTrp trapping in the raphe nuclei within the right midbrain (t13 = 6.66, k = 67 voxels, coordinates x, y, z = 6, −20, −22 mm) independent of treatment modality (Fig. 3).
Fig. 3. Positive correlation between baseline K* values and OCD symptom improvement.

Statistical parametric maps (SPM12), with an anatomical MRI overlay, demonstrating brain regions where pre-treatment K* values correlated positively with ΔY-BOCS in the whole sample of OCD patients (n = 15). The t-map threshold was 3.85, with p = 0.001 and an extent threshold of 50 voxels. A significant cluster was found in the right rostral raphe nuclei (t13 = 6.66, k = 67 voxels, coordinates x, y, z = 6, −20, −22 mm)
Consistent with the global K* value findings in clinical response sub-groups, pre–post treatment changes in global K* values (ΔK*Global) correlated positively with % decrease in Y-BOCS scores (rs = 0.46, p = 0.08), as shown in Fig. 4. Notably, there was a clear outlier in this correlation, and when the outlier was removed, the correlation reached significance (rs = 0.67, p = 0.009). ΔBDI scores did not correlate with pre–post treatment changes in global K* values (rs = −0.01, p = 0.96) or with ΔY-BOCS scores, suggesting that concurrent changes in depressive symptoms were unlikely to have driven the reported results.
Fig. 4. Changes in global K* values vs. changes in OCD symptom severity.
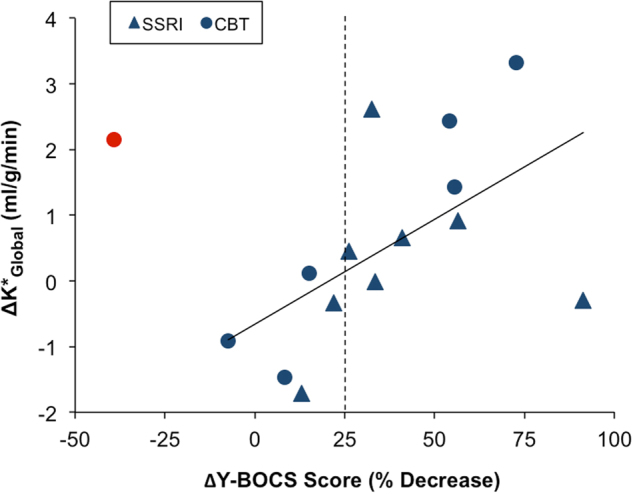
Pre–post treatment changes in global K* values (ΔK*global) correlated positively with % decrease in Y-BOCS scores (rs = 0.46, p = 0.08). Notably, there was a clear outlier in this correlation (with the outlier removed, rs = 0.67, p = 0.009); the outlier (red circle) was not included in the least-squares linear fit to the data shown here. Patients treated with sertraline are represented by triangles, and patients treated with CBT are represented by circles. The ΔY-BOCS score cut-off for responder/partial responder and non-responder subgroups is indicated with the dashed vertical line. Y-BOCS Yale-Brown Obsessive-Compulsive Scale, rs Spearman’s rank correlation coefficient
Discussion
In this study, three distinct observations were made: (i) the SSRI sertraline and specific cognitive behavior therapy markedly reduce obsessive compulsive symptoms, (ii) this effect, though robust and significant, seldom does achieve full remission, and (iii) this effect is associated with a significant pre–post increase in whole-brain 5-HT synthesis capacity in those patients who respond to either treatment. Moreover, in the whole patient sample, increases in global 5-HT synthesis capacity correlated with reductions in OCD symptom severity. Regional changes in absolute α-[11C]MTrp trapping also revealed widespread increases in 5-HT synthesis capacity in responders and partial responders to either CBT or SSRI treatment (Supplementary Figure 1). Collectively, these findings support a primarily brain-wide, rather than localized, enhancement of central 5-HT synthesis capacity during effective cognitive-behavioral or sertraline (SSRI) treatment in OCD.
The reductions in obsessive-compulsive symptoms observed here are in line with previous reports of SSRI or CBT efficacy in OCD patients1,2. However, whereas seven patients achieved symptom remission, as defined by a Y-BOCS score ≤ 1239, nine patients did not remit following 12 weeks of conventional treatment. A greater understanding of the mechanisms that support symptom reduction is critical to treatment optimization, the ultimate goal being to leverage these mechanisms to achieve higher rates of remission in OCD patients. To this end, the current study emphasizes the importance of functional changes to brain 5-HT neurotransmission in the control of obsessive-compulsive symptoms, likely in conjunction with other neurotransmitters.
Independent of treatment modality, greater improvement in OCD symptoms with SSRI or CBT was also associated with higher pre-treatment 5-HT synthesis capacity in the raphe nuclei. The dorsal and median raphe nuclei are midbrain structures that contain the major serotonergic populations40. 5-HT is produced by the raphe nuclei, and ascending serotonergic projections from the dorsal/median raphe project to most of the brain41, including CSTC circuitry implicated in OCD neuropathology. The observed correlation between clinical response and baseline 5-HT neurotransmission therefore prompts speculation that elevated 5-HT synthesis capacity in the raphe nuclei prior to treatment facilitates increases in the terminal regions during clinical improvement.
Taken together with our previous findings of abnormally elevated brain regional 5-HT synthesis capacity in OCD patients at baseline25, the results presented here provide preliminary support for a serotonergic “braking system” operative during successful therapeutics in OCD. The current observations of further increases in 5-HT synthesis capacity with effective treatment support a compensatory, rather than pathological, role of 5-HT neurotransmission in OCD. The serotonergic braking system model posits that activation of the central serotonergic system, prior to treatment, might connote an unsuccessful attempt to inhibit obsessive-compulsive symptoms. CBT or SSRI exposure in OCD patients that respond to treatment could enhance this pre-existing serotonergic braking system, such that it can more effectively inhibit OC symptoms. Considering the observed association between higher pre-treatment 5-HT synthesis capacity in the raphe nuclei and greater clinical response, standard OCD treatments may provide sufficient support to this braking system in patients with higher serotonergic functioning at baseline, therefore enabling a therapeutic response. In line with the present findings, long-term treatment with SSRIs has been found to increase serotonergic neurotransmission in animals42–44, and, more specifically, long-term administration of sertraline has been shown to increase 5-HT synthesis in the dorsal raphe nucleus of the rat45. Furthermore, as reductions in brain 5-HTT expression are associated with increased serotonergic neurotransmission46,47, findings of reduced 5-HTT availability in OCD patients at baseline, and further reductions in 5-HTT availability with clomipramine or escitalopram treatment17,48, are also consistent with a serotonergic braking system.
In psychiatry, it is often assumed that abnormal brain processes will normalize to those of healthy controls with successful treatment; for example, in depression, 5-HT metabolism has been shown to be abnormally low at baseline24 and to normalize (increase) with antidepressant treatment49. Earlier brain functional imaging studies of OCD patients have also demonstrated normalization (via reduction) of glucose metabolism in OCD patients who respond to either behavioral or drug therapy6,7. These findings are not necessarily incongruous with the current 5-HT metabolism observations; for example, pharmacological manipulations that decrease glucose metabolism have been associated with increases in 5-HT metabolism in rodents50. Alternatively, the link between previous glucose metabolism findings and the current 5-HT metabolism findings in OCD could be mediated by other neural mechanisms and neurotransmitters.
Indeed, neurotransmitters seldom act in isolation. Other mechanisms and neurotransmitters, including dopamine and glutamate, have been implicated in OCD51,52, and likely interact with the serotonergic system in OCD11,53,54. Dopamine, for example, is also an important neurotransmitter in the CSTC circuit, and hyperactive dopaminergic functioning within the striatum has been associated with OCD11 and with compulsive behaviors in animal models of OCD55. Although the serotonergic findings reported here were brain-wide, our ROI analyses revealed significant increases in 5-HT synthesis capacity within CSTC circuitry following successful treatment, including in the bilateral caudate (see Supplementary Figure 1). It is possible that the 5-HT braking system counteracts dopaminergic hyperactivity within CSTC circuitry through serotonin–dopamine interactions. Specifically, increased 5-HT synthesis capacity associated with successful CBT or SSRI treatment may result in augmented 5-HT tonic inhibition of dopamine activity, and thus a reduction in compulsive symptoms. Accordingly, clinical response to SSRI therapy in OCD has been associated with reduced dopaminergic activity in the basal ganglia56, and can be improved using dopamine antagonist augmentation strategies57. Another potential mechanism underlying the observed link between elevated 5-HT metabolism and greater therapeutic response could be the known trophic properties of 5-HT in the regulation of cell proliferation, differentiation, and maturation58. In support of a potential neurogenic mechanism mediating the relationship between 5HT metabolism and effective treatment, 5-HT1A receptor knockout mice showed impaired neurogenesis and were insensitive to the behavioral effects of the SSRI fluoxetine59. It is also conceivable that non-response to SSRI or CBT may invoke mechanisms and/or neuromodulators other than serotonin. In such treatment-refractory patients, alternative therapies such as deep brain stimulation60 or SSRI augmentation with an antipsychotic57 might be beneficial.
Some limitations of the current study should be considered. (I) Although well within the range of similar studies in the field, the sample size is modest, thus replication of these findings is critical. (II) OCD research is often confounded by clinical and biological heterogeneity. Here, considerable attention was focused on preventing contamination of the biological measure of interest by non-specific factors; yet, controlling for all the non-specific factors, known (sleep, mood, motor activity, biological rhythms) or not yet known, is always difficult in clinical behavioral research, in particular with widespread neurotransmitters, such as serotonin. (III) Patients with OCD may require longer-term treatment with specialty CBT in order to optimize treatment response (see Sookman27 for review). Important differences in clinical and physiological indices between the treatment groups may have emerged following longer treatment duration. (IV) Several patients had been previously treated with SSRIs and/or behavioral therapy, although these patients were free of treatment for 3–90 months prior to beginning the study. Thus, we cannot formally exclude the possibility that some of the observed modifications after CBT or SSRI treatment were facilitated by previous treatments. (V) The significance of the α-[11C]MTrp/PET method has been discussed in some detail, and it has been suggested that the method might measure the blood–brain barrier transport of tryptophan rather than the synthesis of serotonin61. These reservations have been addressed in several studies and reviews from our group19,32,33,62–65 and others20,66,67, and cross-validation studies support the general consensus that brain regional α-[11C]MTrp trapping provides an acceptable proxy for 5-HT synthesis. (VI) It is unlikely that the observed pre–post treatment differences in regional K* values could be attributed to changes in cerebral blood flow due to treatment, since tracers with a low plasma–brain rate constant, such as α-[11C]MTrp, are insensitive to variations in cerebral blood flow68.
In conclusion, the present study did not identify region-specific changes in 5-HT synthesis capacity following treatment with either sertraline or CBT for OCD. Yet, the evidence that elevations in brain-wide serotonergic function co-varied with clinical response raises the intriguing possibility that these increases in OCD are compensatory. In this model, a serotonergic braking system, which is unable to sufficiently inhibit dysfunctional mechanisms prior to treatment, could become more engaged over the course of successful treatment with either SSRI or CBT in OCD, allowing OCD symptoms to be more effectively controlled.
Electronic supplementary material
Acknowledgements
This study was supported by funding from Fonds de la Recherche du Quebec – Sante (FRQS). We thank Rick Fukusawa, Gary Sauchuk, DEC, Dean Jolly, Shadreck Mzengeza, PhD, Mirjana Kovacevic, and Gail Rauw, PhD (McConnell Brain Imaging Center, Montreal Neurological Institute, McGill University), for their excellent technical assistance. We are grateful to the Department of Mathematics, UC Davis, for providing an office so that Dr. Nordahl may more directly participate in mathematical activity now that he is Professor Emeritus of the Department of Psychiatry and Behavioral Sciences.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41398-018-0128-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Öst LG, Havnen A, Hansen B, Kvale G. Cognitive behavioral treatments of obsessive–compulsive disorder. A systematic review and meta-analysis of studies published 1993–2014. Clin. Psychol. Rev. 2015;40:156–169. doi: 10.1016/j.cpr.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Bloch MH, McGuire J, Landeros-Weisenberger A, Leckman JF, Pittenger C. Meta-analysis of the dose-response relationship of SSRI in obsessive-compulsive disorder. Mol. Psychiatry. 2010;15:850–855. doi: 10.1038/mp.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benkelfat C, et al. Clomipramine in obsessive-compulsive disorder: further evidence for a serotonergic mechanism of action. Arch. Gen. Psychiatry. 1989;46:23. doi: 10.1001/archpsyc.1989.01810010025004. [DOI] [PubMed] [Google Scholar]
- 4.Murphy DL, Pato MT, Pigott TA. Obsessive-compulsive disorder: treatment with serotonin-selective uptake inhibitors, azapirones, and other agents. J. Clin. Psychopharmacol. 1990;10:91S–100S. doi: 10.1097/00004714-199006001-00016. [DOI] [PubMed] [Google Scholar]
- 5.Aouizerate B, et al. Pathophysiology of obsessive-compulsive disorder: a necessary link between phenomenology, neuropsychology, imagery and physiology. Prog. Neurobiol. 2004;72:195–221. doi: 10.1016/j.pneurobio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Benkelfat C, et al. Local cerebral glucose metabolic rates in obsessive-compulsive disorder: patients treated with clomipramine. Arch. Gen. Psychiatry. 1990;47:840–848. doi: 10.1001/archpsyc.1990.01810210048007. [DOI] [PubMed] [Google Scholar]
- 7.Baxter LR, et al. Caudate glucose metabolic rate changes with both drug and behavior therapy for obsessive-compulsive disorder. Arch. Gen. Psychiatry. 1992;49:681–689. doi: 10.1001/archpsyc.1992.01820090009002. [DOI] [PubMed] [Google Scholar]
- 8.Apostolova I, et al. Effects of behavioral therapy or pharmacotherapy on brain glucose metabolism in subjects with obsessive-compulsive disorder as assessed by brain FDG PET. Psychiatry Res. 2010;184:105–116. doi: 10.1016/j.pscychresns.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Stengler-Wenzke K, Müller U, Angermeyer MC, Sabri O, Hesse S. Reduced serotonin transporter-availability in obsessive-compulsive disorder (OCD) Eur. Arch. Psychiatry Clin. Neurosci. 2004;254:252–255. doi: 10.1007/s00406-004-0489-y. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto R, et al. Reduced serotonin transporter binding in the insular cortex in patients with obsessive-compulsive disorder: a [11C]DASB PET study. Neuroimage. 2010;49:121–126. doi: 10.1016/j.neuroimage.2009.07.069. [DOI] [PubMed] [Google Scholar]
- 11.Perani D, et al. In vivo PET study of 5HT2A serotonin and D2 dopamine dysfunction in drug-naive obsessive-compulsive disorder. Neuroimage. 2008;42:306–314. doi: 10.1016/j.neuroimage.2008.04.233. [DOI] [PubMed] [Google Scholar]
- 12.Pogarell O, et al. Elevated brain serotonin transporter availability in patients with obsessive-compulsive disorder. Biol. Psychiatry. 2003;54:1406–1413. doi: 10.1016/S0006-3223(03)00183-5. [DOI] [PubMed] [Google Scholar]
- 13.Simpson HB, et al. Serotonin transporters in obsessive-compulsive disorder: a positron emission tomography study with [(11)C]McN 5652. Biol. Psychiatry. 2003;54:1414–1421. doi: 10.1016/S0006-3223(03)00544-4. [DOI] [PubMed] [Google Scholar]
- 14.Simpson HB, et al. Serotonin 2A receptors in obsessive-compulsive disorder: a positron emission tomography study with [11C]MDL 100907. Biol. Psychiatry. 2011;70:897–904. doi: 10.1016/j.biopsych.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorén P, et al. Clomipramine treatment of obsessive-compulsive disorder: II. Biochemical aspects. Arch. Gen. Psychiatry. 1980;37:1289–1294. doi: 10.1001/archpsyc.1980.01780240087010. [DOI] [PubMed] [Google Scholar]
- 16.Insel TR, Mueller EA, Alterman I, Linnoila M, Murphy DL. Obsessive-compulsive disorder and serotonin: is there a connection? Biol. Psychiatry. 1985;20:1174–1188. doi: 10.1016/0006-3223(85)90176-3. [DOI] [PubMed] [Google Scholar]
- 17.Zitterl W, et al. Changes in thalamus-hypothalamus serotonin transporter availability during clomipramine administration in patients with obsessive-compulsive disorder. Neuropsychopharmacology. 2008;33:3126–3134. doi: 10.1038/npp.2008.35. [DOI] [PubMed] [Google Scholar]
- 18.Descarries L, Soucy JP, Laeaille F, Mrini A, Tanguay R. Evaluation of three transporter ligands as quantitative markers of serotonin innervation density in rat brain. Synapse. 1995;21:131–139. doi: 10.1002/syn.890210206. [DOI] [PubMed] [Google Scholar]
- 19.Diksic M, Young SN. Study of the brain serotonergic system with labeled α-methyl-L-tryptophan. J. Neurochem. 2001;78:1185–1200. doi: 10.1046/j.1471-4159.2001.00536.x. [DOI] [PubMed] [Google Scholar]
- 20.Chugani DC, Muzik O. α[C-11]methyl-L-tryptophan PET maps brain serotonin synthesis and kynurenine pathway metabolism. J. Cereb. Blood. Flow. Metab. 2000;20:2–9. doi: 10.1097/00004647-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Diksic M, Nagahiro S, Sourkes TL, Yamamoto YL. A new method to measure brain serotonin synthesis in vivo. I. Theory and basic data for a biological model. J. Cereb. Blood Flow Metab. 1990;10:1–12. doi: 10.1038/jcbfm.1990.1. [DOI] [PubMed] [Google Scholar]
- 22.Chugani DC, et al. Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann. Neurol. 2001;45:287–295. doi: 10.1002/1531-8249(199903)45:3<287::AID-ANA3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 23.Leyton M, et al. Brain regional α-[11C]methyl-L-tryptophan trapping in impulsive subjects with borderline personality disorder. Am. J. Psychiatry. 2001;158:775–782. doi: 10.1176/appi.ajp.158.5.775. [DOI] [PubMed] [Google Scholar]
- 24.Rosa-Neto P, et al. Measurement of brain regional α-[11C]methyl-L-tryptophan trapping as a measure of serotonin synthesis in medication-free patients with major depression. Arch. Gen. Psychiatry. 2004;61:556–563. doi: 10.1001/archpsyc.61.6.556. [DOI] [PubMed] [Google Scholar]
- 25.Berney A, et al. Brain regional α-[11C]methyl-L-tryptophan trapping in medication-free patients with obsessive-compulsive disorder. Arch. Gen. Psychiatry. 2011;68:732–741. doi: 10.1001/archgenpsychiatry.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.First, M. B., Spitzer, R. L., Gibbon, M. & Williams, J. B. Structured clinical interview for DSM-IV axis I disorders (SCID I/P, Version2.0). 1995.
- 27.Sookman D. Specialized Cognitive Behavior Therapy for Obsessive Compulsive Disorder: An Expert Clinician Guidebook. New York (NY): Routledge; 2016. [Google Scholar]
- 28.Fernstrom JD, et al. Diurnal variations in plasma concentrations of tryptophan, tryosine, and other neutral amino acids: effect of dietary protein intake. Am. J. Clin. Nutr. 1979;32:1912–1922. doi: 10.1093/ajcn/32.9.1912. [DOI] [PubMed] [Google Scholar]
- 29.Maswood S, Truitt W, Hotema M, Caldarola-Pastuszka M, Uphouse L. Estrous cycle modulation of extracellular serotonin in mediobasal hypothalamus: role of the serotonin transporter and terminal autoreceptors. Brain. Res. 1999;831:146–154. doi: 10.1016/S0006-8993(99)01439-0. [DOI] [PubMed] [Google Scholar]
- 30.Jovanovic H, et al. PET study of 5-HT1A receptors at different phases of the menstrual cycle in women with premenstrual dysphoria. Psychiatry Res. Neuroimaging. 2006;148:185–193. doi: 10.1016/j.pscychresns.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Mzengeza S, Venkatachalam TK, Diksic M. Asymmetric radiosynthesis of α-[11C]methyl-L-tryptophan for PET studies. Nucl. Med. Biol. 1995;22:303–307. doi: 10.1016/0969-8051(94)00116-2. [DOI] [PubMed] [Google Scholar]
- 32.Nishizawa S, et al. Validation of a less-invasive method for measurement of serotonin synthesis rate with α-[11C]methyl-tryptophan. J. Cereb. Blood. Flow. Metab. 1998;18:1121–1129. doi: 10.1097/00004647-199810000-00009. [DOI] [PubMed] [Google Scholar]
- 33.Okazawa H, Leyton M, Benkelfat C, Mzengeza S, Diksic M. Statistical mapping analysis of serotonin synthesis images generated in healthy volunteers using positron-emission tomography and α-[11C]methyl-L-tryptophan. J. Psychiatry Neurosci. 2000;25:359–370. [PMC free article] [PubMed] [Google Scholar]
- 34.Patlak CS, Blasberg RG, Fenstermacher JD. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J. Cereb. Blood Flow Metab. 1983;3:1–7. doi: 10.1038/jcbfm.1983.1. [DOI] [PubMed] [Google Scholar]
- 35.Rosa-Neto P, Diksic M, Leyton M, Mzengeza S, Benkelfat C. Stability of α-[11C]methyl-L-tryptophan brain trapping in healthy male volunteers. Eur. J. Nucl. Med. Mol. Imaging. 2005;32:1199–1204. doi: 10.1007/s00259-005-1829-5. [DOI] [PubMed] [Google Scholar]
- 36.Collins DL, Neelin P, Peters TM, Evans AC. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J. Comput. Assist. Tomogr. 1994;18:192–205. doi: 10.1097/00004728-199403000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Collins DL, Evans AC. Animal: validation and applications of nonlinear registration-based segmentation. Int. J. Pattern Recogn. 1997;11:1271–1294. doi: 10.1142/S0218001497000597. [DOI] [Google Scholar]
- 38.Collins, D. L, Zijdenbos, A. P., Baaré, W. F. & Evans, A. C. ANIMAL+INSECT: improved cortical structure segmentation. In Information Processing in Medical Imaging. IPMI 1999. Lecture Notes in Computer Science, Vol. 1613 (eds. Kuba, A., Šáamal, M. & Todd-Pokropek, A.) 210–223 (Springer, Berlin, Heidelberg, 1999).
- 39.Mataix-Cols D, et al. Towards an international expert consensus for defining treatment response, remission, recovery and relapse in obsessive-compulsive disorder. World Psychiatry. 2016;15:80–81. doi: 10.1002/wps.20299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dahlström A, Fuxe K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol. Scand. Suppl. 1964;232:1–55. [PubMed] [Google Scholar]
- 41.Azmitia EC, Segal M. An autoradiographic analysis of the differential ascending projections of the dorsal and median raphe nuclei in the rat. J. Comp. Neurol. 1978;179:641–667. doi: 10.1002/cne.901790311. [DOI] [PubMed] [Google Scholar]
- 42.Chaput Y, de Montigny C, Blier P. Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments: an in vivo electrophysiologic study in the rat. Neuropsychopharmacology. 1991;5:219–229. [PubMed] [Google Scholar]
- 43.Briley M, Moret C. Neurobiological mechanisms involved in antidepressant therapies. Clin. Neuropharmacol. 1993;16:387–400. doi: 10.1097/00002826-199310000-00002. [DOI] [PubMed] [Google Scholar]
- 44.El Mansari M, Bouchard C, Blier P. Alteration of serotonin release in the guinea pig orbito-frontal cortex by selective serotonin reuptake inhibitors: relevance to treatment of obsessive-compulsive disorder. Neuropsychopharmacology. 1995;13:117–127. doi: 10.1016/0893-133X(95)00045-F. [DOI] [PubMed] [Google Scholar]
- 45.Kim SW, Park SY, Hwang O. Up-regulation of tryptophan hydroxylase expression and serotonin synthesis by sertraline. Mol. Pharmacol. 2002;61:778–785. doi: 10.1124/mol.61.4.778. [DOI] [PubMed] [Google Scholar]
- 46.Mathews TA, et al. Gene dose-dependent alterations in extraneuronal serotonin but not dopamine in mice with reduced serotonin transporter expression. J. Neurosci. Methods. 2004;140:169–181. doi: 10.1016/j.jneumeth.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Z, Zhang HT, Bootzin E, Millan MJ, O’Donnell JM. Association of changes in norepinephrine and serotonin transporter expression with the long-term behavioral effects of antidepressant drugs. Neuropsychopharmacology. 2008;34:1467–1481. doi: 10.1038/npp.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim E, et al. Altered serotonin transporter binding potential in patients with obsessive-compulsive disorder under escitalopram treatment: [11C]DASB PET study. Psychol. Med. 2016;46:357–366. doi: 10.1017/S0033291715001865. [DOI] [PubMed] [Google Scholar]
- 49.Berney A, et al. An index of 5-HT synthesis changes during early antidepressant treatment: α-[11C]methyl-l-tryptophan PET study. Neurochem. Int. 2008;52:701–708. doi: 10.1016/j.neuint.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 50.Vahabzadeh A, Boutelle MG, Fillenz M. Effects of changes in rat brain glucose on serotonergic and noradrenergic neurons. Eur. J. Neurosci. 1995;7:175–179. doi: 10.1111/j.1460-9568.1995.tb01053.x. [DOI] [PubMed] [Google Scholar]
- 51.Denys D, Zohar J, Westenberg HG. The role of dopamine in obsessive-compulsive disorder: preclinical and clinical evidence. J. Clin. Psychiatry. 2004;65:11–17. [PubMed] [Google Scholar]
- 52.Wu K, Hanna GL, Rosenberg DR, Arnold PD. The role of glutamate signaling in the pathogenesis and treatment of obsessive–compulsive disorder. Pharmacol. Biochem. Behav. 2012;100:726–735. doi: 10.1016/j.pbb.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goodman WK, McDougle CJ, Price LH, Riddle MA. Beyond the serotonin hypothesis: a role for dopamine in some forms of obsessive compulsive disorder? J. Clin. Psychiatry. 1990;51(Suppl.):36–43. [PubMed] [Google Scholar]
- 54.Sasaki-Adams DM, Kelley AE. Serotonin-dopamine interactions in the control of conditioned reinforcement and motor behavior. Neuropsychopharmacology. 2001;25:440–452. doi: 10.1016/S0893-133X(01)00240-8. [DOI] [PubMed] [Google Scholar]
- 55.Berridge KC, Aldridge JW, Houchard KR, Zhuang X. Sequential super-stereotypy of an instinctive fixed action pattern in hyper-dopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette’s. BMC Biol. 2005;3:4. doi: 10.1186/1741-7007-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moresco RM, et al. Fluvoxamine treatment and D2 receptors: a PET study on OCD drug-naïve patients. Neuropsychopharmacology. 2007;32:197–205. doi: 10.1038/sj.npp.1301199. [DOI] [PubMed] [Google Scholar]
- 57.Dold M, Aigner M, Lanzenberger R, Kasper S. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int. J. Neuropsychopharmacol. 2013;16:557–574. doi: 10.1017/S1461145712000740. [DOI] [PubMed] [Google Scholar]
- 58.Azmitia EC. Modern views on an ancient chemical: serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res. Bull. 2001;56:413–424. doi: 10.1016/S0361-9230(01)00614-1. [DOI] [PubMed] [Google Scholar]
- 59.Santarelli L, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301:805–809. doi: 10.1126/science.1083328. [DOI] [PubMed] [Google Scholar]
- 60.Alonso P, et al. Deep brain stimulation for obsessive-compulsive disorder: a meta-analysis of treatment outcome and predictors of response. PLoS ONE. 2015;10:e0133591. doi: 10.1371/journal.pone.0133591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoaf SE, et al. The suitability of [11C]-α-methyl-L-tryptophan as a tracer for serotonin synthesis: studies with dual administration of [11C] and [14C] labeled tracer. J. Cereb. Blood Flow Metab. 2000;20:244–252. doi: 10.1097/00004647-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 62.Leyton M, Diksic M, Benkelfat C. Brain regional α-[11C]methyl-L-tryptophan trapping correlates with post-mortem tissue serotonin content and [11C]5-hydroxytryptophan accumulation. Int. J. Neuropsychopharmacol. 2005;8:1–2. doi: 10.1017/S1461145705005420. [DOI] [PubMed] [Google Scholar]
- 63.Diksic M, Tohyama Y, Takada A. Brain net unidirectional uptake of α-[14C]methyl-L-tryptophan (α-MTrp) and its correlation with regional serotonin synthesis, tryptophan incorporation into proteins, and permeability surface area products of tryptophan and α-MTrp. Neurochem. Res. 2000;25:1537–1546. doi: 10.1023/A:1026654116999. [DOI] [PubMed] [Google Scholar]
- 64.Diksic M. Labelled (alpha)-methyl-L-tryptophan as a tracer for the study of the brain serotonergic system. J. Psychiatry Neurosci. 2001;26:293. [PMC free article] [PubMed] [Google Scholar]
- 65.Tohyama Y, Takahashi S, Merid MF, Watanabe A, Diksic M. The inhibition of tryptophan hydroxylase, not protein synthesis, reduces the brain trapping of α-methyl-L-tryptophan: an autoradiographic study. Neurochem. Int. 2002;40:603–610. doi: 10.1016/S0197-0186(01)00132-2. [DOI] [PubMed] [Google Scholar]
- 66.Muzik O, Chugani DC, Chakraborty P, Mangner T, Chugani HT. Analysis of [C-11]alpha-methyl-tryptophan kinetics for the estimation of serotonin synthesis rate in vivo. J. Cereb. Blood Flow Metab. 1997;17:659–669. doi: 10.1097/00004647-199706000-00007. [DOI] [PubMed] [Google Scholar]
- 67.Chugani DC, Chugani HT. PET: mapping of serotonin synthesis. Adv. Neurol. 2000;83:165–171. [PubMed] [Google Scholar]
- 68.Fenstermacher JD, Blasberg RG, Patlak CS. Methods for quantifying the transport of drugs across brain barrier systems. Pharmacol. Ther. 1981;14:217–248. doi: 10.1016/0163-7258(81)90062-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.