

Summary
Human neutrophils are terminally differentiated cells that do not replicate and yet express a number of enzymes, notably cell cycle‐dependent kinases (CDKs), that are associated normally with control of DNA synthesis and cell cycle progression. In neutrophils, CDKs appear to function mainly to regulate apoptosis, although the mechanisms by which they regulate this process are largely unknown. Here we show that the CDK2 inhibitor, purvalanol A, induces a rapid decrease in myeloid cell leukaemia factor‐1 (Mcl‐1) levels in human neutrophils and peripheral blood mononuclear cells (PBMCs), but only induces apoptosis in neutrophils which are dependent upon expression on this protein for survival. This rapid decrease in cellular Mcl‐1 protein levels was due to a purvalanol A‐induced decrease in stability, with the half‐life of the protein decreasing from approximately 2 h in control cells to just over 1 h after addition of the CDK2 inhibitor: it also blocked the granulocyte–macrophage colony‐stimulating factor (GM‐CSF)‐dependent stabilization of Mcl‐1. Purvanalol A blocked GM‐CSF‐stimulated activation of extracellular‐regulated kinase (Erk) and signal transducer and activator of transcription (STAT)‐3, and stimulated an additive activation of protein kinase B (Akt) with GM‐CSF. Purvalanol A alone stimulated a rapid and sustained activation of p38‐mitogen‐activated protein kinase (MAPK) and the pan p38‐MAPK inhibitor, BIRB796, partly blocked the purvalanol A‐induced apoptosis and Mcl‐1 loss. These novel effects of purvalanol A may result, at least in part, from blocking GM‐CSF‐mediated Erk activation. In addition, we propose that purvalanol A‐induced activation of p38‐MAPK is, at least in part, responsible for its rapid effects on Mcl‐1 turnover and acceleration of neutrophil apoptosis.
Keywords: apoptosis, cell activation, neutrophils, protein kinases/phophatases
Introduction
Human neutrophils exhibit high rates of spontaneous apoptosis and, as a consequence, the bone marrow produces and releases billions of mature neutrophils into the bloodstream on a daily basis 1, 2. As in many other cells and tissues, neutrophil apoptosis is regulated by B cell lymphoma‐2 (Bcl‐2) family members and caspases, whose activities and subcellular localization are regulated by intrinsic and extrinsic factors including growth factors, cytokines and death receptor signalling. Human neutrophils are somewhat unusual, however, in that myeloid cell leukaemia factor‐1 (Mcl‐1) [rather than Bcl‐2 or B cell lymphoma‐extra large (Bcl‐XL)] is the major anti‐apoptotic protein that regulates apoptosis 3. Mcl‐1 is an unusual member of the Bcl‐2 family, in that it possesses a large N‐terminal region that contains PEST (proline, glutamic acid, serine and threonine) domains and other motifs that undergo post‐translational modifications such as phosphorylation and ubiquitination that regulate its function or stability 3, 4. For example, phosphorylation on residues Thr163, Thr92 and Ser121 can enhance stability or function, while phosphorylation on residues Ser155 and Ser159 can decrease stability and accelerate its rate of turnover via the proteasome 4, 5, 6. These rapid and reversible modifications to Mcl‐1 act as a versatile ‘molecular switch’ so that Mcl‐1 levels and functions, and hence neutrophil apoptosis, can be up‐ or down‐regulated rapidly in response to pro‐ or anti‐inflammatory signals.
Despite the fact that mature, human neutrophils do not proliferate or divide, they express a number of proteins that are associated normally with cell cycle progression and mitosis 7. These include survivin 7, proliferating cell nuclear antigen (PCNA) 8 and cell cycle‐dependent kinases (CDKs) 9. Human neutrophils express CDKs −2, −7 and −9 (plus several others at low expression levels), and inhibition of these kinases can result in accelerated apoptosis 10, 11. For example, R‐roscovitine, which blocks phosphorylation of RNA polymerase II by CDKs −7 and −9 12 decreased neutrophil transcriptional activity, decreased Mcl‐1 protein levels and induced apoptosis in human neutrophils 12. Similarly, the CDK‐9 inhibitor, flavopiridol, which also blocks transcription, decreased Mcl‐1 levels and induced neutrophil apoptosis 11. Such targeting of CDKs may have clinical benefit in inflammatory diseases, as the CDK inhibitor, AT7519, can reverse the delayed apoptosis observed in neutrophils of patients with sepsis‐related acute respiratory distress syndrome (ARDS) ex vivo 13.
These reports demonstrate clearly the importance of CDKs in the regulation of neutrophil apoptosis by transcriptional control, which when inhibited will lead to a rapid decrease in Mcl‐1 because of its naturally rapid turnover rate 5. In this report, we show that the purvalanol A, a 2,6,9‐tri substituted purine that binds the ATP‐binding site of human CDK‐2 and some other CDKs 14, 15 also accelerates neutrophil apoptosis, in parallel with decreasing Mcl‐1 protein levels. While this inhibitor similarly decreased Mcl‐1 protein levels in human PBMCs it did not induce apoptosis in these cells, presumably because, unlike in human neutrophils, PBMCs also express other anti‐apoptotic Bcl‐2 family proteins such as Bcl‐2 and Bcl‐XL, whose protein levels were largely unaffected by purvalanol A. We show that purvalanol A increased the rate of Mcl‐1 protein turnover and also activated p38‐mitogen‐activated protein kinase (MAPK), which appeared to play a role in this accelerated protein turnover. These novel data indicate that CDKs may play roles in regulation of neutrophil function and apoptosis that are independent of their ability to regulate transcription and act via regulation of kinase cascades that, in part, regulate Mcl‐1 turnover.
Methods and methods
Materials
RPMI‐1640, fetal bovine serum, penicillin–streptomycin and Hanks's balanced salt solution (HBSS) were from Gibco (Paisley, UK); Rapid Romanowsky stain was from HD Supplies (Aylesbury, UK); Guava ViaCount reagent was from Millipore (Watford, UK); acrylamide solution was from Severn Biotech (Kidderminster, UK); polyvinylidene fluoride membrane was from Millipore; enhanced chemiluminescence detection reagents and Hyperfilm were from Amersham Life Science (Little Chalfont, UK). Biotinylated protein ladder detection pack, anti‐human Mcl‐1 (#4572), anti‐human Bcl‐2 (#2870), anti‐human Bcl‐XL (#2764), anti‐human p‐p44/42 MAPK [extracellular signal‐regulated kinase (Erk)1/2] (Thr202/Tyr204) (E10) (#9106), anti‐human p44/42 MAPK (Erk1/2) (#9102), anti‐human p‐signal transducer and activator of transcription 3 (STAT‐3) (Tyr705) (3E2) (#9138), anti‐human STAT‐3 (124 h6) (#9139), anti‐human protein kinase B (Akt) (Ser473, #9271), anti‐human Akt (#9272), anti‐human p‐p38 MAPK (Thr180/Tyr182) (28B10, #9216) and anti‐human p38 MAPK (#9212) were from Cell‐Signalling Technology (Danvers, MA, USA). Anti‐human β‐actin (#ab8226) was from Abcam (Cambridge, UK) and purvalanol A was from Tocris Bioscience (Bristol, UK). All other reagents were from Sigma (Poole, UK).
Isolation of human immune cells from whole blood
The study of adult heathy controls was approved by the University of Liverpool Committee for Research Ethics (CORE) and donors gave written consent. Whole blood (20 ml) was collected into heparinized vacuette containers by venepuncture and processed within 2 h. Neutrophils and PBMCs were isolated by centrifuging over Ficoll‐PaqueTM, as described in the manufacturer's instructions. Briefly, whole blood was first incubated in a 5 : 1 ratio with HetaSepTM for 30 min at 37°C to precipitate the erythrocytes. The supernatant was collected and diluted in RPMI‐1640 medium with 25 mM Hepes plus L‐glutamine (2 mM) before centrifuging at 400 g for 10 min at room temperature. The cell pellet was resuspended in 10 ml media and layered in a 1 : 1 ratio onto Ficoll‐PaqueTM before centrifuging at 500 g for 30 min at room temperature. The PBMC upper layer and neutrophil pellet were then removed and resuspended in fresh media and centrifuged at 500 g for 3 min before being resuspended in fresh media. Contaminating erythrocytes were removed from the neutrophil pellet by incubation in hypotonic lysis buffer [ammonium chloride lysis buffer: 155 mM NH4CL, 13·4 mM KHCO3 and 96·7 mM ethylenediamine tetraacetic acid (EDTA)] at a ratio of 1 : 9. After 3 min incubation, the cell suspension was centrifuged at 500 g for 3 min and neutrophils resuspended in fresh media and counted before being adjusted a final concentration of 5 × 106 cells/ml. The purity of the samples was assessed by light microscopy and cytospins. Neutrophils and PBMCs were incubated in a 5% CO2 incubator at 37°C with gentle agitation under experimental conditions, as described in the text.
Flow cytometry to determine primary immune cell apoptosis
Apoptosis of neutrophils and PBMCs was measured by flow cytometry. Phosphatidylserine expressed on the membrane of early apoptotic cells was stained by annexin V‐fluorescein isothiocyanate (FITC), while propidium iodide (PI) permeability was used to detect late apoptotic cells. Cells at a concentration of 2 × 105 in 200 µl HBSS were incubated with annexin V‐FITC (10 µl/ml) for 15 min at room temperature. Prior to the flow analysis, PI (final concentration of 100 µg/ml) was added to the cells; 5000 gated cells were then analysed on a Guava Easycyte flow cytometer.
In all experiments using the flow cytometer, the absolute number of cells was counted routinely to account for any loss of cells due to lysis. The % and actual number of cells were measured and compared routinely, but under these experimental conditions they were identical.
Preparation of protein lysates
A total of 106 cells per sample were centrifuged and washed with phosphate‐buffered saline (PBS) to obtain cell pellets, which were resuspended in 100 μl hot Laemmli lysis buffer [10% (v/v) glycerol, 1 M Tris‐HCl (pH 6·8) and 3% (w/v) sodium dodecyl sulphide (SDS)] to a final concentration of 1 × 104 cells/μl before boiling for 5 min. All protein extract samples were then analysed for total protein levels by the bicinchoninic acid (BCA) assay and adjusted to the same protein concentrations using Laemmli buffer containing bromophenol blue and dithiothreitol (100 mM, final concentration). Samples were then stored at −20°C until analysis.
Western blotting
Thawed protein extracts were reheated and separated by SDS‐polyacrylamide gel electrophoresis (PAGE) containing 10% (v/v) polyacrylamide; 15 µl of protein extract was added to each well of the gel, with prestained marker proteins loaded in the first well. Gels were electrophoresed at 180 V for 55 min in SDS running buffer (25 mM Tris, 0·1% (w/v) SDS and 192 mM glycine) and then separated proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane by electrophoresis at 100 V for 80 min in transfer buffer (20% (v/v) methanol, 12·5 mM Tris and 95 mM glycine). The membrane was checked to determine if the proteins had been transferred successfully onto the membrane, by immersing into Ponceau S stain [0·1% (w/v) Ponceau S and 5% (v/v) acetic acid]. After washing off the Ponceau stain by washing buffer [150 mM NaCl, 10 mM Tris HCl (pH 8) and 0·1% (v/v) Tween‐20], the membrane was blocked by blocking solution [5% non‐fat dried skimmed milk (Marvel, Premier Foods, St Albans, UK) in Tris‐buffered saline (TBS)‐T (0·05% Tween 20 in Tris‐buffered saline, pH 8)] at room temperature for at least 1 h on an orbital shaker in order to prevent non‐specific antibody binding. After blocking, the membrane was rinsed with washing buffer for 30 s and then incubated with the specific primary antibody (dilutions as specified by the manufacturer's datasheet) at 4°C overnight on an orbital shaker.
The membrane was then washed with wash buffer for 3 × 10 min and then incubated with secondary horseradish peroxidase (HRP)‐linked antibody for 1 h. After washing again with wash buffer for 3 × 10 min, the membrane was covered with enhanced chemiluminescence (ECL) reagents for 1 min and then exposed to photosensitive film for periods of 1–10 min depending on intensity of the signal, taking care to avoid saturation of the film.
After developing the blot, the membrane was stripped by immersing in stripping buffer [50 mM glycine, 150 mM NaCl, 0·1% (v/v) Tween‐20 and HCl to pH 2·5] for 15 min with gentle agitation and then washed. The membrane was reblocked and reprobed with actin antibody to ensure equal loading of wells. The membrane was developed as indicated above. Exposed films were then analysed by the AQM advance 6 Kinetic Imaging System (Zeiss Ltd, Cambridge, UK) and calculated densitometry values are presented after correction for the β‐actin signal.
Isolation of RNA and quantitative PCR (qPCR)
RNA was isolated from a minimum of 107 cells using Trizol : chloroform (Thermo Fisher Scientific, Paisley, UK), precipitated in isopropanol and cleaned using the RNeasy kit (Qiagen, Manchester, UK), including a DNase digestion step. cDNA was synthesized from total RNA using the SuperScript III First‐Strand cDNA Synthesis kit (Thermo Fisher Scientific) using equal concentrations of RNA across samples, as per the manufacturer's instructions. Real‐time polymerase chain reaction (PCR) analysis was carried out using the QuantiTect SYBR Green PCR kit (Qiagen), as per the manufacturer's instructions. Analysis was carried out on a LightCycler 480 (Roche Diagnostics, Burgess Hill, UK) in a 96‐well plate using a 20 µl reaction volume. Mcl‐1 gene expression was quantified using mean normalized against either β‐actin as a housekeeping gene. Specific primers were Mcl‐1 (forward, 5‐GGGCAGGATTGTGACTCTCATT‐3; reverse 5‐GATGCAGCTTTCTTGGTTTATGG‐3); β‐actin (forward, 5‐ AGGATTCCTATGTGGGCGAC‐3; reverse, 5‐CGCGGTTGGCC‐TTGGGGTTCA‐3).
Statistics
All data sets are expressed as mean ± standard error of the mean (s.e.m.), and differences are considered significant for P‐values of ≤ 0·05. Comparisons are made using a combination of parametric and non‐parametric statistical analyses, as specified and as appropriate to the data sets. Paired two‐tailed Student's t‐test or one‐way analysis of variance (ANOVA) followed by post‐hoc comparison tests were used for normally distributed data.
Results
Effects of purvalanol A on apoptosis of neutrophils and PBMC
Our first experiments set out to determine if purvalanol A, like other CDK inhibitors, induced apoptosis in human leucocytes. Initial experiments were performed in which neutrophils were incubated with purvalanol A at concentrations up to 30 µM (or vehicle control), and apoptosis was determined by flow cytometry at 6 and 20 h: apoptosis at 6 h was maximal at 20 µM purvalanol A, while apoptosis at 20 h was maximal at 30 µM purvalanol A (data not shown). This latter concentration of purvalanol A was therefore used in all subsequent experiments. Neutrophils and PBMCs were isolated from the blood of healthy volunteers, incubated in the absence or presence of purvalanol A for 18 h and early and late apoptosis were measured by annexin V and PI staining, respectively. Figure 1a shows that purvalanol A treatment resulted in a significant increase in apoptosis of neutrophils, compared to untreated values under these culture conditions, but this increase did not reach statistical significance in PBMC, which were much more resistant to apoptosis. As approximately 60% of untreated neutrophils exhibited morphological features of apoptosis by this incubation time, we then decided to determine if purvalanol A had any shorter‐term effects on these cells. Untreated neutrophils underwent apoptosis slowly during culture, with approximately 10% of cells showing apoptotic properties by 6 h (Fig. 1b). However, by 6 h incubation with purvalanol A, approximately 30% of the population were apoptotic (P < 0·05, n = 3). Under these experimental conditions, no significant changes in number of apoptotic PBMCs were observed in the absence or presence of this inhibitor (Fig. 1c).
Figure 1.
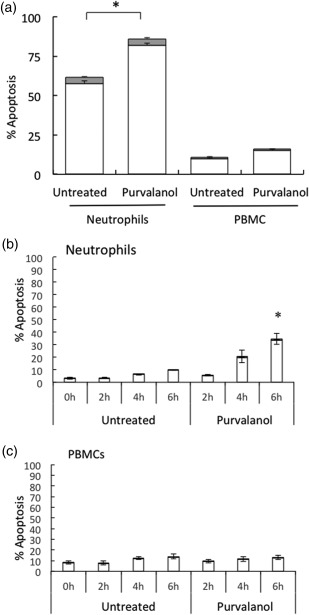
Effects of purvalanol A on white blood cell apoptosis. (a) Neutrophils and peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors and incubated for 18 h in the absence (untreated) or presence of purvalanol A (30 μM). In (b) neutrophils and (c) PBMCs were incubated for 0–6 h in the absence (untreated) or presence of 30 μM purvalanol A. Early and late apoptosis was assessed by flow cytometry using annexin V (white bar) and propidium iodide (PI) (shaded bar), respectively. Data are shown as mean (± standard error of the mean, n = 3), *P ≤ 0·05 (paired two‐tailed Student's t‐test compared to untreated control at the same time‐point).
Effects of purvalanol A on Bcl‐2 family protein expression in neutrophils and PBMCs
The anti‐apoptotic protein Mcl‐1 plays a key role in the regulation of neutrophil apoptosis and survival, and its expression levels are regulated dynamically by post‐translational modifications 3, 5. Therefore, we incubated neutrophils for up to 6 h in the presence or absence of purvalanol A and determined levels of expression of this key survival protein. Figure 2a,b shows that Mcl‐1 declined slowly as control neutrophils were cultured over this 6‐h period, but in the presence of purvalanol A this decline was greatly accelerated: by 6 h incubation with purvalanol A, levels of this protein were only approximately 40% of those at time zero (P < 0·05, n = 3). In cultured PBMCs, levels of Mcl‐1 were stable over this 6‐h incubation period but, again, purvalanol A treatment resulted in a significant decrease in levels of this protein (Fig. 2c,d; P < 0·05, n = 3). Thus, purvalanol A treatment resulted in marked decreases in the levels of this anti‐apoptotic protein by 6 h in both cell types, but induced apoptosis only in neutrophils. The reasons for this differential effect on apoptosis in these different cell types were explored further.
Figure 2.
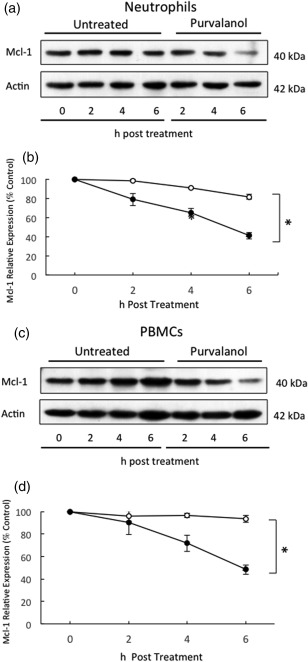
Effects of purvalanol A on Mcl‐1 expression in neutrophils and peripheral blood mononuclear cells (PBMCs). Cells were incubated in the absence (untreated or ○) or presence of 30 µM purvalanol A (purvalanol or ●) for 2, 4 and 6 h before preparation for Western blotting for myeloid cell leukaemia factor‐1 (Mcl‐1) (40 kDa) and β‐actin (42 kDa), quantified by densitometry. (a,c) Typical Western blots; (b,d) densitometric analyses, expressed as a % of untreated control samples at time zero (± standard error of the mean, n = 3), *P ≤ 0·05 (paired two‐tailed Student's t‐test). Western blots shown are representative of n = 3 experiments.
Human neutrophils do not express Bcl‐2 or Bcl‐XL proteins, but these proteins are expressed highly in PBMCs 16. Figure 3 shows that levels of these two anti‐apoptotic proteins changed very little, if at all, during 6 h culture of PBMCs in the presence or absence of purvalanol A. As neutrophils rely mainly upon Mcl‐1 for survival, whereas PBMCs have multiple anti‐apoptotic Bcl‐2 family proteins to regulate their survival, the mechanisms underpinning these differential effects of purvalanol A on apoptosis on these two cell types are explained: this inhibitor knocks down expression of Mcl‐1 in both cell types, but as PBMCs express other survival proteins they do not undergo apoptosis, even though Mcl‐1 is ablated. In contrast, neutrophils cannot survive in the absence of appropriate levels of Mcl‐1.
Figure 3.
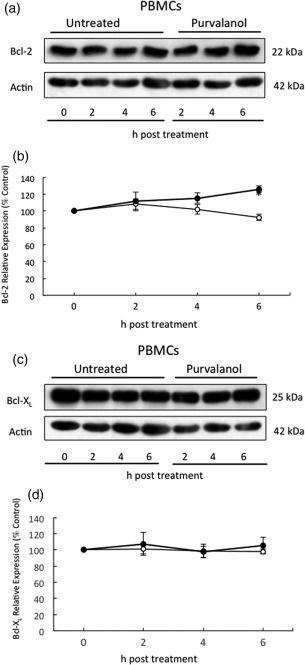
Effects of purvalanol A on B cell lymphoma‐2 (Bcl‐2) or B cell lymphoma‐extra large (Bcl‐XL) expression in peripheral blood mononuclear cells (PBMCs). Cells were incubated in the absence (untreated or ○) or presence of 30 µM purvalanol A (purvalanol or ●) for 2, 4 and 6 h before preparation for Western blotting for Bcl‐2 (22 kDa) and Bcl‐XL (25 kDa) and β‐actin (42 kDa), quantified by densitometry. (a,c) Typical Western blots; (b,d) densitometric analyses, expressed as a % of untreated control samples at time zero (± standard error of the mean, n = 3), *P ≤ 0·05 (paired two‐tailed Student's t‐test). Western blot shown is representative of n = 3 experiments.
Neutrophil apoptosis is delayed significantly in the presence of the proinflammatory cytokine, granulocyte–macrophage colony‐stimulating factor (GM‐CSF), which induces a small increase in transcription of Mcl‐1 but a large increase in the stability of this protein 6, 17. We therefore determined if purvalanol A could interfere with GM‐CSF‐mediated protection of neutrophil apoptosis. Neutrophils were incubated with purvalanol A and GM‐CSF, alone and together during 6‐ and 20‐h incubation periods. After 6 h incubation, purvalanol A treatment alone increased neutrophil apoptosis significantly, but also blocked the protective effect of GM‐CSF (Fig. 4a). After 20 h incubation, these effects were more marked: GM‐CSF protected against neutrophil apoptosis but purvalanol A negated completely this protective effect of GM‐CSF (Fig. 4b). These effects on apoptosis were also mirrored by measurements of Mcl‐1 protein levels (Fig. 4c,d): GM‐CSF protected against Mcl‐1 loss during culture, but could not protect against the purvalanol A accelerated loss of this protein.
Figure 4.
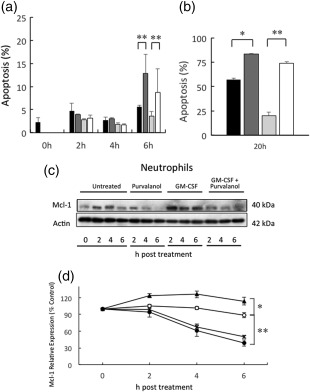
Effects of purvalanol A and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) on neutrophil myeloid cell leukaemia factor‐1 (Mcl‐1) expression and apoptosis. Neutrophils were isisolated from three donors and incubated for 0–6 h (a,c,d) or 20 h (b) in the absence ( , untreated, ○) or presence of 50ng/ml GM‐CSF (
, untreated, ○) or presence of 50ng/ml GM‐CSF ( , GM‐CSF, ▲), 30 μM purvalanol A (
, GM‐CSF, ▲), 30 μM purvalanol A ( , purvalanol, ●) and both together (
, purvalanol, ●) and both together ( , GM‐CSF + purvalanol, x). In (a,b), apoptosis was assessed by flow cytometry using annexin V and propidium iodide (PI), while Mcl‐1 expression was measured by Western blotting (c,d) and expression levels quantified by densitometry (d). Data are shown as mean (± standard error of the mean, n = 3). *P ≤ 0·05; **P ≤ 0·01 [one‐way analysis of variance (ANOVA) followed by Bonferroni's post‐hoc comparison tests]. Western blot shown is representative of n = 3 experiments.
, GM‐CSF + purvalanol, x). In (a,b), apoptosis was assessed by flow cytometry using annexin V and propidium iodide (PI), while Mcl‐1 expression was measured by Western blotting (c,d) and expression levels quantified by densitometry (d). Data are shown as mean (± standard error of the mean, n = 3). *P ≤ 0·05; **P ≤ 0·01 [one‐way analysis of variance (ANOVA) followed by Bonferroni's post‐hoc comparison tests]. Western blot shown is representative of n = 3 experiments.
Previous experiments have shown that CDK inhibitors can block transcription in neutrophils, and this can result in rapid decreases in Mcl‐1 protein because of its naturally high rate of turnover 11, 12, 13. However, we did not detect any effect of purvalanol A on Mcl‐1 mRNA levels during a 4‐h incubation period (Fig. 5a). Therefore, we hypothesized that these rapid changes in Mcl‐1 protein induced by purvalanol A treatment of neutrophils were explained by changes in its rate of turnover 18. We incubated neutrophils in the presence of cycloheximide to block protein translation and in the absence (control) and presence of purvalanol A. In the absence of purvalanol A, the half‐life of Mcl‐1 measured under these conditions was approximately 2 h, in line with previous measurements 6, but in the presence of purvalanol A this half‐life was decreased to just over 1 h (Fig. 5b,c). This change in protein stability was due probably to a post‐translational modification of Mcl‐1 induced directly or indirectly by purvalanol A treatment.
Figure 5.

Effect of purvalanol A on the half‐life of myeloid cell leukaemia factor‐1 (Mcl‐1) in neutrophils. In (a) neutrophils were incubated in the absence (●) or presence (○) of purvalanol A for the indicated times. Levels of MCL1 mRNA were then quantified by quantitative polymerase chain reaction (qPCR) after correction for ACTB mRNA levels. (b,c) Neutrophils were preincubated for 10 min with 10 µg/ml cycloheximide prior to 0, 1, 2, 3 and 4 h incubation in the absence (untreated, ●) or presence of 30 μM purvalanol A (purvalanol, ○). Protein extracts were prepared at the times indicated and analysed by Western blotting for levels of Mcl‐1 (40 kDa) and actin (42 kDa). (b) Typical Western blot; (c) densitometric analyses, expressed as a % of untreated control samples at time zero (± standard error of the mean, n = 3), *P ≤ 0·05 (paired two‐tailed Student's t‐test). Western blot shown is representative of n = 3 experiments.
Purvalanol A‐induced activation of intracellular signalling pathways in neutrophils
A number of intracellular signalling pathways have been implicated in the regulation of Mcl‐1 turnover and control of neutrophil apoptosis and survival 4, 6, 17, 19. We therefore measured the effects of purvalanol A on activation of a number of these pathways and determined if this inhibitor could interfere with the anti‐apoptotic signalling pathways induced by GM‐CSF. Erk was inactive in unstimulated neutrophils, and its activation status did not change during culture for 1 h and, in line with previous observations 6, it was activated rapidly and transiently in response to GM‐CSF (Fig. 6a,b). Purvalanol A alone resulted in a very small, insignificant increase in Erk activation but it ablated completely the activation induced by GM‐CSF. pSTAT‐3 was also activated rapidly and transiently by GM‐CSF, and again this activation was blocked when purvalanol A was added together with GM‐CSF (Fig. 6c,d). GM‐CSF also activated a transient increase in phosphorylation of Akt (Fig. 7a,b), but purvalanol A alone also activated this signalling molecule with similar kinetics to that induced by GM‐CSF: the combined effects of these two agents on Akt activation were additive. GM‐CSF treatment activates p38‐MAPK with unusual kinetics that are sustained over a 5‐h incubation period 6. Purvalanol A treatment resulted in a remarkable level of activation of p38‐MAPK that was sustained over a 1‐h incubation period, and the high level of activation of this signalling molecular induced by this kinase inhibitor was unaffected by co‐incubation with GM‐CSF (Fig. 7c,d).
Figure 6.

Effects of purvalanol A and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) on extracellular‐regulated kinase (Erk) and signal transducer and activator of transcription (STAT)‐3 activation in neutrophils. Cells were incubated in the absence (untreated or ○) or presence of 30 µM purvalanol A (purvalanol or ●), 50 ng/ml GM‐CSF (GM‐CSF or ▲) and both (GM‐CSF + purvalanol or x) for 15, 30 and 60 min before preparation for Western blotting for pErk/pan‐Erk (42–44 kDa) and pSTAT‐3/pan‐STAT‐3 (80 kDa) quantified by densitometry. (a,c) Typical Western blots; (b,d) densitometric analyses, expressed as a % of untreated control samples at time zero (± standard error of the mean, n = 3). *P ≤ 0·05; **P ≤ 0·01 [one‐way analysis of variance (ANOVA) followed by Bonferroni's post‐hoc comparison tests]. Western blot shown is representative of n = 3 experiments.
Figure 7.
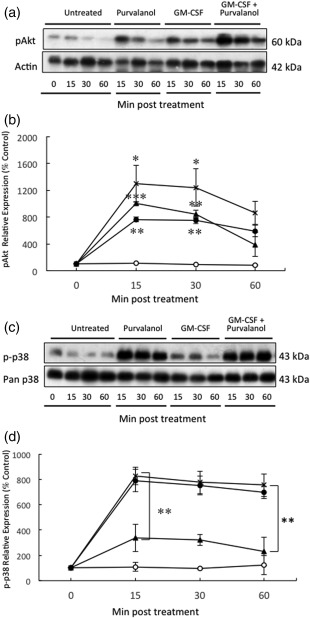
Effects of purvalanol A and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) on protein kinase B (Akt) and p38‐mitogen‐activated protein kinase (MAPK) activation in neutrophils. Cells were incubated in the absence (untreated or ○) or presence of 30 µM purvalanol A (purvalanol or •), 50 ng/ml GM‐CSF (GM‐CSF or ▲) and both (GM‐CSF + purvalanol or x) for 15, 30 min and 1 h before preparation for Western blotting for pAkt (60 kDa) and β‐actin (42 kDa), or pp‐38‐MAPK/pan p38‐MAPK (43 kDa) quantified by densitometry. (a,c) Typical Western blots; (b,d) densitometric analyses, expressed as a % of untreated control samples at time zero. *P ≤ 0·05; **P ≤ 0·01 [one‐way analysis of variance (ANOVA) followed by Bonferroni's post‐hoc comparison tests). Western blot shown is representative of n = 3 experiments.
Thus, purvalanol A inhibits activation of Erk and STAT‐3 signalling induced by GM‐CSF that may partly explain the mechanism by which it blocks the effects of GM‐CSF on neutrophil survival. However, this CDK inhibitor alone induced a substantial and sustained increase in p38‐MAPK activation which may, at least in part, explain its mechanism of action in increasing Mcl‐1 turnover and acceleration of apoptosis.
We then tested the effects of a pan p38‐MAPK inhibitor on purvalanol A‐induced acceleration of neutrophil apoptosis and increased Mcl‐1 turnover. p38‐MAPK can exist in four isoforms (α–δ), but human neutrophils express only p38α and p38δ 20, 21. The inhibitor BIRB796 inhibits all four isoforms of this enzyme 22, so its effects were investigated. Neutrophils were therefore incubated for up to 6 h in the absence (control) or in the presence of purvalanol A and BIRB796, alone or together. As shown previously (Fig. 4), levels of Mcl‐1 declined slowly during culture, but this was accelerated greatly in the presence of purvalanol A (Fig. 8a,b). BIRB796 itself had no effect on Mcl‐1 levels, but it partly blocked the loss in Mcl‐1 levels induced by purvalanol A treatment (P < 0·05). Similarly, BIRB796 partly blocked the purvalanol A induced acceleration of neutrophil apoptosis (Fig. 8c, P < 0·05).
Figure 8.
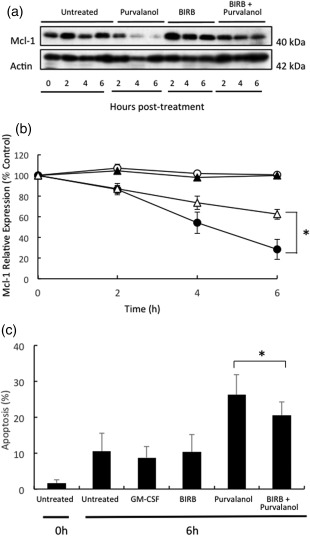
Effects of purvalanol A and p38‐mitogen‐activated protein kinase (MAPK) inhibition on myeloid cell leukaemia factor‐1 (Mcl‐1) expression and apoptosis in neutrophils. Neutrophils were incubated in the absence (untreated or ○) or presence of 30 µM purvalanol A (purvalanol or •), 10 µM BIRB796 (BIRB or ▲) and both (BIRB + purvalanol or △) for 2, 4 and 6 h before preparation of extracts for Western blotting for Mcl‐1 (40 kDa) and β‐actin (42 kDa) quantified by densitometry and apoptosis. (a) Typical Western blots; (b) densitometric analyses, expressed as a % of untreated control samples at time zero. (c) shows effects on apoptosis after 6 h incubation as described above. *P ≤ 0·05 [one‐way analysis of variance (ANOVA) followed by Bonferroni's post‐hoc comparison tests]. Western blot shown is representative of n = 3 experiments.
Discussion
Although circulating human blood neutrophils do not divide and replicate, they express a number of enzymes and processes that are associated normally with DNA synthesis and cell division 7, 8, 9, 10, 11, 12, 13, 14. Neutrophil apoptosis is a key event in the regulation of the function of these cells during inflammatory activation, and rapidly switches cell fate from constitutive apoptosis and a short half of circulating cells, into an extended lifespan when cells are recruited into inflammatory sites 1, 2. This delay in apoptosis is required for inflammatory neutrophils to survive for sufficient time to appropriately perform their function in inflammation or infection, but apoptosis must then be triggered when this function is complete in order for inflammation to resolve. Neutrophils are exquisitely adapted for this rapid switch from apoptosis to survival by virtue of the composition and control of their apoptotic constituents. They express a range of pro‐apoptotic members of the Bcl‐2 family, but their survival is highly dependent upon the function of the anti‐apoptotic protein, Mcl‐1, whose levels within neutrophils can be up‐ or down‐regulated rapidly by post‐translational modifications that include reversible phosphorylation of key resides in the PEST domains 23, 24. Many of the enzymes expressed in neutrophils that are associated normally with cell cycle control and progression appear to interfere, directly or indirectly, with the control of apoptosis.
In the present study, we have made the novel discovery that the CDK‐2 inhibitor, purvalanol A, triggers a rapid onset of apoptosis in human neutrophils, but not in PBMCs. We also found that this inhibitor induces a rapid loss of Mcl‐1 protein, but this inhibitor had no effect on Mcl‐1 mRNA levels (measured by qPCR) during a 4‐h incubation compared to untreated control levels. This rapid loss in Mcl‐1 protein levels was found to be due to an increase in the turnover rate of the protein that was induced by purvalanol A, the half‐life of the protein being decreased from approximately 2 h to approximately 1 h. Unlike PBMCs, which also express the anti‐apoptotic proteins, Bcl‐2 and Bcl‐XL, this loss of Mcl‐1 induced by purvalanol A induced rapid cell death in neutrophils because of their high dependency upon this protein for survival.
It has been shown that the apoptosis‐delaying effects of cytokines such as GM‐CSF on neutrophils are due largely to effects on protein stability, rather than changes in transcriptional levels 6. Conversely, agents that accelerate neutrophil apoptosis can decrease Mcl‐1 stability 18. Several kinase signalling pathways are involved in the regulation of Mcl‐1 levels by GM‐CSF, which rapidly activates pathways such as Erk and Akt, while inhibitors of Erk and phosphatidylinositide 3‐kinases (PI3K) block the protective effects of GM‐CSF on Mcl‐1 stability 6. We therefore determined the effects of purvalanol A on Mcl‐1 stability in the presence of GM‐CSF to determine if the CDK‐2 inhibitor could overcome the stabilizing effects of these cytokine. We found that purvalanol A partly abrogated the protective effects of GM‐CSF on neutrophil apoptosis and Mcl‐1 stability.
We then sought to determine the effects of purvalanol A on a number of signalling cascades implicated in neutrophil survival and Mcl‐1 stability to provide insights into the mechanism of action of the CDK‐2 inhibitor. Remarkably, we found that purvalanol A completely abrogated the activation of Erk and STAT‐3 induced by GM‐CSF, while it activated Akt: its effects on Akt activation in the presence of GM‐CSF were additive. Because GM‐CSF alone activates Akt, which is required, at least in part, for GM‐CSF‐enhanced Mcl‐1 stability 6, we feel it is unlikely that purvalanol A‐induced Akt activation itself plays a major role in promotion of apoptosis by this CDK2 inhibitor. Erk has been shown previously to be required for the GM‐CSF‐mediated increase in Mcl‐1 stability. Hence, abrogation of this GM‐CSF mediated Erk activation may be responsible, at least in part, for its ability to abrogate the protective effect of this cytokine on apoptosis and Mcl‐1 stability.
Surprisingly, we found that purvalanol A induced a very rapid and sustained activation of p38‐MAPK that was unaffected by co‐incubation with GM‐CSF. The role of p38‐MAPK in the regulation of neutrophil survival and apoptosis is somewhat controversial, with some reports suggesting a protective effect, while others suggest a pro‐apoptotic effect 25, 26, 27, 28. Human neutrophils express two of the four known isoforms of p38‐MAPK, namely p38α and p‐38δ 20, so we determined the effects of the pan p38‐MAPK inhibitor, BIRB796, which inhibits all four isoforms of this enzyme on apoptosis and Mcl‐1 levels in the absence or presence of purvalanol A. We show that BIRB796 partially blocks the effects of purvalanol A on apoptosis and Mcl‐1 degradation, suggesting strongly that the effects of purvalanol A in human neutrophils are at least partly dependent upon activation of this kinase, activation of which, directly or indirectly, promotes Mcl‐1 turnover and subsequent apoptosis. One possibility, therefore, is that CDK2 may regulate the activation of p38‐MAPK negatively, but further work is necessary to confirm or refute this hypothesis.
In summary, we show that a CDK‐2 kinase inhibitor induces a rapid and extensive promotion of apoptosis in human neutrophils, and partly negates the effects of the anti‐apoptotic protein, GM‐CSF. This promotion in apoptosis is mediated by a marked increase in the turnover rate of the anti‐apoptotic protein Mcl‐1. We also show that this effect of purvalanol A is mediated by its direct or indirect activation of p38‐MAPK, inhibition of which partly reverses the effects of the CDK‐2 inhibitor on apoptosis. This is another example of an enzyme, CDK‐2, involved normally in cell cycle regulation, having profound effects on neutrophil function that are completely independent of known effects on cell cycle control. These data reveal a role of CDK‐2 playing a role in regulation of the kinase signalling processes that regulate Mcl‐1 post‐translational modifications to alter its stability and turnover rate.
Disclosure
The authors declare no conflicts of interest.
Author contributions
P. P. co‐designed the study, performed the experiments and co‐wrote the manuscript. A. C. and L. G.‐A. performed experiments and reviewed the manuscript. H. L. W. analysed the data and reviewed the manuscript, while S. W. E. designed the study and wrote the manuscript.
Acknowledgements
H. L. W. was supported by the Faculty of Health and Life Sciences, University of Liverpool.
References
- 1. Edwards SW. Biochemistry and physiology of the neutrophil. New York: Cambridge University Press, 1994. [Google Scholar]
- 2. Akgul C, Moulding DA, Edwards SW. Molecular control of neutrophil apoptosis. FEBS Lett 2001; 487:318–22. [DOI] [PubMed] [Google Scholar]
- 3. Edwards SW, Derouet M, Howse M, Moots RJ. Regulation of neutrophil apoptosis by Mcl‐1. Biochem Soc Trans 2004; 32:489–92. [DOI] [PubMed] [Google Scholar]
- 4. Milot E, Filep JG. Regulation of neutrophil survival/apoptosis by Mcl‐1. Sci World J 2011; 11:1948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas LW, Lam C, Edwards SW. Mcl‐1; the molecular regulation of protein function. FEBS Lett 2010; 584:2981–9. [DOI] [PubMed] [Google Scholar]
- 6. Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. Granulocyte macrophage colony‐stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl‐1. J Biol Chem 2004; 279:26915–21. [DOI] [PubMed] [Google Scholar]
- 7. Altznauer F, Martinelli S, Yousefi S et al Inflammation‐associated cell cycle‐independent block of apoptosis by survivin in terminally differentiated neutrophils. J Exp Med 2004; 199:1343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Witko‐Sarsat V, Mocek J, Bouayad D et al Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J Exp Med 2010; 207:2631–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rossi AG, Sawatzky DA, Walker A et al Cyclin‐dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 2006; 12:1056–64. [DOI] [PubMed] [Google Scholar]
- 10. Leitch AE, Lucas CD, Marwick JA, Duffin R, Haslett C, Rossi AG. Cyclin‐dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ 2012; 19:1950–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang K, Hampson P, Hazeldine J et al Cyclin‐dependent kinase 9 activity regulates neutrophil spontaneous apoptosis. PLOS ONE 2012; 7:e30128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leitch AE, Riley NA, Sheldrake TA et al The cyclin‐dependent kinase inhibitor R‐roscovitine down‐regulates Mcl‐1 to override pro‐inflammatory signalling and drive neutrophil apoptosis. Eur J Immunol 2010; 40:1127–38. [DOI] [PubMed] [Google Scholar]
- 13. Dorward DA, Felton JM, Robb CT et al The cyclin‐dependent kinase inhibitor AT7519 accelerates neutrophil apoptosis in sepsis‐related acute respiratory distress syndrome. Thorax 2017; 72:182–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Villerbu N, Gaben AM, Redeuilh G, Mester J. Cellular effects of Purvalanol A: a specific inhibitor of cyclin‐dependent kinase activities. Int J Cancer 2002; 97:761–9. [DOI] [PubMed] [Google Scholar]
- 15. Hikita T, Oneyama C, Okada M. Purvalanol A, a CDK inhibitor, effectively suppresses Src‐mediated transformation by inhibiting both CDKs and c‐Src. Genes Cells 2010; 15:1051–62. [DOI] [PubMed] [Google Scholar]
- 16. Moulding DA, Akgul C, Derouet M, White MR, Edwards SW. BCL‐2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol 2001; 70:783–92. [PubMed] [Google Scholar]
- 17. Akgul C, Turner PC, White MR, Edwards SW. Functional analysis of the human MCL‐1 gene. Cell Mol Life Sci 2000; 57:684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Derouet M, Thomas L, Moulding DA et al Sodium salicylate promotes neutrophil apoptosis by stimulating caspase‐dependent turnover of Mcl‐1. J Immunol 2006; 176:957–65. [DOI] [PubMed] [Google Scholar]
- 19. El Kebir D, Damlaj A, Filep JG, Gao H. Toll‐like receptor 9 signaling delays neutrophil apoptosis by increasing transcription of Mcl‐1. PLOS ONE 2014; 9:e87006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nick JA, Avdi NJ, Young SK et al Selective activation and functional significance of p38alpha mitogen‐activated protein kinase in lipopolysaccharide‐stimulated neutrophils. J Clin Invest 1999; 103:851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuenda A, Rousseau S. p38 MAP‐kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 2007; 1773:1358–75. [DOI] [PubMed] [Google Scholar]
- 22. Kuma Y, Sabio G, Bain J, Shpiro N, Márquez R, Cuenda A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo . J Biol Chem 2005; 280:19472–9. [DOI] [PubMed] [Google Scholar]
- 23. Akgul C. Mcl‐1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci 2009; 66:1326–36.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moulding DA, Quayle JA, Hart CA, Edwards SW. Mcl‐1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood 1998; 92:2495–502. [PubMed] [Google Scholar]
- 25. Frasch SC, Nick JA, Fadok VA, Bratton DL, Worthen GS, Henson PM. p38 mitogen‐activated protein kinase‐dependent and ‐independent intracellular signal transduction pathways leading to apoptosis in human neutrophils. J Biol Chem 1998; 273:8389–97. [DOI] [PubMed] [Google Scholar]
- 26. Alvarado‐Kristensson M, Melander F, Leandersson K, Rönnstrand L, Wernstedt C, Andersson T. p38‐MAPK signals survival by phosphorylation of caspase‐8 and caspase‐3 in human neutrophils. J Exp Med 2004; 199:449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aoshiba K, Yasui S, Hayashi M, Tamaoki J, Nagai A. Role of p38‐mitogen‐activated protein kinase in spontaneous apoptosis of human neutrophils. J Immunol 1999; 162:1692–700. [PubMed] [Google Scholar]
- 28. Choi KS, Park JT, Dumler JS. Anaplasma phagocytophilum delay of neutrophil apoptosis through the p38 mitogen‐activated protein kinase signal pathway. Infect Immun 2005; 73:8209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]