

Summary
The immune system and the kidneys are closely linked. In health the kidneys contribute to immune homeostasis, while components of the immune system mediate many acute forms of renal disease and play a central role in progression of chronic kidney disease. A dysregulated immune system can have either direct or indirect renal effects. Direct immune‐mediated kidney diseases are usually a consequence of autoantibodies directed against a constituent renal antigen, such as collagen IV in anti‐glomerular basement membrane disease. Indirect immune‐mediated renal disease often follows systemic autoimmunity with immune complex formation, but can also be due to uncontrolled activation of the complement pathways. Although the range of mechanisms of immune dysregulation leading to renal disease is broad, the pathways leading to injury are similar. Loss of immune homeostasis in renal disease results in perpetual immune cell recruitment and worsening damage to the kidney. Uncoordinated attempts at tissue repair, after immune‐mediated disease or non‐immune mediated injury, result in fibrosis of structures important for renal function, leading eventually to kidney failure. As renal disease often manifests clinically only when substantial damage has already occurred, new diagnostic methods and indeed treatments must be identified to inhibit further progression and promote appropriate tissue repair. Studying cases in which immune homeostasis is re‐established may reveal new treatment possibilities.
Keywords: fibrosis, immune homeostasis, inflammation, kidney disease
Introduction
The kidneys, as a major filter organ for the blood and the key organ responsible for maintaining total body water balance and circulatory pressure, receive a rich blood supply by which they monitor and modify the functional status of multiple organ systems. Besides clearing metabolic waste products, toxins and drugs from our body, the kidneys also clear circulating cytokines and bacterial toxins such as lipopolysaccharide (LPS) and continuously sample blood‐borne proteins, contributing to homeostasis of the immune system. The removal of cytokines from the blood can limit inflammation 1, 2, and the clearance of bacterial components reduces would‐be immune cell activation by pattern recognition receptors (PRRs) 2, 3. In addition, kidney resident dendritic cells (DCs) appear to be very important in maintaining peripheral tolerance 1. As 85% of the water filtered at the glomerulus is reabsorbed immediately by the proximal tubule, cells in the distal nephron downstream experience filtered low molecular weight antigens at concentrations up to 10 times higher than in the blood itself. These antigens are taken up readily by a dense network of DCs; for example, via dendrites protruding directly into the tubular lumen, enabling these filtered antigens to be presented to T cells in renal draining lymph nodes more efficiently than possible elsewhere in the body. Through this regular presentation of innocuous antigens in the absence of danger signals, potentially autoreactive T cells recognizing these low molecular weight antigens are inactivated. Thus, the kidneys, in addition to the spleen, assist in maintaining peripheral tolerance to circulating antigens such as hormones and food proteins 1, 4. The kidneys' important contribution to immune homeostasis becomes especially clear in end stage renal disease (ESRD), where immune function is severely compromised. The retention of uraemic toxins and cytokines activates innate immune cells leading to a vicious cycle of further cytokine and reactive oxygen species production, both contributing to tissue damage and increasing cardiovascular risk. Additionally, lymphocyte number and function decrease, leaving the patient functionally immunocompromised and at risk of infection as well as viral‐associated cancers 2, 5.
Conversely, the kidneys themselves are also very susceptible to immune‐mediated diseases. Loss of immune homeostasis can affect the kidney adversely either directly or indirectly, leading to loss of kidney function. Homeostasis describes the physiological condition of a system under normal conditions. In the immune system, homeostasis therefore refers to its function and maintenance in the uninfected host 6. However, it could be argued that the inflammatory immune response to infections and tissue damage is a normal extension of the immune system's homeostatic function, as long as the immune response resolves. Assuming the latter definition of immune homeostasis, loss of homeostasis includes over‐ or under‐activity of the immune response, but not the initial reaction to infection or tissue injury. The following is a brief overview of immune‐mediated kidney disease which, in accordance with the above definition, will not include congenital kidney diseases or those caused by infections or sterile injury.
Direct immune‐mediated renal disease
Renal diseases associated with loss of immune homeostasis can be grouped according to direct or indirect immune‐mediated kidney injury. Direct immune‐mediated renal disease involves the immune system targeting specific antigens within the kidney, while in indirect immune‐mediated renal disease the kidneys are a bystander victim of processes resulting from systemic dysregulation of the immune system. Self‐antigens found constituently in the kidneys are the targets for several autoimmune diseases, which can inflict either renal limited pathology or additionally involve multiple organ systems.
Overall, disruption of immune homeostasis leads to direct renal disease through autoreactivity of both T and B cells, which can damage the kidneys at different sites. The resulting injury shows a range of histological patterns, which correspond to various underlying mechanisms associated with specific treatment responses and renal prognosis, but the range of clinical presentations is more limited, highlighting the value of renal biopsy in patient management. Continuous insults to epithelial and interstitial cells in the kidney result in scarring and diminished renal function, thus leading to the establishment of chronic kidney disease (CKD).
Direct targets of the immune system located in the glomerulus
Classic examples of direct immune‐mediated renal disease where the target antigen is located in the glomerulus are anti‐glomerular basement membrane disease (anti‐GBM disease) and membranous glomerulonephritis (MGN), both of which present clinically with glomerular abnormalities, such proteinuria and haematuria, and often progress to CKD and ultimately ESRD. Anti‐GBM disease is a form of crescentic glomerulonephritis (GN), presenting most often with acute renal abnormalities, and in some patients also severe alveolar haemorrhage in the lungs (Goodpasture's syndrome). Circulating autoantibodies bind the NC1 domain of the collagen IV α3 chain [α3(IV)NC1] 7. Interestingly, the epitopes recognized by this autoantibody are typically hidden within the quaternary structure of type IV collagen, so that an initial conformational change must occur before the autoantibodies can bind. Potential triggers for the unmasking of the autoantigen are still unknown, but once binding has occurred it leads to further conformational changes and thus perpetuation of antigen–antibody complex formation 8. The resulting linear immune deposits along glomerular basement membranes, composed typically of immunoglobulin (Ig)G antibody and complement components, damage the surrounding endothelial cells and podocytes, leading to robust immune cell infiltration, inflammation and ultimately fibrosis (Fig. 1).
Figure 1.
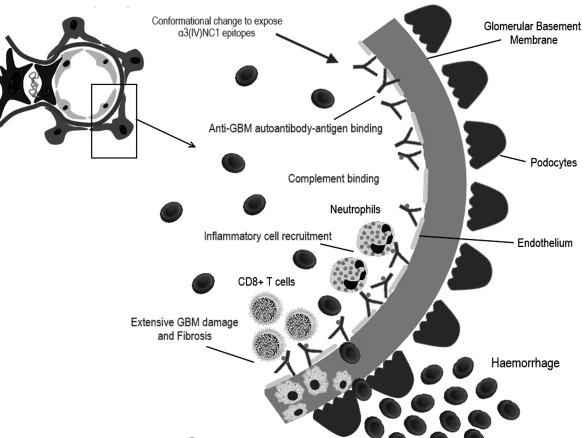
Molecular pathogenesis of anti‐glomerular basement membrane disease. An unknown stimulus directs production of anti‐glomerular basement membrane (GBM) autoantibodies. The principal target for the anti‐GBM antibodies [which are typically immunoglobulin IgG1 and 3 but sometimes IgA or IgM] is the NC1 domain of the alpha‐3 chain of type IV collagen [α3(IV) chain], one of six genetically distinct gene products found in basement membrane collagen. The antigen–antibody binding activates the complement cascade and further inflammatory cell recruitment leads to chronic inflammation and fibrosis.
In contrast, MGN is defined by diffuse thickening of the glomerular capillary wall by subepithelial immune deposits. Although MGN can be caused by immune complex deposition secondary to systemic diseases, such as systemic lupus erythematosus (SLE), infections, such as hepatitis B virus, or in association with malignancy, most cases of MGN are primary and a substantial fraction of these cases are caused by autoantibodies against proteins expressed normally by podocytes in the glomerulus. The binding of these autoantibodies to their respective podocyte membrane antigens activates the classical complement pathway leading to podocyte injury. Podocytes react by altering their cytoskeleton and secreting mediators of fibrosis and proinflammatory cytokines. This local glomerular process results in remodelling of the glomerular basement membrane and can develop into focal glomerular segmental sclerosis; the persistent proteinuria due to impairment of the glomerular filter is blamed for damage and scarring of downstream tubules and interstitium 9. In approximately 75% of primary MGN cases, the autoantibody target is the phospholipase A2 receptor (PLA2R) 9, 10. Autoantibodies against a further podocyte membrane protein – thrombospondin type‐1 domain‐containing 7A – are present in the serum of 8–14% of PLA2R‐negative primary MGN patients 11. It is likely that further autoantigens will be discovered as causes of primary MGN, which may turn out to be simply a collective term for autoimmune diseases targeting renal podocyte membranes.
Direct targets of the immune system located in the renal tubules and interstitium
Anti‐tubular basement membrane (TBM) disease, for example, is an autoantibody‐mediated form of progressive primary tubulointerstitial nephritis (TIN). Damage to the TBM occurs through both autoantibody deposition and activity of autoreactive T cells against the membrane's 3M‐1 glycoprotein 12. Strong linear staining of the proximal TBM with IgG is visible with immunofluorescence microscopy. Light microscopy of these renal biopsies often shows interstitial inflammation with extensive fibrosis and small atrophic tubules 13.
TIN can also result from primary Sjögren's syndrome (pSS), affecting approximately 10% of pSS patients. Although pSS is a systemic autoimmune condition characteristically affecting glandular epithelium, with renal involvement manifested typically by glomerulonephritis due to deposition of circulating immune complexes, development of TIN in pSS seems to result from a renal‐directed immune process. Potential self‐antigens targeted by autoantibodies and autoreactive T cells in pSS include carbonic anhydrase and the hydrogen transporter H+‐ATPase, which are found in both renal tubules and salivary glands 14. pSS‐associated TIN is characterized by lymphocyte infiltration of the tubular and interstitial compartments, accompanied by a large proportion of interstitial plasma cells. Although TIN in general is associated with subsequent tubular atrophy and interstitial fibrosis, often leading to CKD, pSS‐associated TIN usually takes a relatively benign course and often responds well to treatment with rituximab, a monoclonal antibody against the B cell marker CD20 15.
Indirect immune‐mediated renal disease
As for direct immune‐mediated renal disease, the majority of indirect immune‐mediated renal pathologies are due to autoimmunity, but some are the consequence of genetic defects in the complement system or haematological malignancies, such as multiple myeloma or Hodgkin lymphoma. Indirect kidney injury secondary to loss of immune homeostasis is caused by three major mechanisms: circulating immune complex deposition, dysregulation of the alternate complement pathway and deposition of monoclonal immunoglobulins, although others are implicated, such as elevated systemic cytokines in podocytopathies and autoantibodies targeting the systemic vasculature causing thrombotic microangiopathy and downstream ischaemic damage, to which the renal tubular epithelium is especially vulnerable.
Immune complex deposition in the glomerulus occurs in many systemic autoimmune diseases. In addition, immune complexes may also form in the context of infection – most frequently streptococcal and hepatitis B infection 1, 12. Circulating immune complexes consist of several antibodies grouped around their target antigen, which in some cases is another antibody (e.g. rheumatoid factor). These complexes often settle in the glomerulus because of their size and become trapped, according to their charge, within various compartments of the glomerular filtration barrier. In some cases, complexes can form in situ, when a target antigen exogenous to the kidney becomes trapped within the glomerular basement membrane due to its relatively small size and positive charge 9. Similarly, nucleosomes from apoptosed cells can become trapped in the negatively charged GBM 16, where they are subsequently bound by autoantibodies to the nucleosome component of eukaryotic cell chromosomes, which are present in the serum of some SLE patients and have been associated with an increased risk for developing lupus nephritis 17.
Immune complexes damage the glomerular endothelium, epithelium and mesangium by activating circulating immune cells and kidney‐intrinsic cells expressing Fc receptors. Activated cells secrete cytokines and vasoactive substances to create a proinflammatory environment in the region local to immune complex deposition. In addition, the complement cascade is activated via the classical pathway, the end‐point of the cascade being the formation of the membrane attack complex, which injures surrounding cells, leading to further proinflammatory signalling 18. Table 1 lists examples of GN caused by immune complex deposition associated with diseases involving loss of immune homeostasis.
Table 1.
Immune complex‐mediated glomerulonephritis
| Location of glomerular immune complex deposits | Associated glomerulonephritides, with examples | Associated diseases involving loss of immune homeostasis, examples |
|---|---|---|
| Subendothelial |
Membranoproliferative GN IgA nephropathy Lupus nephritis (classes III & IV)* |
Autoimmune disease (e.g. Sjögren's syndrome, scleroderma, SLE) |
| Mesangial |
Membranoproliferative GN IgA nephropathy Lupus nephritis (classes I & II)* |
Autoimmune disease (e.g. coeliac disease, SLE) Inflammatory disease (Crohn's disease) |
| Subepithelial |
Membranoproliferative GN Infection‐related GN Membranous GN Lupus nephritis (class V)* |
Autoimmune disease (e.g. SLE, anti‐PLA2R disease) |
In addition to the listed conditions, other autoimmune diseases uncommonly lead to immune complex deposition. *International Society of Nephrology/Renal Pathology Society 2004 Classification of Lupus Glomerulonephritis 19. GN = glomerulonephritis; SLE = systemic lupus erythematosus; Ig = immunoglobulin; PLA2R = phospholipase A2 receptor.
Over‐activity of the complement system leading to renal damage can be due to genetic mutations in complement regulatory components or autoimmune disease against components of the complement system. The alternative pathway of complement activation is based on the spontaneous breakdown of C3 into C3a and C3b, which can bind to the cell surface. Other complement factors are added to form an enzymatic complex able to cleave further C3 and eventually C5, which is essential for the formation of the membrane attack complex. This system is regulated by several inhibitory factors. The cascade of the alternative pathway leading to the formation of C3 convertase (C3bBb) is controlled by factors H and I. Mutations or autoantibodies against these can lead to increased activation of the C3 convertase, leading to significantly increased levels of activated C3 20. Additionally, the IgG autoantibody, C3 nephritic factor, stabilizes C3 convertase, preventing its inactivation. As a result, damaging C3 deposits can form in the glomerulus, activating downstream inflammatory cascades and promoting leucocyte infiltration. In dense deposit disease (DDD), small band‐like electron dense deposits of complement form along the GBM, leading to GBM thickening and dysfunction 21. In addition to GBM deposits, C3 glomerulopathies also show varying degrees of mesangial C3 deposits, leading to mesangial cell proliferation and matrix expansion. In both DDD and C3 glomerulopathies, immunoglobulin deposits may co‐localize with the complement deposits, although the immune complexes will be present in smaller proportions compared to those seen in true immune complex‐mediated renal diseases 1, 13, 18.
Monoclonal gammopathies are associated with a wide range of renal pathologies. Large amounts of whole or fragmented abnormal immunoglobulins (paraproteins) can be produced in clonal B lymphocyte proliferative disorders and plasma cell dyscrasias/neoplasms, such as multiple myeloma or monoclonal gammopathies of undetermined significance (MGUS), although instances of the latter causing renal pathology are better described as monoclonal gammopathies of renal significance (MGRS) 22. The type of renal lesions caused by monoclonal immunoglobulin deposits is determined by the quantity and structural characteristics of the paraprotein. In contrast to immune complex‐mediated disorders, where deposits most often affect glomeruli and extra‐glomerular deposits are seen in select circumstances, in MRGS the distribution of paraprotein deposits in the kidney is often broader, affecting any compartment of the kidney alone or in combination – glomeruli, renal vasculature, tubules and/or interstitium. Depending on the type and location of the monoclonal deposits, different mechanisms of direct or indirect damage to the renal parenchyma may occur, as exemplified below 23.
Light chain cast nephropathy, the most common monoclonal immunoglobulin‐associated disorder seen on renal biopsy, is a serious complication of multiple myeloma. The high concentration of light chains, which are filtered freely in the glomerulus, exceeds the reabsorptive capacity of the proximal convoluted tubule, so that the light chains in the filtrate reach the thick ascending loop of Henle, where they bind to uromodulin, forming casts occluding the tubular lumen and causing acute loss of renal function. These casts damage the surrounding tubular epithelial cells causing local irritation and inflammation, and obstruction of the tubule may lead to rupture and thus inflammation and later fibrosis of the interstitium 23. In Randall‐type monoclonal immunoglobulin deposition disease, glomerular and tubular basement membranes become thickened by linear deposits of monoclonal light chains alone, intact monoclonal immunoglobulins or rarely monoclonal heavy chains alone, interfering with the selective filtration barrier of the glomerulus, leading to proteinuria. In addition, deposited light chains interact with light chain receptors on mesangial cells, which respond by increasing mesangial matrix deposition 23, 24. Furthermore, monoclonal immunoglobulin deposits can cause various other forms of renal disease and injury, such as glomerulonephritis, vascular occlusion, and renal tubular pathologies, such as light chain proximal tubulopathy, which is a cause of Fanconi syndrome 23, 24.
Injury of the renal vascular tree also can be mediated by conditions related to loss of immune homeostasis. Primary anti‐phospholipid syndrome (APS) is an autoimmune disease characterized by circulating anti‐phospholipid antibodies (aPLs), recurrent venous or arterial thrombosis and pregnancy‐related problems. The aPLs, which include lupus anti‐coagulant (LA), anti‐cardiolipin (aCL) and anti‐β2‐glycoprotein I (anti‐β2GPI), create a procoagulant state, which leads to thrombosis in the presence of a further prothrombotic factor, such as oxidative stress, surgical intervention or infection. Anti‐β2GPI, for example, binds β2GPI on the endothelial cell surface, inducing a procoagulant and proinflammatory phenotype. aPLs also up‐regulate tissue factor expression, promote endothelial leucocyte adhesion, cytokine secretion, prostaglandin E2 synthesis and promote platelet aggregation and activation. They may further affect fibrinolysis and the natural anti‐coagulant annexin A5, thus increasing the procoagulant effect by decreasing natural anti‐coagulants 25. Approximately 10% of patients with primary APS develop renal involvement 26. Thrombosis can occur at any level of the renal vascular tree, from renal artery stenosis to renal vein thrombosis. The various intrarenal vascular lesions and their subsequent chronic effects on kidney tissue are grouped under the term ‘APS nephropathy’. Damage occurs from chronic narrowing of arteries by arteriosclerosis and acute or chronic occlusion of smaller arteries, arterioles and capillaries by thrombotic microangiopathy, both of which can lead to focal cortical atrophy or focal segmental glomerulosclerosis 27. True immune complex‐type deposits are not seen in the lesions, and injury to the renal parenchyma is mediated mainly by ischaemia 25, 26.
Similar‐appearing thrombotic microangiopathy lesions can be caused by haemolytic uraemic syndrome (HUS), either the classical diarrhoea‐associated type or atypical non‐diarrhoea‐associated HUS (aHUS). Classic HUS is caused by Shiga‐toxin, which is produced by Shigella and certain enterohaemorrhagic Eschrichia coli bacteria. The rarer aHUS is a consequence of dysregulation of the alternative complement pathway. aHUS is associated frequently with mutations in the CFH gene encoding complement regulatory protein, Factor H (FH), which normally binds to cell surfaces to protect from complement‐mediated lysis and regulates activity of C3 negatively in the fluid phase. In aHUS, C‐terminal mutations in CFH interfere with the ability of FH to bind endothelial cell surfaces, leaving them vulnerable to complement‐mediated destruction and ensuing microvascular thrombosis 28. In contrast, C3 glomerulonephritis (C3GN) often involves N‐terminal mutations in CFH, leading to disinhibited activation of C3 in the fluid phase and subsequent accumulation of alternate complement pathway components in glomeruli 29. Interestingly, mice deficient in FH demonstrate a characteristic C3GN pattern of renal disease from uncontrolled C3 activation although, upon introduction of a transgenic mouse FH molecule with C‐terminal mutations analogous to those found in human aHUS, switch disease phenotype from C3GN to aHUS 30.
Pauci‐immune focal necrotizing GN is a renal complication of systemic small vessel vasculitides often caused by autoantibodies to neutrophil cytoplasmic antigens (ANCA). ANCA‐GN results in significant damage to the glomerular vasculature and can demonstrate associated arterial damage, although by a different mechanism from APS and aHUS. ANCA‐GN runs a rapidly progressive clinical course and displays a crescentic GN morphology on renal biopsy, with severe necrotizing destruction of the glomerular tuft 31. The pathological mechanisms of ANCA‐GN are not clear. It may be initiated by glomerular injury, which activates neutrophils leading to degranulation and expulsion of neutrophil extracellular traps (NETs) as the first step of the tissue repair process. The expulsion of NETs in the glomerular capillaries releases the ANCA‐associated antigens [myeloperoxidase (MPO), proteinase 3 (PR3) and lysosome‐associated membrane glycoprotein 2], which are then bound by circulating ANCA autoantibodies 1. Furthermore, in‐vivo studies have shown that PR3‐ANCA can activate circulating neutrophils directly 31. Autoreactive T cells have also been implicated as important factors in the pathogenesis of ANCA‐GN 32. Together, reactive oxygen species released by activated neutrophils, autoantibody binding to NETs and consequently broader activation of leucocytes cause localized inflammation and complement activation within glomerular capillaries. This vascular inflammation leads to localized necrosis, resulting in destruction of the glomerular filter with clinically evident haematuria and rapidly diminishing renal function 1.
While the inciting immunopathological events of indirect immune‐mediated renal disease vary, there is considerable overlap in the final pathological process of renal fibrosis leading to loss of renal function. As in direct immune‐mediated renal disease, continuous renal insult leads eventually to CKD.
Transplantation
The immune‐mediated effects on the kidney after transplantation are extraordinary. Not only is organ transplantation an artificial setting – one that the immune system was not designed to deal with in terms of tissue surveillance or peripheral tolerance 33 – the alloimmune response underlying transplant rejection is incredibly robust.
Recipient immune responses directed against the allograft can be viewed in our previous construct of direct immune‐mediated renal injury resulting from loss of immune homeostasis. Allograft rejection can be mediated by alloreactive antibodies or T cells. Acute rejection occurs typically within days or weeks of transplantation, when a major immune reaction occurs. Conversely, chronic rejection is a slow process, during which low‐level immune activity causes continuous damage to the renal epithelium and vasculature leading to fibrosis and progressively declining kidney function 34.
Antibodies and T cells target the foreign major histocompatibility complex (MHC) and MHC‐like molecules expressed on the graft endothelium and epithelium. Donor‐reactive T cells can be generated by pre‐exposure to foreign MHC molecules; for example, through a previous blood transfusion or pregnancy, by generation of pathogen‐specific memory T cells cross‐reactive to epitopes on donor cells or by de‐novo activation of naive T cells against graft antigens, achieved by interaction with DCs derived either from the donor graft or from the recipient. Donor‐derived DCs activate recipient T cells directly via the foreign MHC that they express, regardless of the presented peptide antigen. Recipient DCs present, on self‐MHC, peptides derived from donor‐specific proteins and thus activate the T cells against donor cells indirectly 33. In addition to the multiple adaptive immune pathways that can contribute to the development of the clinical rejection, the innate immune system probably also contributes to the initiation of transplant rejection 35. Due to these complex mechanisms and the fact that the T cells respond so strongly to allogeneic MHC molecules, current immunosuppressive therapy to prevent transplant rejection is often not 100% effective 33, 34. Furthermore, this therapy is complicated by the side effects of immunosuppression, and a difficult balance has to be struck between immune‐mediated transplant loss and immunosuppression‐related malignancies and opportunistic infections.
Post‐transplant immunosuppression is often achieved with calcineurin inhibitors, which are toxic to the kidneys themselves. Additionally, the immunosuppressed state can lead to another form of disease which would fall under our heading of indirect immune‐mediated renal disease, namely post‐transplantation lymphoproliferative disorders (PTLD). PTLD includes several subtypes of lymphoma and is estimated to occur in up to 20% of transplant recipients 36. The most common forms of PTLD are B cell lymphomas. They develop due to immortalization of B cells, often in association with Epstein–Barr virus (EBV). Under normal conditions the abnormal cells are removed by cytotoxic T cells; however, in the immunocompromised patient this regulation cannot take place and lymphoma can develop. PTLD can occur after all types of transplant and in most tissues. Sometimes the tumour is located in the kidney, where its expansion separates the nephron elements, leading to decreased renal function. Local inflammation may occur but fibrosis and necrosis are uncommon 13, 36.
An important function of the immune system is the prevention of malignancy. Natural killer cells and cytotoxic T cells recognize and remove abnormal cells, but the level of surveillance is reduced during immunosuppression. The advances in immunosuppression and allograft survival have also improved the survival of transplant recipients. However, due to the length of immunosuppression, there is an increased risk of occurrence of tumours in general. Renal transplant recipients therefore appear to be at risk not only of PTLD but also of a variety of carcinomas 37. More specific immunosuppressive drugs are therefore required, so that a positive transplant outcome does not lead to serious malignancy.
Immune involvement in the progression of renal disease
The examples above illustrate how dysregulation of the immune system contributes to the initiation of renal disease. There is considerable overlap in the mechanisms by which both direct and indirect immune‐mediated renal diseases are caused, and the progression of each eventually leads to chronic renal failure. The immune system plays a central role in this progression of renal diseases, including those initiated by non‐immunological mechanisms such as hypoperfusion or obstructive pyelonephritis.
During acute kidney injury (AKI) following sterile tissue injury, for example after renal artery infarction or through toxins, intrarenal immune cells are activated. Damaged renal epithelial cells activate their stress response pathways, which lead to the secretion of cytokines and vasoactive factors 5. In addition, resident macrophages and DCs are activated by danger‐associated molecular patterns (DAMPs), which are released by activated renal epithelial and necrotic cells. Together the activated cells recruit further leucocytes and initiate an immune response to clear debris and necrotic tissue before tissue repair can take place 5. The immune response, although detrimental in the long term if sustained, is essential for the repair of tissue. Severe damage can rarely be restored, but the kidneys will often heal after moderate ischaemic injury. The mechanisms by which the immune system regulates the healing process are not understood fully. DCs, macrophages and regulatory T cells, as well as the cytokines IL‐10 and IL‐22, have been implicated 38, 39. A phenotypic shift in the renal mononuclear phagocytes from the proinflammatory lymphocyte antigen 6 complex, locus C (Ly6Chigh) to the Ly6Clow anti‐inflammatory‐associated phenotype can be observed as part of normal resolution following the initial responses to AKI (Fig. 2) 40. It remains uncertain how this switch is triggered.
Figure 2.
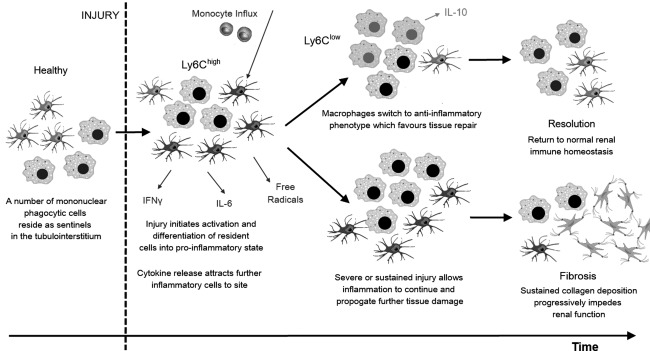
Progression versus resolution of inflammatory processes in the kidney. Pathways following acute kidney injury and activation of the renal sentinel immune cells. Initial switch to inflammatory mononuclear phagocyte phenotype characterized by marked expression of the surface glycoprotein lymphocyte antigen 6 complex locus C (Ly6Chigh). Phenotypic switch to the anti‐inflammatory phenotype (Ly6Clow) favours tissue repair and return to immune homeostasis. However, repeat or prolonged inflammation leads to necrosis of cells, fibrotic change and chronic kidney disease.
In the absence of anti‐inflammatory factors or after severe tissue damage, the immune system contributes further to the development and progression of CKD. As a consequence of any initial renal pathology, immune cells may infiltrate the damaged tissue in response to DAMPs and chemotactic factors. These cells appear to be activated in a way that does not promote healing and instead perpetuates the inflammatory process. Tissue damage also releases previously ‘hidden’ antigens, which may be recognized by autoreactive T cells. This de‐novo formation of autoimmunity has been proposed as a mechanism sustaining the immune response 38. Progressive remodelling leads eventually to tubular atrophy and interstitial scarring, which manifest clinically as worsening renal function 1.
Treatment for AKI and CKD is supportive, with the aim of slowing the progression to ESRD, which requires renal replacement therapy. Targeted treatments have not yet been developed, and are also not available for many of the immune‐mediated renal diseases. The latter are commonly treated by general immunosuppression, for example with corticosteroids. However, several of the target antigens of primary renal autoimmune diseases have been identified, meaning that directed treatment could, theoretically, be possible. For example, a murine model of anti‐GBM disease was used to demonstrate that disease activity could be reduced by induction of mucosal tolerance through nasal administration of an immunodominant peptide from the anti‐GBM target α3(IV)NC1 41. However, for many diseases the exact target antigen and its major epitopes are still unknown. In addition, clinical manifestation of the disease often occurs relatively late in the pathogenesis, when substantial damage has already occurred. Although knowledge of genetic susceptibility has increased, environmental factors are still essential to the initiation of autoimmune diseases, so that prophylactic treatment is not possible or feasible at this stage.
It would be interesting to determine whether or not the progression to ESRD can be stopped by a single intervention, regardless of the initial aetiology of renal disease. Perhaps the immune system itself can be harnessed to promote healing. Several studies in experimental ischaemic AKI suggest that cellular therapy with DCs and regulatory T cells could promote tissue repair 42, 43, 44. These treatments appear effective during the initial injury, but early administration in normal clinical conditions is usually not possible 38. Further research is therefore needed to find treatment options which could be used in clinical practice. First steps could include further investigation of the immune responses and tissue repair in patients with AKI which does not progress to CKD. The differentiation of T cell populations might be of particular interest. Moreover, anti‐GBM disease can sometimes be self‐limiting and regulatory cell populations have been described in the later phases 45. Additional research into these T cells might aid further in the understanding of the decision point between regulatory or effector T cell activation.
To conclude, the immune system plays a central role in the initiation, progression and resolution of renal disease (summarized in Fig. 3). Our understanding of renal resident immune cells and of the varied immune mechanisms underlying renal disease is increasing, and may aid in the design of future targeted immune‐based therapies to restore homeostasis of the immune system, rather than simply suppress it, in order to promote appropriate healing and prevent progression to CKD and ESRD.
Figure 3.
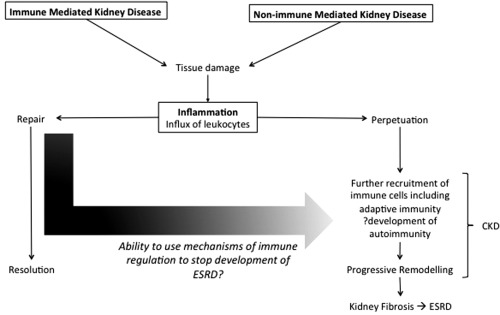
The central role of the immune system in renal pathology. CKD = chronic kidney disease; ESRD = end stage renal disease.
Disclosure
The authors have no conflicts of interest.
References
- 1. Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol 2013; 13:738–53. [DOI] [PubMed] [Google Scholar]
- 2. Betjes MG. Immune cell dysfunction and inflammation in end‐stage renal disease. Nat Rev Nephrol 2013; 9:255–65. [DOI] [PubMed] [Google Scholar]
- 3. Hato T, Dagher PC. How the innate immune system senses trouble and causes trouble. Clin J Am Soc Nephrol 2015; 10:1459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soos TJ, Sims TN, Barisoni L et al CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int 2006; 70:591–6. [DOI] [PubMed] [Google Scholar]
- 5. Yatim KM, Lakkis FG. A brief journey through the immune system. Clin J Am Soc Nephrol 2015; 10:1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murphy K, Travers P, Walport M. Janeway's immunobiology, 7th edn New York: Garland Science, 2008. [Google Scholar]
- 7. Saus J, Wieslander J, Langeveld JP, Quinones S, Hudson BG. Identification of the Goodpasture antigen as the alpha 3(IV) chain of collagen IV. J Biol Chem 1988; 263:13374–80. [PubMed] [Google Scholar]
- 8. Pedchenko V, Bondar O, Fogo AB et al Molecular architecture of the Goodpasture autoantigen in anti‐GBM nephritis. N Engl J Med 2010; 363:343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sinico RA, Mezzina N, Trezzi B, Ghiggeri GM, Radice A. Immunology of membranous nephropathy: from animal models to humans. Clin Exp Immunol 2016; 183:157–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beck LH Jr, Bonegio RG, Lambeau G et al M‐type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009; 361:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tomas NM, Beck LH Jr, Meyer‐Schwesinger C et al Thrombospondin type‐1 domain‐containing 7A in idiopathic membranous nephropathy. N Engl J Med 2014; 371:2277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neilson EG, Sun MJ, Kelly CJ et al Molecular characterization of a major nephritogenic domain in the autoantigen of anti‐tubular basement membrane disease. Proc Natl Acad Sci USA 1991; 88:2006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonsib SM. Atlas of medical renal pathology. New York: Springer, 2013. [Google Scholar]
- 14. Francois H, Mariette X. Renal involvement in primary Sjogren syndrome. Nat Rev Nephrol 2016; 12:82–93. [DOI] [PubMed] [Google Scholar]
- 15. Bitik B, Gonul II, Haznedaroglu S, Goker B, Tufan A. Granulomatous interstitial nephritis associated with primary Sjogren's syndrome. Z Rheumatol 2017; 76:458–60. [DOI] [PubMed] [Google Scholar]
- 16. Waldman M, Madaio MP. Pathogenic autoantibodies in lupus nephritis. Lupus 2005; 14:19–24. [DOI] [PubMed] [Google Scholar]
- 17. Bigler C, Lopez‐Trascasa M, Potlukova E et al Antinucleosome antibodies as a marker of active proliferative lupus nephritis. Am J Kidney Dis 2008; 51:624–9. [DOI] [PubMed] [Google Scholar]
- 18. Nangaku M, Couser WG. Mechanisms of immune‐deposit formation and the mediation of immune renal injury. Clin Exp Nephrol 2005; 9:183–91. [DOI] [PubMed] [Google Scholar]
- 19. Weening JJ, D'Agati VD, Schwartz MM et al The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004; 15:241–50. [DOI] [PubMed] [Google Scholar]
- 20. Richards A, Kavanagh D, Atkinson JP. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory states: the examples of vascular damage in atypical hemolytic uremic syndrome and debris accumulation in age‐related macular degeneration. Adv Immunol 2007; 96:141–77. [DOI] [PubMed] [Google Scholar]
- 21. Lionaki S, Gakiopoulou H, Boletis JN. Understanding the complement‐mediated glomerular diseases: focus on membranoproliferative glomerulonephritis and C3 glomerulopathies. APMIS 2016; 124:725–35. [DOI] [PubMed] [Google Scholar]
- 22. Leung N, Bridoux F, Hutchison CA et al Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 2012; 120:4292–5. [DOI] [PubMed] [Google Scholar]
- 23. Al Hussain T, Hussein MH, Al Mana H, Akthar M. Renal involvement in monoclonal gammopathy. Adv Anat Pathol 2015; 22:121–34. [DOI] [PubMed] [Google Scholar]
- 24. Bridoux F, Leung N, Hutchison CA et al Diagnosis of monoclonal gammopathy of renal significance. Kidney Int 2015; 87:698–711. [DOI] [PubMed] [Google Scholar]
- 25. Marcantoni C, Emmanuele C, Scolari F. Renal involvement in primary antiphospholipid syndrome. J Nephrol 2016; 29:507–15. [DOI] [PubMed] [Google Scholar]
- 26. Sinico RA, Cavazzana I, Nuzzo M et al Renal involvement in primary antiphospholipid syndrome: retrospective analysis of 160 patients. Clin J Am Soc Nephrol 2010; 5:1211–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang A. Thrombotic microangiopathy and the kidney: a nephropathologist's perspective. Diagn Histopathol 2013; 19:158–90. [Google Scholar]
- 28. Pérez‐Caballero D, González‐Rubio C, Gallardo ME et al Clustering of missense mutations in the C‐terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet 2001; 68:478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pickering MC, Cook HT, Warren J et al Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor. Nat Genet 2002; 31:424–8. [DOI] [PubMed] [Google Scholar]
- 30. Pickering MC, Goicoechea de Jorge E, Martínez‐Barricarte R et al Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med 2007; 204:1249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jennette JC, Falk RJ, Gasim AH. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Curr Opin Nephrol Hypertens 2011; 20:263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kerstein A, Müller A, Kabelitz D, Lamprecht P. Effector memory T‐cells in the pathogenesis of ANCA‐associated vasculitis. Z Rheumatol 2017; 76:14. [DOI] [PubMed] [Google Scholar]
- 33. Lakkis FG, Lechler RI. Origin and biology of the allogeneic response. Cold Spring Harb Perspect Med 2013; 3:a014993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nankivell BJ, Alexander SI. Rejection of the kidney allograft. N Engl J Med 2010; 363:1451–62. [DOI] [PubMed] [Google Scholar]
- 35. Zecher D, van Rooijen N, Rothstein DM, Shlomchik WD, Lakkis FG. An innate response to allogeneic nonself mediated by monocytes. J Immunol 2009; 183:7810–6. [DOI] [PubMed] [Google Scholar]
- 36. Morscio J, Tousseyn T. Recent insights in the pathogenesis of post‐transplantation lymphoproliferative disorders. World J Transplant 2016; 6:505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bieniasz M, Chmura A, Kwapisz M et al Renal tumor in allogeneic kidney transplant recipient. Transplant Proc 2016; 48:1849–54. [DOI] [PubMed] [Google Scholar]
- 38. Rabb H. The promise of immune cell therapy for acute kidney injury. J Clin Invest 2012; 122:3852–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rogers NM, Ferenbach DA, Isenberg JS, Thomson AW, Hughes J. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol 2014; 10:625–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2014; 2:1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reynolds J, Abbott DS, Karegli J, Evans DJ, Pusey CD. Mucosal tolerance induced by an immunodominant peptide from rat alpha3(IV)NC1 in established experimental autoimmune glomerulonephritis. Am J Pathol 2009; 174:2202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li L, Huang L, Ye H et al Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest 2012; 122:3931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun P, Liu J, Li W et al Human endometrial regenerative cells attenuate renal ischemia reperfusion injury in mice. J Transl Med 2016; 14:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lai LW, Yong KC, Lien YHH. Pharmacologic recruitment of regulatory T cells as a novel therapy for ischemic acute kidney injury. Kidney Int 2012; 81:983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salama AD, Chaudhry AN, Holthaus KA et al Regulation by CD25+ lymphocytes of autoantigen‐specific T‐cell responses in Goodpasture's (anti‐GBM) disease. Kidney Int 2003; 64:1685–94. [DOI] [PubMed] [Google Scholar]