

Abstract
It is well established that age‐related decline of a womanˈs fertility is related to the poor developmental potential of her gametes. The age‐associated decline in female fertility is largely attributable to the oocyte aging caused by ovarian aging. Age‐associated oocyte aging results in a decrease in oocyte quality. In contrast to ovarian aging, there is a concept of postovulatory oocyte aging. Postovulatory aging of oocytes, not being fertilized for a prolonged time after ovulation, is known to significantly affect the development of oocytes. Both categories of oocyte aging have similar phenotypes of reproductive failure. However, the mechanisms of the decline in oocyte quality are not necessarily equivalent. An age‐dependent increase in aneuploidy is a key determinant of oocyte quality. The reduced expression of molecules regulating cell cycle control during meiosis might be involved in the age‐dependent increase in aneuploidy. The mechanism of age‐associated oocyte aging might be involved in mitochondrial dysfunction, whose etiologies are still unknown. Alternatively, the mechanism of postovulatory oocyte aging might be involved in reactive oxygen species‐induced mitochondrial injury pathways followed by abnormal intracellular Ca2+ regulation of the endoplasmic reticulum. We suggest that future research into the mechanism of oocyte aging will be necessary to develop a method to rescue the poor developmental potential of aged oocytes.
Keywords: Calcium regulation, Oocyte aging, Ovarian aging, Oxidative stress, Postovulatory oocyte aging
Introduction
The problem of infertility in the developed countries has increased in the past 30 years [1]. It is difficult to accurately calculate the number of infertility patients. To date, the numbers of treatment cycles in assisted reproductive technology (ART) have steadily increased [2]. There are several factors involved in the increase in infertility patients. The most important factor is that the average age of patients seeking infertility treatment has increased [3]. Since the development of effective contraception in the 1960s, women have been able to delay childbearing at their own discretion, and the average maternal age has increased by approximately 5 years during the last 30 years [4, 5]. Decreased fecundity with increasing female age has been recognized from demographic and epidemiological studies [6]. Thus, delayed childbearing reduces the chance of achieving spontaneous pregnancy [7]. The age‐associated decline in female fertility is largely attributable to the decrease in oocyte quality due to ovarian aging [8, 9]. Because the mechanisms of decline in oocyte quality remain unknown, there are no treatments available for patients whose infertility arises from this cause, except for oocyte donation programs [10, 11].
In contrast to ovarian aging, there is a concept of postovulatory oocyte aging. In mammals, ovulated oocytes are arrested at the metaphase stage of the second meiotic division until they are fertilized. The optimal period for oocyte fertilization lasts less than 10 h [12, 13, 14]. Fertilization within this narrow window of opportunity results in normal embryo development. Postovulatory aging of oocytes, not being fertilized for a prolonged time after ovulation, is known to significantly affect the development of oocytes [15, 16]. Postovulatory oocyte aging also impairs oocyte quality and results in reproductive failures [15, 16]. However, it is not well known whether the mechanism for the impairment of oocyte quality by these two categories of oocyte aging is similar. The aim of the present paper is to review in detail the mechanisms of these two categories of oocyte aging.
Defining oocyte aging
Terminology and definition of “oocyte aging” are very confusing. There are two categories of oocyte aging; one is preovulatory and the other is postovulatory oocyte aging [15]. Classification of oocyte aging is shown in Table 1. Preovulatory oocyte aging can be further differentiated into two different types. One of the two types of preovulatory oocyte aging is a consequence of ovarian aging. Ovarian aging is seen in reproductive aged women over 35 years old [17]. The other type of preovulatory oocyte aging is seen when ovulation does not occur in timely fashion, and the oocytes stay in the ovarian follicles after LH or hCG stimulation [15]. In other words, it represents an “intrafollicular oocyte aging” after ovulation stimulation. This type of preovulatory oocyte aging can be induced by treatment of females with a sodium pentobarbital [18], a gonadotropin‐releasing hormone antagonist [19], some progestins [20], or inosine monophosphate dehydrogenase inhibitors [21]. It is quite different from the concept of preovulatory oocyte aging with maternal aging, and is rather similar to postovulatory oocyte aging.
Table 1.
Classification of oocyte aging mechanisms
| A. Preovulatory oocyte aging |
| Oocyte aging caused by ovarian aging (maternal aging) |
| Intrafollicular oocyte aging a |
| B. Postovulatory oocyte aging |
| In vivo oocyte aging |
| In vitro oocyte aging |
aSee details in the text
The second category, postovulatory oocyte aging, can be classified into two types: in vivo‐ and in vitro‐postovulatory oocyte aging. If fertilization does not occur during an optimal period after ovulation, an unfertilized oocyte that remains in the oviduct (in vivo‐postovulatory oocyte aging) or in in vitro culture (in vitro‐postovulatory oocyte aging) goes through a time‐dependent aging process [15, 22, 23, 24]. There is no consensus on the duration of postovulatory oocyte aging. In this review, we discuss these types of oocyte aging individually.
Effects of oocyte aging on female reproduction
Preovulatory oocyte aging
Ovarian aging
Preovulatory oocyte aging has been considered to be equivalent to ovarian aging [25]. In general, ovarian aging is associated with chronological aging (maternal aging), except in cases of premature ovarian insufficiency (POI) [17]. It has been shown that female fertility declines in mammals including humans with increasing maternal age [1, 7, 17]. In humans, it is well established from demographic and epidemiological studies that female fertility begins to decline many years prior to the onset of menopause, despite continued regular ovulatory cycles [1, 4, 6]. Although there is no strict definition, advanced reproductive age in women is generally accepted as over 35 years [8, 9]. Under natural conditions, 75% of women who first try to conceive at age 30 years will have a conception ending in a live birth within 1 year, 66% at age 35 years, and 44% at aged 40 years. Within 4 years the success rates will be 91, 84, and 64%, respectively [26]. The age associated decline in female fertility and increased risk of spontaneous abortion are largely due to oocyte aging [8]. This is demonstrated by the fact that the age‐associated decline in female fertility can be overcome by oocyte donation from younger women [10, 11].
Intrafollicular oocyte aging
The other type of preovulatory oocyte aging, an intrafollicular aging of the oocytes, also impairs female reproduction in experimental and domestic animals [15]. The female rats treated with sodium pentobarbital results in delayed ovulation and the oocytes exhibit decreased fertilization, polyspermy, chromosomal anomalies, abnormal embryo development, and increased fetal mortality [27, 28]. These detrimental effects by intrafollicular aging of oocytes are very similar to those caused by postovulatory aging of oocytes (discussed later).
Postovulatory oocyte aging
Numerous investigators have reported that in vivo‐ and in vitro‐aged oocytes frequently exhibit lower fertilization rates, polyspermy, digyny, chromosomal anomalies, and abnormal embryo development [15]. These abnormalities of early embryo development result in decreased litter size in animals, lower pregnancy rate and an increased spontaneous miscarriage in humans [24, 29]. As well as abnormalities of early embryo development, postovulatory aging of oocytes is associated with retarded sensorimotor integration during pre‐weaning development, higher spontaneous motor activity, and higher emotionality in adulthood in the mouse [30]. A recent study demonstrated that postovulatory aging affects epigenetic changes in the mouse oocytes [31].
Cellular and molecular changes in aged oocytes
Preovulatory oocyte aging
Ovarian aging
The morphological changes in the ovary associated with aging have been extensively analyzed [32]. It is well known that ovarian aging results in ovarian follicle loss. Although the mechanism of ovarian follicle loss is still elusive, it is widely accepted that apoptosis is a driving force underlying age‐related ovarian follicle loss [33, 34, 35, 36, 37, 38]. However, little is known about the morphological changes in the oocyte caused by ovarian aging. There are several reports about aging‐related morphological changes in the oocytes, such as cellular fragmentation, milky or dark cytoplasm, and presence of cellular remains enclosed by the zona pellucida [39, 40]. Oocytes with cellular fragmentation in aged mice indicate apoptosis of oocytes [39, 40]. In contrast with the results of previous reports, we cannot distinguish between the oocytes collected from aged mice and those from young mice under a microscope. While ovulation number of oocytes decreases in aged mice compared to young mice, these oocytes from both mice are apparently similar in terms of morphology (Takahashi and Kurachi, unpublished data).
On the other hand, numerous data support that oocyte aneuploidy increases with ovarian aging in mammals, including humans [41, 42, 43]. Increasing evidence shows that aneuploidy by ovarian aging results from a defective cell cycle control during meiosis I, especially metaphase I to anaphase I [25]. Ovarian aging affects some key molecules for cell cycle control in oocytes: there is a reduced number of transcripts for ‘cohesion proteins’ such as SMCβ1 and for ‘spindle check point proteins’ such as MAD2 [44, 45, 46].
Intrafollicular oocyte aging
Intrafollicular oocyte aging affects morphological and cellular changes in oocytes treated with sodium pentobarbital, resulting in irregularities in the oolemma, decreased number of cortical granules, disruption of cytoskeleton arrangement, and changes in mitochondrial structure [18, 27, 47, 48, 49]. These morphological changes in oocytes by intrafollicular oocyte aging are very similar to those caused by postovulatory oocyte aging [15].
Postovulatory oocyte aging
It is well established that in vivo‐ and in vitro‐postovulatory aging of oocytes is associated with changes in various morphological, biochemical, and molecular pathways involved in intracellular signaling [15, 16]. In vivo‐ and in vitro‐postovulatory aging of oocytes share many common properties.
Morphological and cellular changes
Postovulatory oocyte aging affects numerous morphological and cellular changes: changes in structure of oolemma, zona pellucida, cortical granules, mitochondria, cytoskeleton, meiotic spindle, and chromosome alignment [15, 16]. The lining of the oocyte cortex in the fresh oocytes is composed of thick and thin microfilament domains. Aged oocytes show disruption or loss of the thick microfilament domain beneath the oolemma [50, 51, 52, 53, 54]. The zona pellucida in fresh oocytes appears as a granulofibrillar, interconnected reticulum with pores, while the zona pellucida in aged oocytes shows a ‘cobblestone’ appearance [55]. Chymotrypsin‐mediated dissolution of the zona pellucida takes more time in aged oocytes compared to fresh oocytes [16, 56, 57]. This indicates that zona pellucida hardening occurs naturally in aged oocytes. The reason for zona hardening in aged oocytes is that cortical reaction, exocytosis of cortical granules, is easily triggered spontaneously without fertilization [47, 56, 58]. Aged oocytes show an increase in the number of phosphatase positive organelles (lysosomes), aggregation of tubuli of smooth endoplasmic reticulum, and aggregation of small mitochondria‐vesicle complexes [51, 59]. Meiotic spindle assembly is a cellular structure important for accurate chromosomal distribution [60]. The meiotic spindle consists of a central region of chromosomes and of microtubules radiating from the chromosomes to the two opposite spindle poles, which consist of foci of pericentriolar material [61]. The pericentriolar material consists of a network of 12–15 nm filaments with which the other components associate [62]. One well‐characterized component of the pericentriolar material is the γ‐tubulin ring complex [63]. There are many reports that postovulatory aging of oocytes results in disruption and loss of the meiotic spindle assembly in experimental animals and humans [50, 64, 65, 66, 67, 68]. In mouse studies, although the meiotic spindle is barrel‐shaped and microtubules are clearly detected in fresh oocytes, microtubules become gradually lost from the spindle in aged oocytes [16]. In vitro‐aged human oocytes, like those of experimental animals, show aberrant expression of γ‐tubulin, which indicates disruption of centrosome structure at the meiotic poles [60, 69]. These changes in aged oocytes lead to premature chromosomal separation, which is strongly associated with aneuploidy [44, 67].
Biochemical and molecular changes
Postovulatory aging of oocytes affects various biochemical and molecular changes in mammalian oocytes [15, 16]. After fertilization, recruitment of maternal mRNAs occurs and results in changes in the spectrum of polypeptides synthesized in oocytes [70]. The newly synthesized protein expression patterns change in fertilized aged oocytes [56]. The intracytoplasmic level of glutathione (GSH), which plays a major role in protection against reactive oxygen species (ROS), decreases in aged oocytes [71]. The level of lipid peroxidation, which indicates the degree of oxidative stress, increases in in vivo‐aged oocytes [72]. Moreover, the amount of ROS increases in aged oocytes with increasing time of in vitro culture [73]. The intracytoplasmic level of ATP decreases in aged oocytes [74, 75]. The inactivation of MPF, which consists of two subunits of p34cdc2 and cyclin B, and MAPK occurs earlier in aged oocytes [56, 76, 77, 78]. The expression of BCL2, anti‐apoptotic protein, is decreased in in vivo‐and in vitro‐aged oocytes [73, 78, 79]. Postovulatory aging of oocytes impairs intracellular Ca2+ regulation, which is most important for early events after fertilization and also for subsequent embryo development [80, 81, 82, 83]. Abnormal intracellular Ca2+ regulation in aged oocytes will be discussed later in the section regarding mechanisms of oocytes aging.
Mechanisms of oocyte aging
We describe the mechanisms of two categories of oocyte aging: oocyte aging caused by ovarian aging and postovulatory oocyte aging.
Ovarian aging
Ovarian aging impairs both the quantity and the quality of oocytes [3, 5, 8]. The decrease in oocyte quantity is a part of ovarian follicle loss. As mentioned above, although the mechanism of ovarian follicle loss remains unknown, it is widely accepted that the age‐dependent decline of ovarian follicles is involved in apoptotic pathways [33, 36, 38]. Activation of apoptotic pathways results in ovarian follicle atresia. Although every primordial follicle has the potential to grow, mature, and ovulate, this is not the case in reality [84]. The mechanism of follicle recruitment from the primordial follicle pool is still unknown. A recent study using genetically targeted mice reveals that PTEN/PI3K signaling pathways within oocytes are important for follicle recruitment [85, 86, 87, 88]. The balance between follicle recruitment and follicle atresia might be important for ovarian follicle loss. Premature ovarian insufficiency is defined as cessation of ovarian function before the age of 40 years and affects about 1% of women in the general population [89]. POI cases without an obvious cause provide a model for the study of genetic mechanisms of ovarian aging [17, 90]. Many candidate genes have been reported to be involved in POI (see review in [17, 90]). A recent genome‐wide association study reveals new loci of single‐nucleotide polymorphisms in natural menopause cohorts [91, 92].
However, the mechanism of age‐dependent decline of oocyte quality remains unknown [25]. Age‐dependent increase in aneuploidy is a key determinant of oocyte quality. Little is known of how aneuploidy originates by increase in maternal age [3, 93]. As mentioned above, ovarian aging affects expression of some key molecules involved in cell cycle control, such as SMC1β and MAD2 [45, 46, 94]. The impairment of cell cycle control during meiosis might be involved in the age‐dependent increase in aneuploidy.
Microarray methods reveal that ovarian aging changes the expression patterns of accumulation of maternal RNAs required for oocyte‐specific processes and metabolism in mouse and human oocytes [95, 96, 97]. Ovarian aging negatively affects the expression of oocyte genes involved in mitochondrial functions, oxidative stress, cell cycle regulation, and DNA and chromosome stability [95, 96, 97]. Because mitochondria are organelles producing ATP via oxidative phosphorylation, they are most important for maintenance of oocyte quality. Ovarian aging changes mitochondrial morphology and functions in oocytes. Aberrant mitochondrial arrangement has been observed in aged‐mouse oocytes [39]. Ovarian aging results in abnormal mitochondrial ultrastructure with high density of the matrix, vacuolization, and swelling in aged‐human oocytes [98]. Wilding et al. [99] reported that the mitochondrial membrane potential, which indicates mitochondrial function of oocytes from reproductive‐aged women is decreased compared to that of oocytes from young women. Moreover, oocytes from reproductive‐aged women present an accumulation of the mitochondrial DNA point mutations and higher levels of mitochondrial DNA deletions [100, 101]. These results suggest that age‐dependent impairment of mitochondrial function might be a cause for a decline of oocyte quality. A question that arises from these results is what mechanism of impairment of mitochondrial functions is involved? One relevant idea about aging involves the accumulation of damage exerted by increased levels of ROS, a condition known as oxidative stress [15, 102]. In fact, the levels of GSH and GSH‐transferase activity, which play an important role in cellular defense against ROS, decreased in oocytes from aged mice [103]. Moreover, the genes involved in protecting against oxidative stresses, such as Sod1 and the thioredoxin family (Txn1 and Apacd) are downregulated in aged‐mouse oocytes [95, 96]. However, there is no evidence that ovarian aging directly affects oxidative stress to the oocytes. Further studies are needed to determine whether oxidative stress is involved in the age‐dependent decline of oocyte quality.
Recent evidence suggests that epigenetic mechanisms in oocytes may be altered by ovarian aging. In mouse oocytes, several transcripts encoding proteins involved in epigenetic modifications, such as chromatin remodeling and DNA methylation, are affected by aging, including the DNA methyltransferases (Dnmt)‐1, 1o, 3a, 3L and 3b, and DNMT‐associated protein‐1 (Dmap1) [95, 104].
Postovulatory oocyte aging
As described in the section regarding cellular and molecular changes in postovulatory aged oocytes, these morphological and biochemical changes are translated into detrimental early and late phases of embryo development, such as lower fertilization rate, polyspermy, dingy, chromosomal anomalies, abnormal embryo development, and post‐implantation mortality [15]. Although the mechanism of poor embryo development by postovulatory aging of oocytes remains unknown, there are some clues to help researchers puzzle out the mechanism. Unfertilized aged oocytes undergo spontaneous cytoplasmic fragmentation [105]. Like unfertilized oocytes, embryos derived from aged oocytes exhibit fragmentation after fertilization [73]. The fact that these fragmented oocytes and embryos show TUNEL‐positive staining [40, 73, 105] suggests the activation of apoptosis pathways during the period of postovulatory aging. Mammalian oocytes express several caspases and anti‐ and pro‐apoptotic members of the BCL2 gene family [106, 107]. Pro‐apoptotic molecules such as Bax induce the release of cytochrome c, which activates caspases, while anti‐apoptotic molecules such as BCL2 prevent it [108]. In mouse and pig oocytes, the expression of BCL2 is decreased and the percentage of TUNEL‐positive unfertilized oocytes is increased with in vitro‐aging [78, 79]. In addition, we and Gordo et al. have reported that the expression of BCL2 protein is decreased whereas that of BAX protein is unchanged in oocytes aged in vitro [73, 109]. We also confirmed that the expression of BCL2 was decreased in in vivo‐aged mouse oocytes (Takahashi and Kurachi, unpublished data). These results suggest that the postovulatory aged oocytes are prone to undergo apoptosis due to the decreased BCL2 expression.
In mammalian oocytes at fertilization, sperm induces drastic changes in intracellular Ca2+ concentration ([Ca2+]i), which consists of a single long‐lasting rise in [Ca2+]i, followed by short repetitive changes in [Ca2+]i lasting for several hours. These temporal changes in [Ca2+]i are termed as “Ca2+ oscillations” [110]. The increase in [Ca2+]i plays important roles in fertilization, cortical granule exocytosis, resumption of meiosis, pronucleus formation, and subsequent embryo development [111, 112, 113, 114]. In addition, the patterns of Ca2+ oscillations, such as amplitude and frequency of Ca2+ oscillations, affect early and post‐implantation embryo development [115]. We have demonstrated that in vivo‐and in vitro‐postovulatory aging alter the patterns of Ca2+ oscillations at fertilization in mouse oocytes [73, 81]. Frequency of Ca2+ oscillations at fertilization in aged oocytes is higher than that in freshly ovulated oocytes, while the amplitude of individual Ca2+ oscillations is lower in the former than in the latter [73, 81]. Jones and Whittingham [83] also reported that both the amplitude and rate of rise of individual Ca2+ oscillations at fertilization are decreased in in vivo‐aged mouse oocytes. We and other groups have reported the Ca2+ release from the inositol 1,4,5‐triphosphate (InsP3)‐sensitive Ca2+ stores is decreased in in vivo‐aged oocytes compared to that in fresh oocytes [82, 83]. Furthermore, we have reported that both the Ca2+ reuptake by Ca2+‐ATPases and Ca2+ stores in the endoplasmic reticulum (ER) in in vivo‐aged oocytes are decreased compared to those in fresh oocytes [81, 82]. We have reported that Ca2+ stores in the ER in in vitro‐aged oocytes are also decreased [73]. Collectively, these results indicate the impaired Ca2+ homeostasis in postovulatory aged oocytes.
The abnormal intracellular Ca2+ handling in aged oocytes leads to the apoptosis of oocytes [14]. Gordo et al. [116] reported that injection of Ca2+ oscillators, such as sperm cytosolic factor and adenophostin A, a potent agonist of InsP3 receptor, into in vitro‐aged oocytes causes increase in the fragmentation and caspase activity of oocytes. They also reported that injection of Ca2+ oscillators into in vitro‐aged oocytes induces abnormal Ca2+ oscillations with low amplitude and abrupt cessation [116]. Moreover, we and other groups reported that decrease in the ER Ca2+ stores of fresh oocytes by thapsigargin, which is a specific inhibitor of smooth endoplasmic reticulum Ca2+‐ATPases (SERCA), results in abnormal Ca2+ oscillations, with low amplitude and high frequency at fertilization compared to those observed in the vehicle‐treated fresh oocytes [73, 83, 117]. Moreover, we have reported that thapsigargin treatment of fresh oocytes causes lower fertilization rate, lower blastocyst formation rate, and higher rate of fragmented embryo after in vitro fertilization compared to those in the vehicle‐treated fresh oocyte [73]. These results suggest that abnormal Ca2+ handling in the postovulatory aged oocytes might be related to poor embryo development after fertilization.
What mechanisms are involved in the impairment of Ca2+ homeostasis in postovulatory aged oocytes? In mammals, the ER is the major intracellular Ca2+ storage site and the type I InsP3 receptor of the ER membrane mediates Ca2+ oscillations in oocytes [118, 119]. As mentioned above, we have reported that the activity of SERCA is decreased in in vivo‐aged mouse oocytes [81] and the Ca2+ store of the ER is decreased in in vivo‐ and in vitro‐aged mouse oocytes [73, 82]. The Ca2+ stores are maintained within the ER by the replenishment of Ca2+ from the cytosol through the activity of the SERCA [120, 121]. The activity of SERCA is highly dependent on availability of intracellular ATP [122]. In fact, Chi et al. [74] reported that in vitro culture decreases ATP content in unfertilized mouse oocytes. In addition, we have reported that the ATP content of fertilized in vivo aged oocytes is significantly decreased compared to that in fertilized fresh oocytes [75]. Mitochondrial ATP production is prerequisite for Ca2+ oscillations at fertilization and Ca2+ homeostasis in oocytes [123, 124]. Thus, impairment of fertilization‐triggered mitochondrial ATP production possibly links to impairment of Ca2+ homeostasis and abnormal patterns of Ca2+ oscillations at fertilization in aged oocytes. On the other hand, reduced amount of BCL2 may negatively affect the function of SERCA. Overexpression of BCL2 maintains Ca2+ stores of the ER and prevents thapsigargin‐induced apoptosis in lymphoma cells [125]. In addition, BCL2 prevents Ca2+ store of the ER by upregulating SERCA [126]. In fact, as mentioned above, the expression of BCL2 is decreased in aged oocytes [73, 109]. Taken together, these results suggest that reduction in both ATP production and BCL2 expression might be involved in the impairment of Ca2+ homeostasis in aged oocytes.
A second question that arises from these results is what mechanisms are responsible for impairment of mitochondrial function and reduced expression of BCL2 in aged oocytes? Mitochondria are organelles that produce ATP by oxidative phosphorylation to supply energy for various cell functions. Mitochondrial dysfunction has been linked with pathological conditions, including various reproductive failures [127, 128]. ROS, such as superoxide anion radical, hydrogen peroxide (H2O2), and hydroxyl radical, are produced endogenously by proton electrochemical gradient during mitochondrial respiration. Because the mitochondria are a major source of ROS, mitochondria need continuous protection from free radical attack by ROS scavenger systems [129]. Tarin et al. [15] proposed a mechanism based on the “the oxygen radical mitochondrial injury hypothesis of aging” to explain the effects of postovulatory aging on impairment of early and embryo and fetal development. This mechanism is based on the idea that ROS harm mitochondrial DNA, proteins, and lipids [15]. In fact, we have reported that the magnitude of lipid membrane peroxidation in in vivo‐aged oocytes is increased compared to that in fresh oocytes [72]. And we have also reported that the levels of ROS in in vitro‐aged oocytes compare to those in fresh oocytes [73]. Boerjan and de Boer [71] reported that the amount of GSH, which is a ROS scavenger, is decreased in in vivo‐aged mouse oocytes. These results suggest that aged oocytes are prone to oxidative stresses by decrease in ROS scavengers. Furthermore, we have reported that exposure of fresh oocytes to 100 μM H2O2 results in abnormal patterns of Ca2+ oscillations with low amplitude and high frequency, which are similar to those in postovulatory aged oocytes [72]. We have also reported that the H2O2‐pretreated fresh oocytes results in poor embryo development after fertilization [73]. In somatic cells, ROS are important mediators of intracellular signaling for numerous cell functions, including Ca2+ homeostasis through modulating SERCA and InsP3 receptor functions [130, 131]. The increase in ROS production in aged oocytes might directly affect Ca2+ homeostasis and/or impair mitochondrial function followed by ATP depletion. On the other hand, the mechanism for the decrease in BCL2 expression in aged oocytes remains unknown. Although transcriptional control of BCL2 has been reported, increasing evidence suggests that an important component of BCL2 regulation is post‐transcriptional, such as micro RNAs (miR15A and miR16‐1) [132]. There are very few reports about the mechanism of BCL2 regulation in oocytes. We have reported that treatments of H2O2 and thapsigargin to fresh oocytes result in decreased expression of BCL2 [73]. As mentioned above, BCL2 expression is closely related to the Ca2+ store of the ER [125, 126]. Alternatively, ROS control the expression of BCL2 by regulating its phosphorylation and ubiquitination in cancer cells [133]. In addition, ROS downregulate the expression of BCL2 in T cells [134]. These results suggest that the increase in ROS levels in postovulatory aged oocytes might result in both mitochondrial dysfunction and reduced expression of BCL2. We show a model of mechanism of poor embryo development in post‐ovulatory aging of oocytes (Fig. 1).
Figure 1.
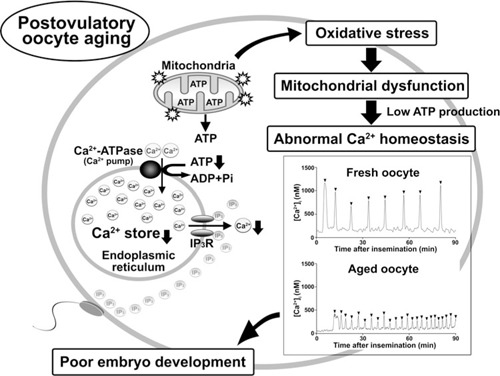
Scheme of the mechanism of poor embryo development in postovulatory oocyte aging. We show the model of the mechanism of poor embryo development in postovulatory‐aged oocytes. Postovulatory aging of oocytes results in increase in mitochondrial oxidative stress. Oxidative stress‐induced mitochondrial dysfunction results in low ATP production followed by impairment of intracellular Ca2+ regulation, such as decrease in the Ca2+ stores of the endoplasmic reticulum (ER) and the Ca2+ release from the ER via inositol 1,4,5‐triphosphate (InsP3) receptor. When postovulatory aged‐oocytes, which are impaired in the intracellular Ca2+ regulations, are fertilized with sperm, the abnormal Ca2+ oscillations occur at fertilization. Representative data show that sperm triggers Ca2+ oscillations in the fresh (14 h after hCG treatment) and the aged (20 h after hCG treatment) mouse oocytes. The patterns of Ca2+ oscillations at fertilization are changed by postovulatory oocyte aging: In the aged oocytes, the abnormal Ca2+ oscillations with lower amplitude and high frequency are shown. The abnormal Ca2+ oscillations may result in poor embryo development in postovulatory‐aged oocytes. Arrowheads indicate the individual Ca2+ oscillations. IP3, InsP3. IP3R, InsP3 receptor
Conclusion
We here review that there are two categories of oocyte aging: oocyte aging caused by ovarian aging and postovulatory oocyte aging. Both categories of oocyte aging have similar phenotypes of reproductive failure. However, the mechanisms for the impairment in oocyte quality are not necessarily equivalent. The mechanism of oocyte aging caused by ovarian aging might be a chronic process of damage to the oocytes and/or ovarian follicle cells, such as thecal and granulosa cells. There are several problems with studying the oocyte aging caused by ovarian aging; it takes over 1 year for experimental animals to grow older and the numbers of oocytes available are very small. Moreover, there is no animal model anaologous to the ovarian aging study. In contrast to the limitations of the data in the ovarian aging study, a model to study the postovulatory aging of oocytes is more easily accessible and more data are available. ROS‐induced mitochondrial injury pathways followed by abnormal intracellular Ca2+ regulation of the ER may be involved in the mechanism of postovulatory oocyte aging. According to this scenario, the antioxidant treatment in vivo and in vitro might prevent the oocyte damage by postovulatory aging. We suggest that future research into the mechanism of oocyte aging will be necessary in order to develop a method to rescue the poor developmental potential of aged oocytes.
Acknowledgments
This study was supported by a Grant‐in‐aid for General Science Research No. 22591815 to Toshifumi Takahashi, 20591905 to Hideki Igarashi, 22390308 to Hirohisa Kurachi, and the Global COE Program for Medical Sciences from the Japan Society for the Promotion of Science.
Conflict of interest
The authors have nothing to disclose.
References
- 1. Balasch J. Ageing and infertility: an overview. Gynecol Endocrinol, 2010, 26, 855–860 10.3109/09513590.2010.501889 [DOI] [PubMed] [Google Scholar]
- 2. Mouzon J, Lancaster P, Nygren KG, Sullivan E, Zegers‐Hochschild F, Mansour R, Ishihara O, Adamson D. World collaborative report on assisted reproductive technology, 2002. Hum Reprod, 2009, 24, 2310–2320 10.1093/humrep/dep098 [DOI] [PubMed] [Google Scholar]
- 3. Baird DT, Collins J, Egozcue J, Evers LH, Gianaroli L, Leridon H, Sunde A, Templeton A, Steirteghem A, Cohen J, Crosignani PG, Devroey P et al. Fertility and ageing. Hum Reprod Updat, 2005, 11, 261–276 10.1093/humupd/dmi006 [DOI] [PubMed] [Google Scholar]
- 4. Leridon H. Can assisted reproduction technology compensate for the natural decline in fertility with age? A model assessment. Hum Reprod, 2004, 19, 1548–1553 10.1093/humrep/deh304 [DOI] [PubMed] [Google Scholar]
- 5. te Velde ER, Pearson PL. The variability of female reproductive ageing. Hum Reprod Updat, 2002, 8, 141–154 10.1093/humupd/8.2.141 [DOI] [PubMed] [Google Scholar]
- 6. Menken J, Trussell J, Larsen U. Age and infertility. Science, 1986, 233, 1389–1394 10.1126/science.3755843 [DOI] [PubMed] [Google Scholar]
- 7. Alviggi C, Humaidan P, Howles CM, Tredway D, Hillier SG. Biological versus chronological ovarian age: implications for assisted reproductive technology. Reprod Biol Endocrinol, 2009, 7, 101 10.1186/1477‐7827‐7‐101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Practice Committee of the American Society for Reproductive Medicine. Aging and infertility in women. Fertil Steril. 2006;86:S248–52. [DOI] [PubMed]
- 9. Heffner LJ. Advanced maternal age–how old is too old?. N Engl J Med, 2004, 351, 1927–1929 10.1056/NEJMp048087 [DOI] [PubMed] [Google Scholar]
- 10. Sauer MV, Kavic SM. Oocyte and embryo donation 2006: reviewing two decades of innovation and controversy. Reprod Biomed Online, 2006, 12, 153–162 10.1016/S1472‐6483(10)60855‐3 [DOI] [PubMed] [Google Scholar]
- 11. Toner JP, Grainger DA, Frazier LM. Clinical outcomes among recipients of donated eggs: an analysis of the U.S. national experience, 1996–1998. Fertil Steril, 2002, 78, 1038–1045 10.1016/S0015‐0282(02)03371‐X [DOI] [PubMed] [Google Scholar]
- 12. Sakai N, Endo A. Effects of delayed mating on preimplantation embryos in spontaneously ovulated mice. Gamete Res, 1988, 19, 381–385 10.1002/mrd.1120190409 [DOI] [PubMed] [Google Scholar]
- 13. Yanagimachi R, Chang MC. Fertilizable life of golden hamster ova and their morphological changes at the time of losing fertilizability. J Exp Zool, 1961, 148, 185–203 10.1002/jez.1401480303 [DOI] [PubMed] [Google Scholar]
- 14. Fissore RA, Kurokawa M, Knott J, Zhang M, Smyth J. Mechanisms underlying oocyte activation and postovulatory ageing. Reproduction, 2002, 124, 745–754 10.1530/rep.0.1240745 [DOI] [PubMed] [Google Scholar]
- 15. Tarin JJ, Perez‐Albala S, Cano A. Consequences on offspring of abnormal function in ageing gametes. Hum Reprod Updat, 2000, 6, 532–549 10.1093/humupd/6.6.532 [DOI] [PubMed] [Google Scholar]
- 16. Miao YL, Kikuchi K, Sun QY, Schatten H. Oocyte aging: cellular and molecular changes, developmental potential and reversal possibility. Hum Reprod Updat, 2009, 15, 573–585 10.1093/humupd/dmp014 [DOI] [PubMed] [Google Scholar]
- 17. Broekmans FJ, Soules MR, Fauser BC. Ovarian aging: mechanisms and clinical consequences. Endocr Rev, 2009, 30, 465–493 10.1210/er.2009‐0006 [DOI] [PubMed] [Google Scholar]
- 18. Peluso JJ, Butcher RL. The effect of follicular aging on the ultrastructure of the rat oocyte. Fertil Steril, 1974, 25, 494–502 [PubMed] [Google Scholar]
- 19. Oussaid B, Lonergan P, Khatir H, Guler A, Monniaux D, Touze JL, Beckers JF, Cognie Y, Mermillod P. Effect of GnRH antagonist‐induced prolonged follicular phase on follicular atresia and oocyte developmental competence in vitro in superovulated heifers. J Reprod Fertil, 2000, 118, 137–144 10.1530/reprod/118.1.137 [PubMed] [Google Scholar]
- 20. Mihm M, Curran N, Hyttel P, Knight PG, Boland MP, Roche JF. Effect of dominant follicle persistence on follicular fluid oestradiol and inhibin and on oocyte maturation in heifers. J Reprod Fertil, 1999, 116, 293–304 10.1530/jrf.0.1160293 [DOI] [PubMed] [Google Scholar]
- 21. Downs SM. Induction of meiotic maturation in vivo in the mouse by IMP dehydrogenase inhibitors: effects on the developmental capacity of ova. Mol Reprod Dev, 1994, 38, 293–302 10.1002/mrd.1080380310 [DOI] [PubMed] [Google Scholar]
- 22. Saito H, Koike K, Saito T, Nohara M, Kawagoe S, Hiroi M. Aging changes in the alignment of chromosomes after human chorionic gonadotropin stimulation may be a possible cause of decreased fertility in mice. Horm Res, 1993, 39 (Suppl 1) 28–31 10.1159/000182754 [DOI] [PubMed] [Google Scholar]
- 23. Smith AL, Lodge JR. Interactions of aged gametes: in vitro fertilization using in vitro‐aged sperm and in vivo‐aged ova in the mouse. Gamete Res, 1987, 16, 47–56 10.1002/mrd.1120160106 [DOI] [PubMed] [Google Scholar]
- 24. Wilcox AJ, Weinberg CR, Baird DD. Post‐ovulatory ageing of the human oocyte and embryo failure. Hum Reprod, 1998, 13, 394–397 10.1093/humrep/13.2.394 [DOI] [PubMed] [Google Scholar]
- 25. Tatone C, Amicarelli F, Carbone MC, Monteleone P, Caserta D, Marci R, Artini PG, Piomboni P, Focarelli R. Cellular and molecular aspects of ovarian follicle ageing. Hum Reprod Updat, 2008, 14, 131–142 10.1093/humupd/dmm048 [DOI] [PubMed] [Google Scholar]
- 26. Templeton A, Morris JK, Parslow W. Factors that affect outcome of in vitro fertilisation treatment. Lancet, 1996, 348, 1402–1406 10.1016/S0140‐6736(96)05291‐9 [DOI] [PubMed] [Google Scholar]
- 27. Butcher RL, Collins WE, Fugo NW. Altered secretion of gonadotropins and steroids resulting from delayed ovulation in the rat. Endocrinology, 1975, 96, 576–586 10.1210/endo‐96‐3‐576 [DOI] [PubMed] [Google Scholar]
- 28. Hamaguchi H, Mikamo K. Morphological and cytogenetic studies on blastocysts following intrafollicular overripeness induced by delayed ovulation (author's transl). Jinrui Idengaku Zasshi, 1974, 19, 88–89 [PubMed] [Google Scholar]
- 29. Huhtinen M, Koskinen E, Skidmore JA, Allen WR. Recovery rate and quality of embryos from mares inseminated after ovulation. Theriogenology, 1996, 45, 719–726 10.1016/0093‐691X(96)00001‐5 [DOI] [PubMed] [Google Scholar]
- 30. Tarin JJ, Perez‐Albala S, Aguilar A, Minarro J, Hermenegildo C, Cano A. Long‐term effects of postovulatory aging of mouse oocytes on offspring: a two‐generational study. Biol Reprod, 1999, 61, 1347–1355 10.1095/biolreprod61.5.1347 [DOI] [PubMed] [Google Scholar]
- 31. Liang XW, Zhu JQ, Miao YL, Liu JH, Wei L, Lu SS, Hou Y, Schatten H, Lu KH, Sun QY. Loss of methylation imprint of Snrpn in postovulatory aging mouse oocyte. Biochem Biophys Res Commun, 2008, 371, 16–21 10.1016/j.bbrc.2008.03.105 [DOI] [PubMed] [Google Scholar]
- 32. Faddy MJ, Gosden RG, Gougeon A, Richardson SJ, Nelson JF. Accelerated disappearance of ovarian follicles in mid‐life: implications for forecasting menopause. Hum Reprod, 1992, 7, 1342–1346 [DOI] [PubMed] [Google Scholar]
- 33. Tilly JL. Commuting the death sentence: how oocytes strive to survive. Nat Rev Mol Cell Biol, 2001, 2, 838–848 10.1038/35099086 [DOI] [PubMed] [Google Scholar]
- 34. Morita Y, Maravei DV, Bergeron L, Wang S, Perez GI, Tsutsumi O, Taketani Y, Asano M, Horai R, Korsmeyer SJ, Iwakura Y, Yuan J et al. Caspase‐2 deficiency prevents programmed germ cell death resulting from cytokine insufficiency but not meiotic defects caused by loss of ataxia telangiectasia‐mutated (Atm) gene function. Cell Death Differ, 2001, 8, 614–620 10.1038/sj.cdd.4400845 [DOI] [PubMed] [Google Scholar]
- 35. Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH, Tilly JL. Aromatic hydrocarbon receptor‐driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat Genet, 2001, 28, 355–360 10.1038/ng575 [DOI] [PubMed] [Google Scholar]
- 36. Pru JK, Tilly JL. Programmed cell death in the ovary: insights and future prospects using genetic technologies. Mol Endocrinol, 2001, 15, 845–853 10.1210/me.15.6.845 [DOI] [PubMed] [Google Scholar]
- 37. Matikainen T, Perez GI, Zheng TS, Kluzak TR, Rueda BR, Flavell RA, Tilly JL. Caspase‐3 gene knockout defines cell lineage specificity for programmed cell death signaling in the ovary. Endocrinology, 2001, 142, 2468–2480 10.1210/en.142.6.2468 [DOI] [PubMed] [Google Scholar]
- 38. Hussein MR. Apoptosis in the ovary: molecular mechanisms. Hum Reprod Updat, 2005, 11, 162–177 10.1093/humupd/dmi001 [DOI] [PubMed] [Google Scholar]
- 39. Tarin JJ, Perez‐Albala S, Cano A. Cellular and morphological traits of oocytes retrieved from aging mice after exogenous ovarian stimulation. Biol Reprod, 2001, 65, 141–150 10.1095/biolreprod65.1.141 [DOI] [PubMed] [Google Scholar]
- 40. Fujino Y, Ozaki K, Yamamasu S, Ito F, Matsuoka I, Hayashi E, Nakamura H, Ogita S, Sato E, Inoue M. DNA fragmentation of oocytes in aged mice. Hum Reprod, 1996, 11, 1480–1483 [DOI] [PubMed] [Google Scholar]
- 41. Kuliev A, Cieslak J, Verlinsky Y. Frequency and distribution of chromosome abnormalities in human oocytes. Cytogenet Genome Res, 2005, 111, 193–198 10.1159/000086889 [DOI] [PubMed] [Google Scholar]
- 42. Pellestor F, Andreo B, Anahory T, Hamamah S. The occurrence of aneuploidy in human: lessons from the cytogenetic studies of human oocytes. Eur J Med Genet, 2006, 49, 103–116 10.1016/j.ejmg.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 43. Pellestor F, Anahory T, Hamamah S. Effect of maternal age on the frequency of cytogenetic abnormalities in human oocytes. Cytogenet Genome Res, 2005, 111, 206–212 10.1159/000086891 [DOI] [PubMed] [Google Scholar]
- 44. Steuerwald NM, Steuerwald MD, Mailhes JB. Post‐ovulatory aging of mouse oocytes leads to decreased MAD2 transcripts and increased frequencies of premature centromere separation and anaphase. Mol Hum Reprod, 2005, 11, 623–630 10.1093/molehr/gah231 [DOI] [PubMed] [Google Scholar]
- 45. Cukurcam S, Betzendahl I, Michel G, Vogt E, Hegele‐Hartung C, Lindenthal B, Eichenlaub‐Ritter U. Influence of follicular fluid meiosis‐activating sterol on aneuploidy rate and precocious chromatid segregation in aged mouse oocytes. Hum Reprod, 2007, 22, 815–828 10.1093/humrep/del442 [DOI] [PubMed] [Google Scholar]
- 46. Hodges CA, Revenkova E, Jessberger R, Hassold TJ, Hunt PA. SMC1beta‐deficient female mice provide evidence that cohesins are a missing link in age‐related nondisjunction. Nat Genet, 2005, 37, 1351–1355 10.1038/ng1672 [DOI] [PubMed] [Google Scholar]
- 47. Gulyas BJ. Cortical granules of mammalian eggs. Int Rev Cytol, 1980, 63, 357–392 10.1016/S0074‐7696(08)61762‐3 [DOI] [PubMed] [Google Scholar]
- 48. Peluso JJ, England‐Charlesworth C, Hutz R. Effect of age and of follicular aging on the preovulatory oocyte. Biol Reprod, 1980, 22, 999–1005 10.1095/biolreprod22.4.999 [DOI] [PubMed] [Google Scholar]
- 49. Gallicano GI, Larabell CA, McGaughey RW, Capco DG. Novel cytoskeletal elements in mammalian eggs are composed of a unique arrangement of intermediate filaments. Mech Dev, 1994, 45, 211–226 10.1016/0925‐4773(94)90009‐4 [DOI] [PubMed] [Google Scholar]
- 50. Szollosi D. Morphological changes in mouse eggs due to aging in the fallopian tube. Am J Anat, 1971, 130, 209–225 10.1002/aja.1001300207 [DOI] [PubMed] [Google Scholar]
- 51. Longo FJ. Ultrastructural changes in rabbit eggs aged in vivo. Biol Reprod, 1974, 11, 22–39 10.1095/biolreprod11.1.22 [DOI] [PubMed] [Google Scholar]
- 52. Webb M, Howlett SK, Maro B. Parthenogenesis and cytoskeletal organization in ageing mouse eggs. J Embryol Exp Morphol, 1986, 95, 131–145 [PubMed] [Google Scholar]
- 53. Pickering SJ, Johnson MH, Braude PR, Houliston E. Cytoskeletal organization in fresh, aged and spontaneously activated human oocytes. Hum Reprod, 1988, 3, 978–989 [DOI] [PubMed] [Google Scholar]
- 54. Kim NH, Moon SJ, Prather RS, Day BN. Cytoskeletal alteration in aged porcine oocytes and parthenogenesis. Mol Reprod Dev, 1996, 43, 513–518 10.1002/(SICI)1098‐2795(199604)43:4<513::AID‐MRD14>3.0.CO;2‐# [DOI] [PubMed] [Google Scholar]
- 55. Longo FJ. Changes in the zones pellucidae and plasmalemma of aging mouse eggs. Biol Reprod, 1981, 25, 399–411 10.1095/biolreprod25.2.399 [DOI] [PubMed] [Google Scholar]
- 56. Xu Z, Abbott A, Kopf GS, Schultz RM, Ducibella T. Spontaneous activation of ovulated mouse eggs: time‐dependent effects on M‐phase exit, cortical granule exocytosis, maternal messenger ribonucleic acid recruitment, and inositol 1,4,5‐trisphosphate sensitivity. Biol Reprod, 1997, 57, 743–750 10.1095/biolreprod57.4.743 [DOI] [PubMed] [Google Scholar]
- 57. Goud AP, Goud PT, Diamond MP, Oostveldt P, Hughes MR. Microtubule turnover in ooplasm biopsy reflects ageing phenomena in the parent oocyte. Reprod Biomed Online, 2005, 11, 43–52 10.1016/S1472‐6483(10)61297‐7 [DOI] [PubMed] [Google Scholar]
- 58. Dodson MG, Minhas BS, Curtis SK, Palmer TV, Robertson JL. Spontaneous zona reaction in the mouse as a limiting factor for the time in which an oocyte may be fertilized. J In Vitro Fertil Embryo Transfer IVF, 1989, 6, 101–106 10.1007/BF01130735 [DOI] [PubMed] [Google Scholar]
- 59. Sundstrom P, Nilsson BO, Liedholm P, Larsson E. Ultrastructural characteristics of human oocytes fixed at follicular puncture or after culture. J In Vitro Fertil Embryo Transfer IVF, 1985, 2, 195–206 10.1007/BF01201797 [DOI] [PubMed] [Google Scholar]
- 60. Sun QY, Schatten H. Centrosome inheritance after fertilization and nuclear transfer in mammals. Adv Exp Med Biol, 2007, 591, 58–71 10.1007/978‐0‐387‐37754‐4_4 [DOI] [PubMed] [Google Scholar]
- 61. Sun QY, Schatten H. Regulation of dynamic events by microfilaments during oocyte maturation and fertilization. Reproduction, 2006, 131, 193–205 10.1530/rep.1.00847 [DOI] [PubMed] [Google Scholar]
- 62. Sathananthan AH, Selvaraj K, Girijashankar ML, Ganesh V, Selvaraj P, Trounson AO. From oogonia to mature oocytes: inactivation of the maternal centrosome in humans. Microsc Res Tech, 2006, 69, 396–407 10.1002/jemt.20299 [DOI] [PubMed] [Google Scholar]
- 63. Palacios MJ, Joshi HC, Simerly C, Schatten G. Gamma‐tubulin reorganization during mouse fertilization and early development. J Cell Sci, 1993, 104 (Pt 2) 383–389 [DOI] [PubMed] [Google Scholar]
- 64. Eichenlaub‐Ritter U, Stahl A, Luciani JM. The microtubular cytoskeleton and chromosomes of unfertilized human oocytes aged in vitro. Hum Genet, 1988, 80, 259–264 10.1007/BF01790094 [DOI] [PubMed] [Google Scholar]
- 65. Wissen B, Bomsel‐Helmreich O, Debey P, Eisenberg C, Vautier D, Pennehouat G. Fertilization and ageing processes in non‐divided human oocytes after GnRHa treatment: an analysis of individual oocytes. Hum Reprod, 1991, 6, 879–884 [DOI] [PubMed] [Google Scholar]
- 66. Rodman TC. Chromatid disjunction in unfertilized ageing oocytes. Nature, 1971, 233, 191–193 10.1038/233191a0 [DOI] [PubMed] [Google Scholar]
- 67. Mailhes JB, Young D, London SN. Postovulatory ageing of mouse oocytes in vivo and premature centromere separation and aneuploidy. Biol Reprod, 1998, 58, 1206–1210 10.1095/biolreprod58.5.1206 [DOI] [PubMed] [Google Scholar]
- 68. Zenzes MT, Casper RF. Cytogenetics of human oocytes, zygotes, and embryos after in vitro fertilization. Hum Genet, 1992, 88, 367–375 10.1007/BF00215667 [DOI] [PubMed] [Google Scholar]
- 69. Schatten H. The mammalian centrosome and its functional significance. Histochem Cell Biol, 2008, 129, 667–686 10.1007/s00418‐008‐0427‐6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cascio SM, Wassarman PM. Program of early development in the mammal: post‐transcriptional control of a class of proteins synthesized by mouse oocytes and early embryos. Dev Biol, 1982, 89, 397–408 10.1016/0012‐1606(82)90328‐1 [DOI] [PubMed] [Google Scholar]
- 71. Boerjan ML, Boer P. First cell cycle of zygotes of the mouse derived from oocytes aged postovulation in vivo and fertilized in vivo. Mol Reprod Dev, 1990, 25, 155–163 10.1002/mrd.1080250208 [DOI] [PubMed] [Google Scholar]
- 72. Takahashi T, Takahashi E, Igarashi H, Tezuka N, Kurachi H. Impact of oxidative stress in aged mouse oocytes on calcium oscillations at fertilization. Mol Reprod Dev, 2003, 66, 143–152 10.1002/mrd.10341 [DOI] [PubMed] [Google Scholar]
- 73. Takahashi T, Igarashi H, Kawagoe J, Amita M, Hara S, Kurachi H. Poor embryo development in mouse oocytes aged in vitro is associated with impaired calcium homeostasis. Biol Reprod, 2009, 80, 493–502 10.1095/biolreprod.108.072017 [DOI] [PubMed] [Google Scholar]
- 74. Chi MM, Manchester JK, Yang VC, Curato AD, Strickler RC, Lowry OH. Contrast in levels of metabolic enzymes in human and mouse ova. Biol Reprod, 1988, 39, 295–307 10.1095/biolreprod39.2.295 [DOI] [PubMed] [Google Scholar]
- 75. Igarashi H, Takahashi T, Takahashi E, Tezuka N, Nakahara K, Takahashi K, Kurachi H. Aged mouse oocytes fail to readjust intracellular adenosine triphosphates at fertilization. Biol Reprod, 2005, 72, 1256–1261 10.1095/biolreprod.104.034926 [DOI] [PubMed] [Google Scholar]
- 76. Kikuchi K, Naito K, Noguchi J, Shimada A, Kaneko H, Yamashita M, Tojo H, Toyoda Y. Inactivation of p34cdc2 kinase by the accumulation of its phosphorylated forms in porcine oocytes matured and aged in vitro. Zygote, 1999, 7, 173–179 10.1017/S0967199499000544 [DOI] [PubMed] [Google Scholar]
- 77. Tian XC, Lonergan P, Jeong BS, Evans AC, Yang X. Association of MPF. MAPK, and nuclear progression dynamics during activation of young and aged bovine oocytes. Mol Reprod Dev, 2002, 62, 132–138 10.1002/mrd.10072 [DOI] [PubMed] [Google Scholar]
- 78. Tatone C, Carbone MC, Gallo R, Delle Monache S, Di Cola M, Alesse E, Amicarelli F. Age‐associated changes in mouse oocytes during postovulatory in vitro culture: possible role for meiotic kinases and survival factor BCL2. Biol Reprod, 2006, 74, 395–402 10.1095/biolreprod.105.046169 [DOI] [PubMed] [Google Scholar]
- 79. Ma W, Zhang D, Hou Y, Li YH, Sun QY, Sun XF, Wang WH. Reduced expression of MAD2, BCL2, and MAP kinase activity in pig oocytes after in vitro aging are associated with defects in sister chromatid segregation during meiosis II and embryo fragmentation after activation. Biol Reprod, 2005, 72, 373–383 10.1095/biolreprod.104.030999 [DOI] [PubMed] [Google Scholar]
- 80. Vincent C, Cheek TR, Johnson MH. Cell cycle progression of parthenogenetically activated mouse oocytes to interphase is dependent on the level of internal calcium. J Cell Sci, 1992, 103 (Pt 2) 389–396 [DOI] [PubMed] [Google Scholar]
- 81. Igarashi H, Takahashi E, Hiroi M, Doi K. Aging‐related changes in calcium oscillations in fertilized mouse oocytes. Mol Reprod Dev, 1997, 48, 383–390 10.1002/(SICI)1098‐2795(199711)48:3<383::AID‐MRD12>3.0.CO;2‐X [DOI] [PubMed] [Google Scholar]
- 82. Takahashi T, Saito H, Hiroi M, Doi K, Takahashi E. Effects of aging on inositol 1,4,5‐triphosphate‐induced Ca(2+) release in unfertilized mouse oocytes. Mol Reprod Dev, 2000, 55, 299–306 10.1002/(SICI)1098‐2795(200003)55:3<299::AID‐MRD8>3.0.CO;2‐G [DOI] [PubMed] [Google Scholar]
- 83. Jones KT, Whittingham DG. A comparison of sperm‐ and IP3‐induced Ca2+ release in activated and aging mouse oocytes. Dev Biol, 1996, 178, 229–237 10.1006/dbio.1996.0214 [DOI] [PubMed] [Google Scholar]
- 84. Adhikari D, Liu K. Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr Rev, 2009, 30, 438–464 10.1210/er.2008‐0048 [DOI] [PubMed] [Google Scholar]
- 85. Adhikari D, Liu K. mTOR signaling in the control of activation of primordial follicles. Cell Cycle, 2010, 9, 1673–1674 10.4161/cc.9.9.11626 [DOI] [PubMed] [Google Scholar]
- 86. Adhikari D, Zheng W, Shen Y, Gorre N, Hamalainen T, Cooney AJ, Huhtaniemi I, Lan ZJ, Liu K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet, 2010, 19, 397–410 10.1093/hmg/ddp483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jagarlamudi K, Reddy P, Adhikari D, Liu K. Genetically modified mouse models for premature ovarian failure (POF). Mol Cell Endocrinol, 2010, 315, 1–10 10.1016/j.mce.2009.07.016 [DOI] [PubMed] [Google Scholar]
- 88. Reddy P, Adhikari D, Zheng W, Liang S, Hamalainen T, Tohonen V, Ogawa W, Noda T, Volarevic S, Huhtaniemi I, Liu K. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum Mol Genet, 2009, 18, 2813–2824 10.1093/hmg/ddp217 [DOI] [PubMed] [Google Scholar]
- 89. Coulam CB, Adamson SC, Annegers JF. Incidence of premature ovarian failure. Obstet Gynecol, 1986, 67, 604–606 [PubMed] [Google Scholar]
- 90. Laissue P, Vinci G, Veitia RA, Fellous M. Recent advances in the study of genes involved in non‐syndromic premature ovarian failure. Mol Cell Endocrinol, 2008, 282, 101–111 10.1016/j.mce.2007.11.005 [DOI] [PubMed] [Google Scholar]
- 91. Stolk L, Zhai G, Meurs JB, Verbiest MM, Visser JA, Estrada K, Rivadeneira F, Williams FM, Cherkas L, Deloukas P, Soranzo N, Keyzer JJ et al. Loci at chromosomes 13, 19 and 20 influence age at natural menopause. Nat Genet, 2009, 41, 645–647 10.1038/ng.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. He C, Kraft P, Chen C, Buring JE, Pare G, Hankinson SE, Chanock SJ, Ridker PM, Hunter DJ, Chasman DI. Genome‐wide association studies identify loci associated with age at menarche and age at natural menopause. Nat Genet, 2009, 41, 724–728 10.1038/ng.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Warburton D. Biological aging and the etiology of aneuploidy. Cytogenet Genome Res, 2005, 111, 266–272 10.1159/000086899 [DOI] [PubMed] [Google Scholar]
- 94. Steuerwald N, Cohen J, Herrera RJ, Sandalinas M, Brenner CA. Association between spindle assembly checkpoint expression and maternal age in human oocytes. Mol Hum Reprod, 2001, 7, 49–55 10.1093/molehr/7.1.49 [DOI] [PubMed] [Google Scholar]
- 95. Hamatani T, Falco G, Carter MG, Akutsu H, Stagg CA, Sharov AA, Dudekula DB, VanBuren V, Ko MS. Age‐associated alteration of gene expression patterns in mouse oocytes. Hum Mol Genet, 2004, 13, 2263–2278 10.1093/hmg/ddh241 [DOI] [PubMed] [Google Scholar]
- 96. Steuerwald NM, Bermudez MG, Wells D, Munne S, Cohen J. Maternal age‐related differential global expression profiles observed in human oocytes. Reprod Biomed Online, 2007, 14, 700–708 10.1016/S1472‐6483(10)60671‐2 [DOI] [PubMed] [Google Scholar]
- 97. Grondahl ML, Yding Andersen C, Bogstad J, Nielsen FC, Meinertz H, Borup R. Gene expression profiles of single human mature oocytes in relation to age. Hum Reprod, 2010, 25, 957–968 10.1093/humrep/deq014 [DOI] [PubMed] [Google Scholar]
- 98. Bruin JP, Dorland M, Spek ER, Posthuma G, Haaften M, Looman CW, te Velde ER. Age‐related changes in the ultrastructure of the resting follicle pool in human ovaries. Biol Reprod, 2004, 70, 419–424 10.1095/biolreprod.103.015784 [DOI] [PubMed] [Google Scholar]
- 99. Wilding M, Dale B, Marino M, di Matteo L, Alviggi C, Pisaturo ML, Lombardi L, Placido G. Mitochondrial aggregation patterns and activity in human oocytes and preimplantation embryos. Hum Reprod, 2001, 16, 909–917 10.1093/humrep/16.5.909 [DOI] [PubMed] [Google Scholar]
- 100. Keefe DL, Niven‐Fairchild T, Powell S, Buradagunta S. Mitochondrial deoxyribonucleic acid deletions in oocytes and reproductive aging in women. Fertil Steril, 1995, 64, 577–583 [PubMed] [Google Scholar]
- 101. Barritt JA, Cohen J, Brenner CA. Mitochondrial DNA point mutation in human oocytes is associated with maternal age. Reprod Biomed Online, 2000, 1, 96–100 10.1016/S1472‐6483(10)61946‐3 [DOI] [PubMed] [Google Scholar]
- 102. Agarwal A, Gupta S, Sharma R. Oxidative stress and its implications in female infertility—a clinician's perspective. Reprod Biomed Online, 2005, 11, 641–650 10.1016/S1472‐6483(10)61174‐1 [DOI] [PubMed] [Google Scholar]
- 103. Tarin JJ, Gomez‐Piquer V, Pertusa JF, Hermenegildo C, Cano A. Association of female aging with decreased parthenogenetic activation, raised MPF, and MAPKs activities and reduced levels of glutathione S‐transferases activity and thiols in mouse oocytes. Mol Reprod Dev, 2004, 69, 402–410 10.1002/mrd.20180 [DOI] [PubMed] [Google Scholar]
- 104. Pan H, Ma P, Zhu W, Schultz RM. Age‐associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev Biol, 2008, 316, 397–407 10.1016/j.ydbio.2008.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Takase K, Ishikawa M, Hoshiai H. Apoptosis in the degeneration process of unfertilized mouse ova. Tohoku J Exp Med, 1995, 175, 69–76 10.1620/tjem.175.69 [DOI] [PubMed] [Google Scholar]
- 106. Exley GE, Tang C, McElhinny AS, Warner CM. Expression of caspase and BCL‐2 apoptotic family members in mouse preimplantation embryos. Biol Reprod, 1999, 61, 231–239 10.1095/biolreprod61.1.231 [DOI] [PubMed] [Google Scholar]
- 107. Spanos S, Rice S, Karagiannis P, Taylor D, Becker DL, Winston RM, Hardy K. Caspase activity and expression of cell death genes during development of human preimplantation embryos. Reproduction, 2002, 124, 353–363 10.1530/rep.0.1240353 [DOI] [PubMed] [Google Scholar]
- 108. Rong Y, Distelhorst CW. Bcl‐2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol, 2008, 70, 73–91 10.1146/annurev.physiol.70.021507.105852 [DOI] [PubMed] [Google Scholar]
- 109. Gordo AC, Rodrigues P, Kurokawa M, Jellerette T, Exley GE, Warner C, Fissore R. Intracellular calcium oscillations signal apoptosis rather than activation in in vitro aged mouse eggs. Biol Reprod, 2002, 66, 1828–1837 10.1095/biolreprod66.6.1828 [DOI] [PubMed] [Google Scholar]
- 110. Miyazaki S, Ito M. Calcium signals for egg activation in mammals. J Pharmacol Sci, 2006, 100, 545–552 10.1254/jphs.CPJ06003X [DOI] [PubMed] [Google Scholar]
- 111. Ozil JP, Huneau D. Activation of rabbit oocytes: the impact of the Ca2+ signal regime on development. Development, 2001, 128, 917–928 [DOI] [PubMed] [Google Scholar]
- 112. Ducibella T, Huneau D, Angelichio E, Xu Z, Schultz RM, Kopf GS, Fissore R, Madoux S, Ozil JP. Egg‐to‐embryo transition is driven by differential responses to Ca(2+) oscillation number. Dev Biol, 2002, 250, 280–291 10.1006/dbio.2002.0788 [PubMed] [Google Scholar]
- 113. Toth S, Huneau D, Banrezes B, Ozil JP. Egg activation is the result of calcium signal summation in the mouse. Reproduction, 2006, 131, 27–34 10.1530/rep.1.00764 [DOI] [PubMed] [Google Scholar]
- 114. Rogers NT, Halet G, Piao Y, Carroll J, Ko MS, Swann K. The absence of a Ca(2+) signal during mouse egg activation can affect parthenogenetic preimplantation development, gene expression patterns, and blastocyst quality. Reproduction, 2006, 132, 45–57 10.1530/rep.1.01059 [DOI] [PubMed] [Google Scholar]
- 115. Ozil JP, Banrezes B, Toth S, Pan H, Schultz RM. Ca2+ oscillatory pattern in fertilized mouse eggs affects gene expression and development to term. Dev Biol, 2006, 300, 534–544 10.1016/j.ydbio.2006.08.041 [DOI] [PubMed] [Google Scholar]
- 116. Gordo AC, Wu H, He CL, Fissore RA. Injection of sperm cytosolic factor into mouse metaphase II oocytes induces different developmental fates according to the frequency of [Ca(2+)](i) oscillations and oocyte age. Biol Reprod, 2000, 62, 1370–1379 10.1095/biolreprod62.5.1370 [DOI] [PubMed] [Google Scholar]
- 117. Kline D, Kline JT. Thapsigargin activates a calcium influx pathway in the unfertilized mouse egg and suppresses repetitive calcium transients in the fertilized egg. J Biol Chem, 1992, 267, 17624–17630 [PubMed] [Google Scholar]
- 118. Miyazaki S, Yuzaki M, Nakada K, Shirakawa H, Nakanishi S, Nakade S, Mikoshiba K. Block of Ca2+ wave and Ca2+ oscillation by antibody to the inositol 1,4,5‐trisphosphate receptor in fertilized hamster eggs. Science, 1992, 257, 251–255 10.1126/science.1321497 [DOI] [PubMed] [Google Scholar]
- 119. Miyazaki S, Shirakawa H, Nakada K, Honda Y. Essential role of the inositol 1,4,5‐trisphosphate receptor/Ca2+ release channel in Ca2+ waves and Ca2+ oscillations at fertilization of mammalian eggs. Dev Biol, 1993, 158, 62–78 10.1006/dbio.1993.1168 [DOI] [PubMed] [Google Scholar]
- 120. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol, 2000, 1, 11–21 10.1038/35036035 [DOI] [PubMed] [Google Scholar]
- 121. East JM. Sarco(endo)plasmic reticulum calcium pumps: recent advances in our understanding of structure/function and biology (review). Mol Membr Biol, 2000, 17, 189–200 10.1080/09687680010009646 [DOI] [PubMed] [Google Scholar]
- 122. Misquitta CM, Mack DP, Grover AK. Sarco/endoplasmic reticulum Ca2+ (SERCA)‐pumps: link to heart beats and calcium waves. Cell Calcium, 1999, 25, 277–290 10.1054/ceca.1999.0032 [DOI] [PubMed] [Google Scholar]
- 123. Dumollard R, Marangos P, Fitzharris G, Swann K, Duchen M, Carroll J. Sperm‐triggered [Ca2+] oscillations and Ca2+ homeostasis in the mouse egg have an absolute requirement for mitochondrial ATP production. Development, 2004, 131, 3057–3067 10.1242/dev.01181 [DOI] [PubMed] [Google Scholar]
- 124. Dumollard R, Duchen M, Sardet C. Calcium signals and mitochondria at fertilisation. Semin Cell Dev Biol, 2006, 17, 314–323 10.1016/j.semcdb.2006.02.009 [DOI] [PubMed] [Google Scholar]
- 125. He H, Lam M, McCormick TS, Distelhorst CW. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl‐2. J Cell biol, 1997, 138, 1219–1228 10.1083/jcb.138.6.1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Kuo TH, Kim HR, Zhu L, Yu Y, Lin HM, Tsang W. Modulation of endoplasmic reticulum calcium pump by Bcl‐2. Oncogene, 1998, 17, 1903–1910 10.1038/sj.onc.1202110 [DOI] [PubMed] [Google Scholar]
- 127. Ramalho‐Santos J, Varum S, Amaral S, Mota PC, Sousa AP, Amaral A. Mitochondrial functionality in reproduction: from gonads and gametes to embryos and embryonic stem cells. Hum Reprod Updat, 2009, 15, 553–572 10.1093/humupd/dmp016 [DOI] [PubMed] [Google Scholar]
- 128. Wallace DC. Mitochondrial diseases in man and mouse. Science, 1999, 283, 1482–1488 10.1126/science.283.5407.1482 [DOI] [PubMed] [Google Scholar]
- 129. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis Int J Program Cell Death, 2007, 12, 913–922 [DOI] [PubMed] [Google Scholar]
- 130. Rohn TT, Hinds TR, Vincenzi FF. Ion transport ATPases as targets for free radical damage. Protection by an aminosteroid of the Ca2+ pump ATPase and Na+/K+ pump ATPase of human red blood cell membranes. Biochem Pharmacol, 1993, 46, 525–534 10.1016/0006‐2952(93)90530‐A [DOI] [PubMed] [Google Scholar]
- 131. Wesson DE, Elliott SJ. The H2O2‐generating enzyme, xanthine oxidase, decreases luminal Ca2+ content of the IP3‐sensitive Ca2+ store in vascular endothelial cells. Microcirculation, 1995, 2, 195–203 10.3109/10739689509146767 [DOI] [PubMed] [Google Scholar]
- 132. Willimott S, Wagner SD. Post‐transcriptional and post‐translational regulation of Bcl2. Biochem Soc Trans, 2010, 38, 1571–1575 10.1042/BST0381571 [DOI] [PubMed] [Google Scholar]
- 133. Li D, Ueta E, Kimura T, Yamamoto T, Osaki T. Reactive oxygen species (ROS) control the expression of Bcl‐2 family proteins by regulating their phosphorylation and ubiquitination. Cancer science, 2004, 95, 644–650 10.1111/j.1349‐7006.2004.tb03323.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl‐2 expression by reactive oxygen species. Proc Natl Acad Sci USA, 2003, 100, 15035–15040 10.1073/pnas.1936213100 [DOI] [PMC free article] [PubMed] [Google Scholar]