

Summary
Clearance of hepatitis C virus (HCV) is dependent on an effective virus‐specific CD8+ T‐cell response, which is dysfunctional in chronic HCV infection. Dysfunction in bulk or non‐HCV‐specific CD8+ T‐cells in HCV infection has also been observed. This may contribute to observed reductions in immunity to other diseases (e.g. cancer, viral co‐infections) in HCV‐infected individuals. Evidence suggests that the HCV core protein (found in blood as free protein) may contribute to this impairment. To determine if HCV core contributes to the impairment of effector functions and survival potential of CD8+ T‐cells, isolated human CD8+ T‐cells from healthy donors were pre‐incubated with recombinant HCV core protein for 72 hr and then stimulated in vitro to evaluate proliferation, survival potential and effector functions. Pre‐incubation of stimulated CD8+ T‐cells with HCV core significantly reduced their proliferation. Perforin production and degranulation were also decreased, but interferon‐γ production was unchanged. Additionally, when CD8+ T‐cells were treated with serum from HCV + individuals, they produced less perforin than cells treated with healthy serum. Up‐regulation of anti‐apoptotic Bcl‐2 was slightly lower in cells treated with HCV core, but signal transducer and activator of transcription 5 (STAT5) activation was increased, suggesting dysregulation downstream of STAT activation. Our study reveals that HCV core reduces the activity and target lysis‐associated functions of CD8+ T‐cells. This may contribute to the generalized impairment of CD8+ T‐cells observed in HCV infection. These findings provide insight for the design of novel counteractive immune‐mediated strategies including the design of effective therapeutic vaccines for use in HCV + individuals.
Keywords: CD8 cell, cell proliferation, cytotoxic T‐cells, cytotoxicity, viral
Abbreviations
- Akt
protein kinase B
- Bcl‐2
B‐cell chronic lymphocytic leukaemia/lymphoma 2
- CFSE
carboxyfluorescein succinimidyl ester
- FITC
fluorescein isothiocyanate
- gC1qR
globular head of the C1q receptor
- HCV
hepatitis C virus
- IFN‐γ
interferon ‐γ
- IL‐2
interleukin‐2
- Lck
lymphocyte‐specific protein tyrosine kinase
- MFI
mean fluorescence intensity
- PD‐1
programmed death‐1
- PI3K
phosphatidylinositol‐3‐kinase
- STAT5
signal transducer and activator of transcription 5
Introduction
Hepatitis C virus (HCV), classified in the Hepacivirus genus in the Flaviviridae family, is a single‐stranded positive‐sense RNA virus that affects approximately 170 million people worldwide.1, 2, 3 A small percentage of those infected clear the virus spontaneously but the remainder (~80%) develop chronic infection, which may eventually lead to end‐stage liver diseases such as cirrhosis and hepatocellular carcinoma.1, 4 New interferon‐free oral direct‐acting antivirals provide promising cure rates,2 but they remain expensive, and the search for a vaccine is ongoing.
Clearance of HCV is dependent on a successful virus‐specific CD8+ T‐cell response (as seen during viral clearance in acute infection), but dysfunction in HCV‐specific CD8+ T‐cells has been widely observed in chronic infection.5, 6, 7 Additionally, generalized or non‐HCV‐specific CD8+ T‐cell dysfunction has also been observed in chronic infection.7, 8 Lucas et al.9 showed that in HCV‐infected patients, cytomegalovirus‐specific CD8+ T‐cells produced less Fas ligand and perforin, and lost markers of maturity but exhibited appropriate proliferative capacity. Expression of anti‐apoptotic B‐cell chronic lymphocytic leukaemia/lymphoma 2 (Bcl‐2) protein and activation of its upstream regulator signal transducer and activator of transcription 5 (STAT5) has also been shown to be lower in bulk CD8+ T‐cells in chronically infected HCV+ individuals, indicating lower survival potential or viability.10 Increased activation‐induced apoptosis and unique pro‐apoptotic gene signature in bulk T‐cells in HCV infection has also been observed.8
Generalized impairment of CD8+ T‐cells may contribute to observed decreases in immunity to other viral infections and cancers during HCV infection. Among various host and pathogen factors that have been posited to be responsible for this generalized dysfunction is the HCV core protein, the first protein in the N‐terminal region of the polyprotein produced by the virus during replication.7, 11, 12 It forms the nucleocaspid, and modulates virion assembly after polyprotein translation and processing.13, 14 HCV core protein can be found in the serum of HCV‐infected patients as part of the whole virus, as infectious non‐enveloped virus particles, as free protein and as damaged virus particles, and so has ample opportunity to potentially interact directly with circulating CD8+ T‐cells.7, 15, 16, 17
In addition to aiding virus assembly, HCV core plays an important role in immune evasion, in part by its modulation of T‐cell function.3, 11, 18 Specifically, it has been shown to interact with the gC1qR receptor (receptor for the complement C1q protein) to reduce T‐cell activity.3, 18, 19 In isolated T‐cells and using the thymidine incorporation assay, Kittlesen et al.18 showed that in a mixed lymphocyte reaction and when stimulated with concanavalin A (a mitogen) the presence of HCV core reduced T‐cell proliferation in a dose‐dependent manner. Blocking gC1qR with antibody appeared to reverse this effect. Yao et al.3 similarly demonstrated reduced anti‐CD3/28‐stimulated proliferation of T‐cells when exposed to HCV core. Furthermore, expression of HCV core protein in transfected T‐cells was shown to significantly reduce interleukin‐2 (IL‐2) production and inhibit co‐stimulatory c‐Jun N‐terminal kinase signalling pathway, suggesting anergy in cells.12 The study also observed increased IL‐4, IL‐10, interferon‐γ (IFN‐γ) and tumour necrosis factor‐α production. In contrast, another study found decreased IFN‐γ production in CD8+ T‐cells when peripheral blood mononuclear cells were treated with HCV core.20
We therefore sought to determine whether HCV core protein directly contributes to CD8+ T‐cell impairment, as is observed in HCV infection.10 We evaluated effects on CD8+ T‐cell activity, survival potential and effector functions. Our study provides novel insights into HCV core protein‐mediated impairment of bulk CD8+ T‐cells, which in turn will contribute to the observed generalized CD8+ T‐cell dysfunction in chronic HCV infection.
Materials and methods
Cells
Human peripheral blood mononuclear cells were isolated from the blood of healthy HCV− donors using Lymphoprep (StemCell Technologies, Vancouver, BC, Canada) density gradient centrifugation, followed by isolation of CD8+ T‐cells using CD8+ T‐cell Positive Magnetic Selection Kit I or II (StemCell Technologies). CD8+ T‐cells were then resuspended in complete RPMI medium (i.e. RPMI‐1640 containing l‐glutamine supplemented with 20% fetal calf serum, 1% penicillin/streptomycin, 1% l‐glutamine; Gibco, Life Technologies, Burlington, ON, Canada) and allowed to rest overnight at 37°, 5% CO2. Cells (5 × 105 cells/ml) were then incubated with recombinant HCV core protein (5 μg/ml; HCV genotype 1b; ViroGen Corporation, Watertown, MA) or medium for 72 hr before stimulation. Several studies have shown that an irrelevant protein prepared in the same manner as HCV core has limited effect on T‐cell functions. Therefore, medium was considered an appropriate control for the experiments.18, 21 This study was approved by The Ottawa Health Science Network Research Ethics Board, and written informed consent was obtained from all individuals.
Proliferation and cell viability
Isolated CD8+ T‐cells were labelled with carboxyfluorescein succinimidyl ester (CFSE, 8 μm; Cell Trace CFSE cell proliferation kit, Molecular Probes; Life Technologies) following established protocol.22 CFSE‐labelled CD8+ T‐cells were incubated with HCV core for 72 hr before stimulation with anti‐CD3/28 (0·0625 μg/ml) for 5 days before analysis by flow cytometry (FC 500 MCL System, Beckman Coulter, Marseille, France). Anti‐CD3 was obtained from the National Cancer Institute (Frederick, MD) and anti‐CD28 (Clone CD28.2) from eBioscience (San Diego, CA). CFSE low or CFSE dilute cells were considered to be dividing cells. For preliminary proliferation and viability time–course studies, cells were incubated with or without HCV core (5 or 2 µg/ml, respectively) for 0–72 hr and then stimulated with anti‐CD3/28 or phytohaemagglutinin (Sigma‐Aldrich, St Louis, MO). Proliferation was analysed by flow cytometry as described above. To study viability, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) (Life Technologies) was added to treated cells for 4 hr before addition of stop solution and measurement by spectrophotometry (Spectra MAX 190; Molecular Devices, Sunnyvale, CA). Cell viability was determined by the relative increase from absorbance of unstimulated cells.
Phosphorylated STAT5 and Bcl‐2
CD8+ T‐cells were incubated with or without HCV core for 72 hr before stimulation with IL‐7 (1 ng/ml; Sigma Aldrich, Oakville, ON, Canada) for 15 min to study STAT5 activation and for 48 hr to study Bcl‐2 production. Cells were fixed and permeabilized before staining with AlexaFluor 488‐conjugated anti‐pSTAT5 antibody (pY694; Clone 47; BD Pharmingen, San Jose, CA) or fluorescein isothiocyanate (FITC)‐conjugated anti‐human Bcl‐2 antibody (Clone Bcl‐2/100; BD Pharmingen) or isotype control before analysis on a flow cytometer.
Perforin, degranulation (CD107a expression) and IFN‐γ production
To study perforin production and cell degranulation, CD8+ T‐cells were incubated with or without HCV core for 72 hr before stimulation with anti‐CD3/28 (1 µg/ml) for 48 hr (perforin and degranulation) or 24 hr (IFN‐γ) before analysis by flow cytometry. To study intracellular perforin production, after 48 hr of stimulation, cells were fixed and permeabilized before staining with FITC‐conjugated anti‐human perforin (Clone δ G9; BD Pharmingen) or isotype control. To study degranulation, cells were incubated with FITC‐conjugated anti‐human CD107a antibody (Clone H4A3; BD Pharmingen) or isotype control and BD Golgi Stop (containing Monensin; BD Pharmingen) for the last 6 hr of the 48 hr of stimulation. For IFN‐γ production, BD GolgiPlug (containing Brefeldin A; BD Pharmingen) was added for the last 5 hr before being fixed, permeabilized and labelled with FITC‐conjugated anti‐human IFN‐γ antibody (5 µl/1 × 105 cells; Clone: 45.15; Beckman Coulter) or isotype control before analysis on a flow cytometer. To quantify secreted cytokines, supernatants were collected from CD8+ T‐cells stimulated with anti‐CD3/28 (1 µg/ml) for 5 days after pre‐incubation with HCV core (5 µg/ml). Supernatants were then analysed for perforin, granzyme‐B and IL‐10 production using Millipore Sigma's Milliplex Human CD8+ T‐cell Magnetic Bead Panel (Millipore, Billerica, MA).
Effect of HCV‐infected serum
To study whether serum from HCV‐infected patients was able to reduce CD8+ T‐cell function, CD8+ T‐cells from healthy donors were cultured in complete RPMI with pooled heat‐inactivated serum (10 µl/1 × 105 cells) from five healthy or five HCV‐infected individuals for 72 hr, and then stimulated with 50 µl of Immunocult Human CD3/28 activator (StemCell Technologies) for 48 hr, before staining and analysis for perforin. Donor characteristics are detailed in Table 1.
Table 1.
Characteristics of donors used for pooled serum experiments
| HCV status | Gender | Age | Genotype | HCV viral load (IU/ml) |
|---|---|---|---|---|
| Untreated HCV+ | Female | 51 | 1b | 6·0 × 106 |
| Male | 64 | 1a | 6·8 × 106 | |
| Male | 52 | 1 | 7·0 × 106 | |
| Male | 59 | 1 | 6·8 × 106 | |
| Male | 75 | 1 | 5·6 × 106 | |
| Healthy HCV− | Male | 57 | Not applicable | Not applicable |
| Male | 49 | |||
| Male | 43 | |||
| Female | 28 | |||
| Female | 50 |
Analysis and statistics
All flow cytometry was performed on the FC 500 MCL System from Beckman Coulter and analysed using FCS Express Research Edition 4.0 (De Novo Software, Los Angeles, CA). Statistical analysis and graphing were completed using graphpad prism 5.0 (San Diego, CA). For flow cytometry analysis, cells were gated on the resting and proliferating lymphocyte population according to forward and side scatter plots. Repeated measures analysis of variance with Bonferroni's Multiple Comparison Test was used for analysis of time–course assays. For all other assays, two‐tailed paired Student's t‐test was conducted with P < 0·05 being considered statistically significant, except where observed results were being validated, in which case a one‐tailed test was used. All results are stated as value ± SD.
Results
HCV core reduces proliferation of CD8+ T‐cells
An important factor in the determination of CD8+ T‐cell function is the capacity to proliferate. Time–course assays revealed that longer incubation of CD8+ T‐cells with HCV core reduced the proliferation of anti‐CD3/28‐stimulated cells, indicating that inhibition by this viral factor was strongest after 72 hr of incubation (repeated measures analysis of variance with Bonferroni's multiple comparison test, Fig. 1a). This also indicated that the 5 µg/ml concentration of recombinant HCV core protein, which is within the range (0·01–10 µg/ml) of HCV core protein concentrations used in previous reports using similar culture systems, enabled the detection of proliferation differences over time.18, 21 This may be explained, in part, by observations in our early studies that indicated that prolonged exposure to HCV core resulted in reduced viability of CD8+ T‐cells stimulated with the mitogen phytohaemagglutinin, although the effects were not statistically significant (Fig. 1b).
Figure 1.
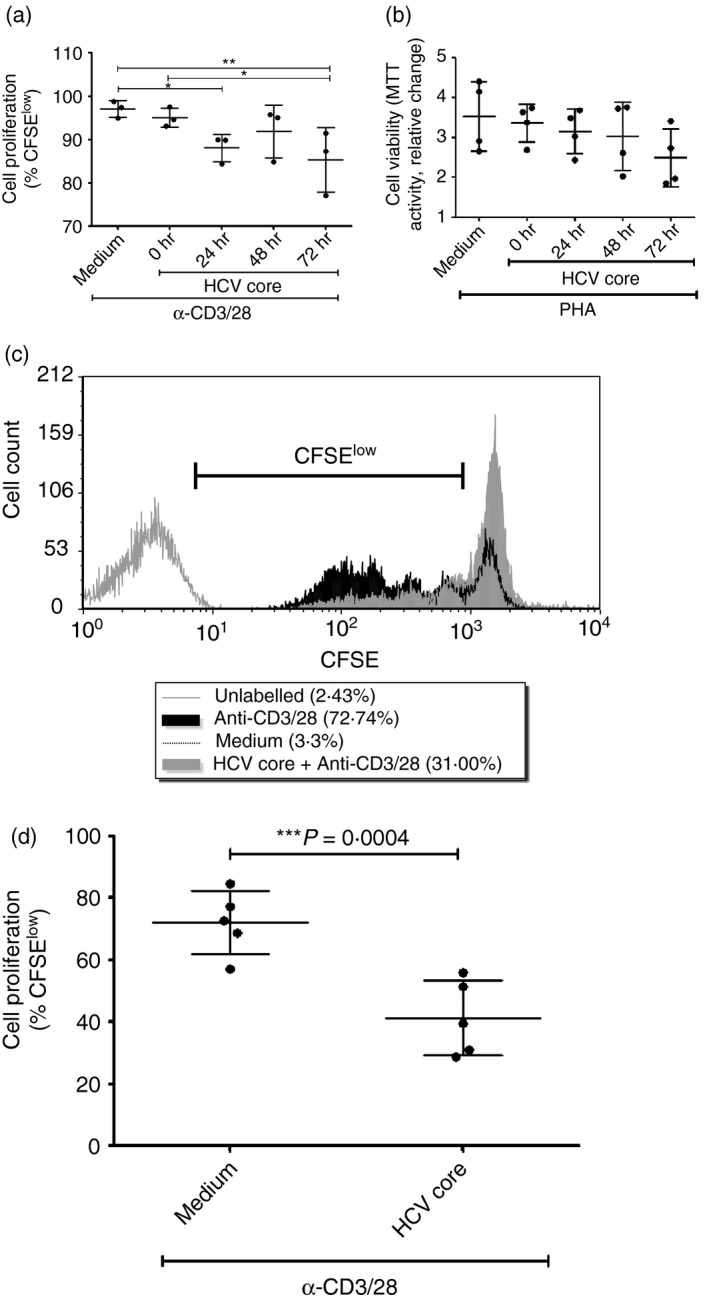
Hepatitis C virus (HCV) core protein reduces CD8+ T‐cell proliferation and viability. (a) A scatter plot graph shows the proliferation of CFSE‐labelled CD8+ T‐cells cultured alone (Medium) or pre‐incubated with HCV core for increasing periods of time (0, 24, 48 and 72 hr) followed by a 5‐day stimulation with anti‐CD3/CD28 (n = 3, repeated measures analysis of variance with Bonferroni's multiple comparison test). *P < .05, **P < 0·01. (b) A second scatter plot graph indicates the viability of stimulated CD8+ T‐cells incubated with HCV core in a time–course experiment, as determined in studies using phytohaemagglutinin as a stimulant (n = 4; relative change in MTT absorbance). To confirm effects on proliferation, CFSE‐labelled CD8+ T‐cells were pre‐incubated with or without HCV core (5 µg/ml) for 72 hr followed by 5 days of stimulation with anti‐CD3/28 (0·0625 µg/ml). (c) A representative histogram shows the proliferation profile of CFSE‐labelled CD8+ T‐cells. The marker indicates the decreasing fluorescence intensity traces of proliferating (i.e. CFSE low) cells. (d) A scatter plot summarizes the proliferation of CD8+ T‐cells pre‐incubated with medium or HCV core for 72 hr followed by stimulation with anti‐CD3/28 for 72 hr (mean ± SD, two‐tailed paired Student's t‐test, n = 5).
To confirm the effect of prolonged exposure to HCV core on cell division, CFSE‐labelled CD8+ T‐cells were incubated with or without HCV core for 72 hr before anti‐CD3/28 stimulation. HCV core significantly reduced the stimulation‐induced proliferation of CD8+ T‐cells (P = 0·0004, n = 5, Fig. 1c,d), compared with cells not treated with HCV core. On average, 72·2% (± 10·2) of CD8+ T‐cells divided upon stimulation, but only 41·4% (± 12·1) of cells pre‐incubated with HCV core divided in response to stimulation.
HCV core reduces IL‐7‐mediated up‐regulation of Bcl‐2 production but increases STAT5 activation
Expression of anti‐apoptotic Bcl‐2 prolongs survival of CD8+ T‐cells, and increased survival will support proliferation‐driven activation of CD8+ T‐cells.23 Upon treatment with IL‐7, CD8+ T‐cells up‐regulated Bcl‐2 production (P < 0·0001, n = 8, Fig. 2). On average, 17·4% (± 6·6) of unstimulated cells were determined to be Bcl‐2high and 67·1% (± 8·4) of IL‐7‐treated cells were Bcl‐2high. Up‐regulation of Bcl‐2 was hampered in HCV core‐treated cells, thereby altering the ratio of Bcl‐2low versus Bcl‐2high cells. On average, 61·4% (± 8·4) of HCV core‐incubated cells were Bcl‐2high after IL‐7 stimulation compared with 67·1% (± 8·4) of cells not incubated with HCV core (P = 0·028, n = 8, Fig. 2).
Figure 2.

Incubation with hepatitis C virus (HCV) core protein decreases interleukin‐7 (IL‐7) ‐mediated Bcl‐2 production in CD8+ T‐cells. CD8+ T‐cells incubated with or without HCV core protein (5 µg/ml) for 72 hr were treated with IL‐7 (1 ng/ml) for 48 hr. Cells were then stained with anti‐human Bcl‐2 antibody before analysis on a flow cytometer. (a) Representative histograms, with marker indicating Bcl‐2hi cells. (b) Average proportion of Bcl‐2hi cells ± SD (two‐tailed paired Student's t‐test, n = 8).
However, STAT5 activation, an important signalling step leading to Bcl‐2 expression, was higher in HCV core‐treated cells. After IL‐7 treatment, the degree of pSTAT5 expression was higher in CD8+ T‐cells incubated with HCV core compared with cells that were not treated with HCV core before IL‐7 treatment (P = 0·001, n = 6, Fig. 3). On average, pSTAT5 expression [by mean fluorescence intensity (MFI] in untreated cells was 3·7 (± 0·4), and in IL‐7‐treated cells was 9·8 (± 2·5). In HCV core‐treated cells MFI was 13·1 (± 3·4).
Figure 3.
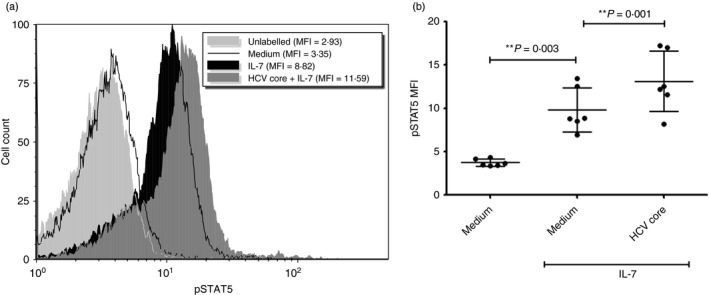
Incubation with hepatitis C virus (HCV) core protein increases signal transducer and activator of transcription 5 (STAT5) activation in CD8+ T‐cells. CD8+ T‐cells were incubated with or without HCV core protein (5 µg/ml) for 72 hr before treatment with interleukin‐7 (IL‐7; 1 ng/ml) for 15 min. Cells were then labelled with anti‐pSTAT5 antibody and analysed on a flow cytometer. (a) Representative histogram depicting effect of HCV core on STAT5 activation. (b) Mean MFI ± SD of pSTAT5 production (i.e. activated STAT5; two‐tailed paired Student's t‐test, n = 6).
HCV core reduces the proportion of perforin+ and CD107a+ CD8+ T‐cells
One of the main lytic functions of CD8+ T‐cells is their ability to up‐regulate perforin production and degranulate. To determine HCV core's effects on these functions, isolated CD8+ T‐cells were incubated and treated as above. Upon anti‐CD3/28 stimulation, perforin production in CD8+ T‐cells was increased (P = 0·0003, n = 9, Fig. 4), with 24·9% (± 9·5) of unstimulated cells containing perforin, and 65·4% (± 20·2) of stimulated cells containing perforin.
Figure 4.
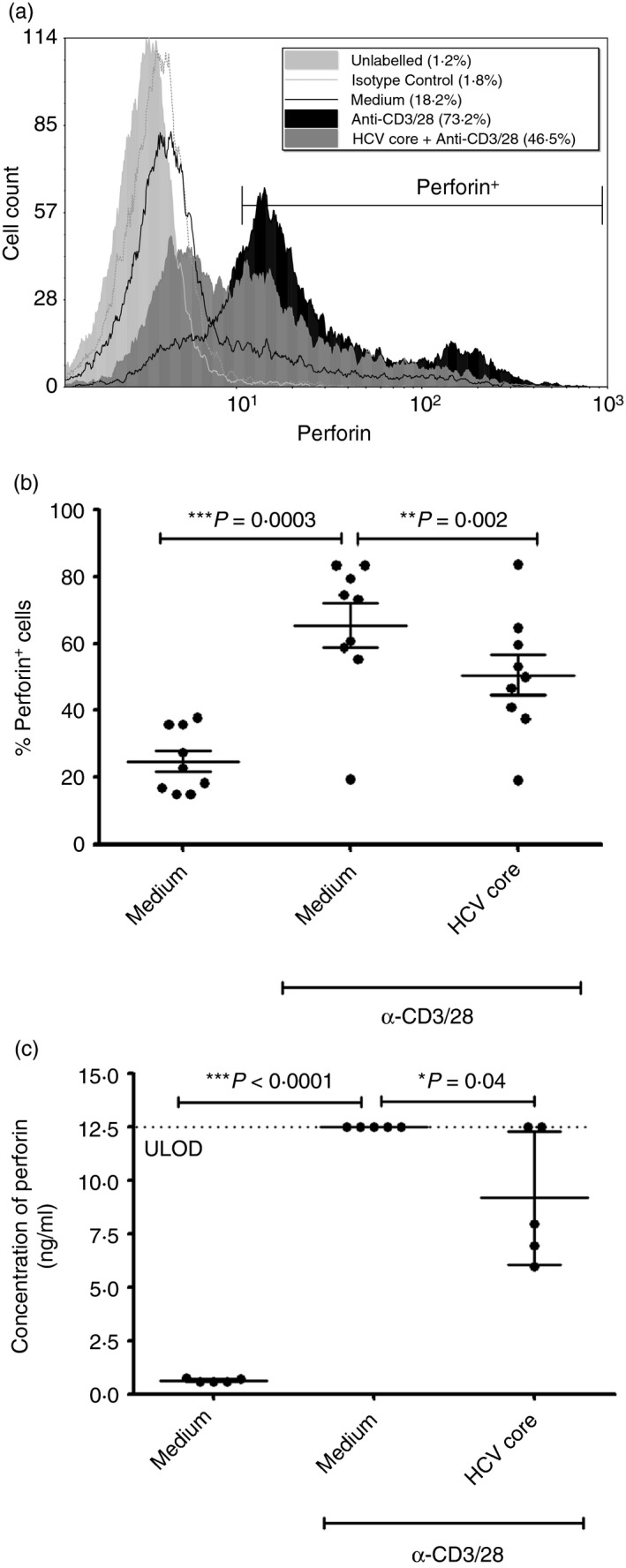
Hepatitis C virus (HCV) core protein reduces perforin production of CD8+ T‐cells. CD8+ T‐cells were incubated with or without HCV core protein (5 µg/ml) for 72 hr before stimulation with anti‐CD3/28 (1 µg/ml) for 48 hr, and then labelled with anti‐human perforin antibody and analysed by flow cytometry, or stimulated for a further 72 hr before collection of supernatants for quantification of perforin release. (a) A representative histogram is shown with marker indicating perforin+ cells. (b) Average proportion of perforin+ cells ± SD (two‐tailed paired Student's t‐test, n = 9). (c) Perforin release from stimulated CD8+ T‐cells as measured from supernatants by Milliplex Bead Array (one‐tailed paired Student's t‐test, n = 5). ULOD, upper limit of detection.
Perforin production in CD8+ T‐cells incubated with HCV core was lower than in cells not treated with core before stimulation (P = 0·002, n = 9, Fig. 4). On average, after anti‐CD3/28 stimulation, 50·6% (± 18·2) of core‐treated cells were perforin+, while 65·4% (± 20·2) of cells not treated with core were perforin+. Similarly, the amount of perforin in HCV core‐treated stimulated cells was reduced by the viral protein relative to stimulated cells alone [0·68 (± 0·14) perforin MFI relative to stimulated cells, P = 0·0004, n = 9, data not shown]. When unstimulated cells were treated with HCV core, the proportion of perforin+ cells was similar to that of unstimulated cells not treated with HCV core. Furthermore, perforin release was found to be significantly lower from CD8+ T‐cells pre‐incubated with HCV core before 5 days of stimulation with anti‐CD3/28 (P = 0·04, compared with CD8+ T‐cells not pre‐incubated with HCV core before stimulation; Fig. 4c). Similarly, Granzyme‐B release was found to be lower from CD8+ T‐cells pre‐incubated with HCV core before 5 days of stimulation with anti‐CD3/28, but this was not statistically significant (see Supplementary material, Fig. S1A).
Furthermore, degranulation of CD8+ T‐cells, indicated by CD107a expression, after anti‐CD3/28 stimulation was lower in HCV core‐treated cells compared with cells not treated with HCV core (P = 0·015, n = 5, Fig. 5). On average, after anti‐CD3/28 stimulation, 17·7% (± 12·9) of HCV core‐treated cells were CD107a+, compared with 23·7% (± 11·3) of cells not treated with core were CD107a+. This reduction was also reflected in the degree of degranulation where, relative to stimulated cells, CD107a expression in HCV core‐treated stimulated cells was significantly less than in cells not exposed to HCV core protein [0·88 (± 0·08) CD107a MFI relative to stimulated cells, P = 0·028, n = 5, data not shown].
Figure 5.
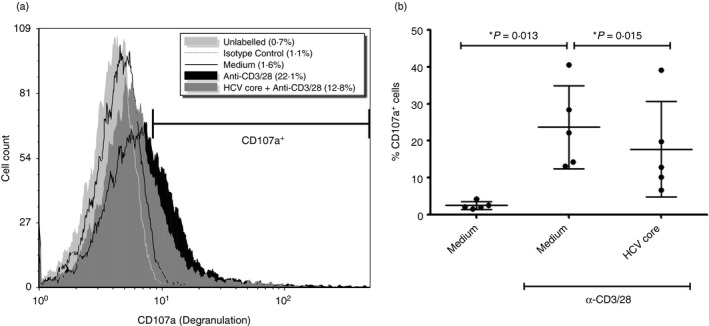
Incubation with hepatitis C virus (HCV) core protein reduces degranulation (as indicated by CD107a expression) of CD8+ T‐cells. CD8+ T‐cells incubated with or without HCV core protein (5 µg/ml) for 72 hr were stimulated with anti‐CD3/28 stimulation (1 µg/ml) for 48 hr before analysis by flow cytometry. For the last 6 hr of stimulation, anti‐human CD107a antibody and BD GolgiPlug (containing monensin) was added. (a) A representative histogram with marker indicating CD107a+ cells. (b) Average proportion of CD107a+ cells ± SD (two‐tailed paired Student's t‐test, n = 5).
HCV core does not affect the proportion of IFN‐γ + CD8+ T‐cells
Another important aspect of CD8+ T‐cell effector functions is the production of pro‐inflammatory IFN‐γ. The production of IFN‐γ by CD8+ T‐cells was increased from 19·5% (± 6·2) to 30·7% (± 6·7) with anti‐CD3/28 stimulation. Interferon‐γ production after anti‐CD3/28 stimulation was slightly higher (but statistically insignificant) in HCV‐core treated CD8+ T‐cells compared with cells not treated with HCV core (P = 0·177, n = 8, Fig. 6). On average, after anti‐CD3/28 stimulation, 38·9% (±14·6) of core‐treated cells were IFN‐γ +, whereas 30·7% (±6·7) of cells not treated with core were IFN‐γ +.
Figure 6.
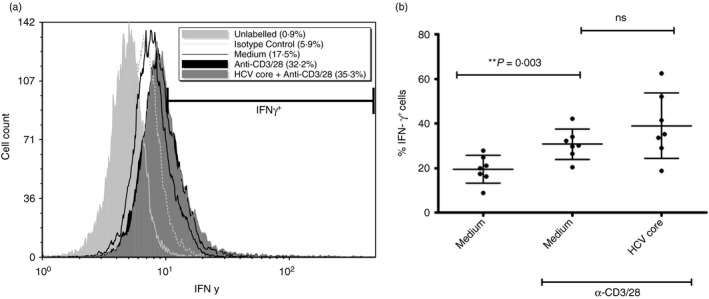
Incubation with hepatitis C virus (HCV) core protein does not affect proportion of IFN‐γ + CD8+ T‐cells. CD8+ T‐cells incubated with or without HCV core protein (5 µg/ml) for 72 hr were stimulated with anti‐CD3/28 (1 µg/ml) for 24 hr before labelling with anti‐human interferon‐ γ (IFN ‐ γ) antibody and analysis by flow cytometry. Cells were treated with BD GolgiPlug (containing Brefeldin‐A) for the last 5 hr of stimulation. (a) A representative histogram, with analysis marker indicating IFN‐γ + cells. (b) Average proportion of IFN‐γ + cells ± SD (two‐tailed paired Student's t‐test, n = 7).
The absence of an HCV core protein effect on IFN‐γ production by stimulated cells was also observed on a per cell basis (1.18 IFN‐γ MFI relative to stimulated cells, P = 0·088, n = 8, data not shown).
Cells treated with serum from HCV‐infected individuals produce less perforin upon stimulation
To confirm this impairment and to attempt to translate these findings to what may be observed in vivo, isolated CD8+ T‐cells were treated with pooled serum collected from viraemic HCV+ individuals (Table 1). These cells treated with serum from viraemic HCV+ individuals produce less perforin upon stimulation compared with cells treated with pooled serum from healthy controls (P = 0·02, n = 6, Fig. 7). Since the effect of HCV core on perforin production was novel and most stark, it was chosen as a marker to verify the effect of HCV+ serum on CD8+ T‐cell function. On average, 46·4% (± 12·8) of stimulated cells treated with healthy serum were perforin+, but only 39·2% (± 9·2) of cells treated with HCV‐infected serum were perforin+. There was no statistical difference in baseline perforin levels in cells treated with HCV− or HCV+ serum.
Figure 7.
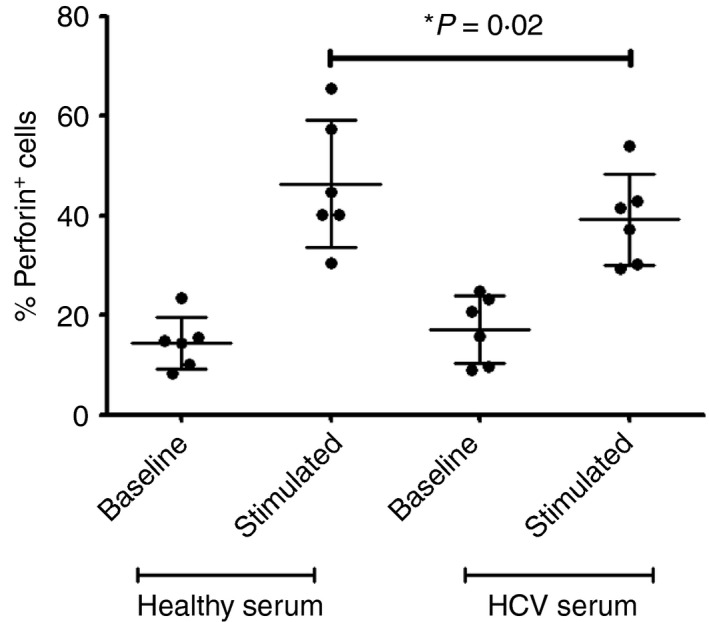
Cells treated with hepatitis C virus (HCV) ‐infected serum produce less perforin upon stimulation. CD8+ T‐cells were incubated with serum from HCV + or healthy HCV − serum for 72 hr before stimulation with anti‐CD3/28 for 48 hr. Cells were then labelled with anti‐human perforin antibody and analysed by flow cytometry. Average proportion of perforin+ cells ± SD (two‐tailed paired Student's t‐test, n = 6).
Discussion
Our study reveals that HCV core protein modulates various aspects of CD8+ T‐cell activity, survival potential and effector functions. It decreases CD8+ T‐cell proliferation, perforin production and degranulation but does not affect IFN‐γ production. Interestingly, while reducing IL‐7‐mediated Bcl‐2 up‐regulation, HCV core increases upstream STAT5 activation. This suggests that HCV core plays a role in bulk CD8+ T‐cell dysfunction observed in HCV‐infected individuals.
HCV core protein reduced the proliferation of CD8+ T‐cells, confirming the observations of others.3, 18 As our study was conducted in an isolated CD8+ T‐cell population, it also confirms that HCV core impairs CD8+ T‐cells by directly interacting with them as opposed to through its effects on other immune cells like macrophages and CD4+ T‐cells, that may then inhibit CD8+ T‐cell activity. Reduced proliferation of CD8+ T‐cells will hamper clonal expansion of cells upon antigen stimulation, and will inhibit an efficient CD8+ T‐cell response. Our data also reveal that HCV core exerts its effects in a time‐dependent manner, and that prolonged exposure to HCV core increasingly exacerbates CD8+ T‐cell activity. This suggests the need to detect and treat HCV infection earlier than is current practice.
We also found that anti‐apoptotic Bcl‐2 up‐regulation is inhibited by HCV core, suggesting that the viral protein may contribute to the reduced survival potential of CD8+ T‐cells observed in chronic HCV infection.10 Interleukin‐7‐mediated Bcl‐2 production enhances the survival of naive CD8+ T‐cells and the development of memory CD8+ T‐cells.24, 25 Hence, HCV core, by reducing Bcl‐2 up‐regulation, may reduce the presence of viable CD8+ T‐cells and impair the development of memory responses. In fact, in chronic HCV infection, CD8+ T‐cells are known to be more prone to activation‐induced apoptosis.8
We have previously shown that increases in Bcl‐2 are dependent on STAT5 activation and we recently observed decreased STAT5 activation coupled with lower Bcl‐2 levels in HCV‐infected individuals.10, 26 Contrary to expectations based on those findings, HCV core protein up‐regulated IL‐7‐mediated STAT5 activation (Fig. 4). This suggests that HCV core dysregulates the signalling pathway leading to Bcl‐2 production after phosphorylation of STAT5. Contribution of other signalling pathways may be required for IL‐7/STAT‐5‐mediated up‐regulation of Bcl‐2, particularly that of the phosphatidylinositol‐3‐kinase–protein kinase B (PI3K‐Akt) pathway.27 Yao et al.3 showed that HCV core reduces Akt activation in CD8+ T‐cells. Therefore, in addition to reducing the activity of CD8+ T‐cells, HCV core is reducing the cell survival potential or viability. This dysregulation of IL‐7 signalling via PI3K inhibition may have even further reaching consequences. We previously demonstrated that inhibition of the PI3K pathway interferes with IL‐7‐mediated proliferation and glucose uptake in CD8+ T‐cells.26 As IL‐7‐mediated signalling is required for the development and maintenance of CD8+ T‐cell responses even in the absence of antigen signalling, this inhibition of IL‐7 signalling by HCV core may have significant impacts on CD8+ T‐cell‐mediated immunity.
Studies on the effects of HCV core on the lysis‐associated effector functions of CD8+ T‐cells have been limited. We were able to show that in response to anti‐CD3/28 stimulation, pre‐incubation with HCV core protein reduces perforin production and degranulation in CD8+ T‐cells, suggesting functional anergy. As perforin is required for other lytic granules to enter and exert their activity, functional efficacy of CD8+ T‐cells is reduced in the continued presence of HCV core in chronic infection. Furthermore, we showed that degranulation (i.e. exocytosis of perforin and granzymes, as measured by CD107a expression) is decreased in HCV core‐incubated cells, further confirming that granule‐induced killing by CD8+ T‐cells is impaired by HCV core protein. Similarly, perforin and granzyme‐B (not significant) release was also found to be lower from CD8+ T‐cells pre‐incubated with HCV core. Lucas et al. demonstrated that, in HCV‐infected individuals, cytomegalovirus‐specific CD8+ T‐cells had lower Fas ligand and perforin production.9 Our research suggests that dysfunctional lytic capacity of CD8+ T‐cells is not restricted to any antigen‐specific CD8+ T‐cells.
Mechanistically, HCV core‐induced reduction in lytic activity can be best explained by lower Akt activation [via reduced activation of ζ‐chain‐associated protein kinase 70 by lymphocyte‐specific protein tyrosine kinase (Lck)].3 However, this does not explain why T‐cell receptor‐stimulated IFN‐γ production in core‐treated cells would not change (or even increase slightly, although this was not statistically significant). Yao et al. observed reduced Lck activation but not that of Fyn kinase; both Src kinases involved in early T‐cell receptor signalling.3, 28 As the role of Fyn is less well understood and has been posited to be supplemental to Lck, Fyn activation may have a compensatory role in increasing/maintaining IFN‐γ production.28 Sustained IFN‐γ production may be explained by the differential requirement of Lck during initial and memory CD8+ T‐cell responses. A study showed that while Lck‐mediated activation is required during the primary response, it is dispensable during memory or recall responses.29 If in our system, IFN‐γ is being primarily produced by memory CD8+ T‐cells upon T‐cell receptor stimulation, and HCV core‐induced anergy is mediated by Lck signalling, then this may reconcile the different effects on perforin production/cell degranulation and IFN‐γ production.
Although this study was conducted in vitro, the findings suggest that HCV core protein plays a major role in inducing CD8+ T‐cell dysfunction in HCV+ individuals. Circulating HCV core in serum has been correlated with reductions in natural killer cell activity.30 And as suggested by our serum experiments, this may be true for CD8+ T‐cells as well: CD8+ T‐cells exposed to serum of HCV+ individuals produce less perforin compared with cells exposed to control serum. This serves as a proof‐of‐concept that factors – including, but not limited to, HCV core protein – in the serum of HCV+ individuals hamper CD8+ T‐cell function. HCV core protein prevalence is correlated with HCV viral RNA load and hence serum from highly viraemic HCV+ individuals was pooled for this study.17 This dysfunction may be mediated by high concentrations of HCV core, or other viral or host factors, like immunoregulatory IL‐10. Interestingly, HCV core protein itself did not appear to significantly alter IL‐10 production from CD8+ T‐cells exposed to HCV core protein (see Supplementary material, Fig. S1B). In fact, the contributions of HCV core to CD8+ T‐cell dysfunction in vivo may be more pronounced than that observed in our experiments. Others have shown that part of the HCV core‐mediated CD8+ T‐cell anergy involves up‐regulation of programmed death‐1 (PD‐1), a co‐inhibitory receptor commonly used as a marker for T‐cell exhaustion in chronic disease and indicating anergic CD8+ T‐cells.7 As our system studied isolated CD8+ T‐cells, the effects of PD‐1 up‐regulation and its engagement by programmed death ligand ‐1 on antigen‐presenting cells were not evaluated.
It is not known whether the dysfunction caused by HCV core protein could persist after treatment and achievement of sustained virological response (i.e. viral clearance 12 weeks after end‐of‐treatment). A wash‐step included in the perforin production experiments before the addition of anti‐CD3/28 stimulation to cells (n = 2, data not shown) revealed that the continued presence of HCV core was not required for the inhibition of perforin production. This suggests that HCV core has either exerted its effect on CD8+ T‐cells during its initial interactions or remains bound to gC1qR or other protein on CD8+ T‐cells. Attempts to block HCV core activity through gC1qR using a commercially available gC1qR‐blocking antibody were inconclusive (the antibodies used in published blocking studies are not commercially available).3
Alternatively, increased STAT5 activation (and possibly higher IFN‐γ production) induced by HCV core protein may be exploited if negative effects of the protein can be attenuated. As gC1qR may not be the only receptor with which HCV core interacts, identification of HCV core epitopes and protein–receptor interactions that mediate positive and negative effects could lead to the development of therapeutic vaccines containing a modified core protein.
These findings suggest that HCV core protein plays a significant role in bulk CD8+ T‐cell dysfunction and anti‐core components in HCV drugs may be highly beneficial in aiding the recovery of generalized immune impairments. Further studies on signalling pathways may offer insights into HCV‐mediated dysfunction of immune responses.
Conflict of interest
The authors declare no conflicts of interest.
Funding
STK is a two‐time recipient of the Canadian Network on Hepatitis C Summer Student Award (2015‐16) and holds an Ontario Graduate Scholarship. WK received the Canadian Network on Hepatitis C Summer Student Award (2014). AMC's salary is provided in part by a Canadian Institutes of Health Research New Investigator Award and a Junior Investigator Development Award (JIDA) from the Ontario HIV Treatment Network (OHTN). AMC's research funding for this work is derived from a J.P. Bickell Foundation Medical Research Award, a Natural Sciences and Engineering Research Council of Canada Discovery Grant, a Canadian Foundation of AIDS Research Operating Grant and operating funds associated with the OHTN JIDA.
Supporting information
Figure S1. Effect of pre‐incubation with hepatitis C virus core protein on Granzyme B and interleukin‐10 release from CD8+ T‐cells.
Acknowledgements
AMC and STK designed the project and wrote the manuscript. STK performed the experiments. WK performed some experiments. CLC provided samples from HCV+ individuals and helped in design of the project. The authors would also like to thank Agatha Vranjkovic for her technical help.
References
- 1. Freeman AJ, Marinos G, Ffrench RA, Lloyd AR. Immunopathogenesis of hepatitis C virus infection. Immunol Cell Biol 2001; 79:515–36. [DOI] [PubMed] [Google Scholar]
- 2. Myers RP, Shah H, Burak KW, Cooper C, Feld JJ. An update on the management of chronic hepatitis C: 2015 Consensus guidelines from the Canadian Association for the Study of the Liver. Can J Gastroenterol Hepatol 2015; 29:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yao ZQ, Eisen‐Vandervelde A, Waggoner SN, Cale EM, Hahn YS. Direct binding of hepatitis C virus core to gC1qR on CD4+ and CD8+ T‐cells leads to impaired activation of Lck and Akt. J Virol 2004; 78:6409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. Hepatology 1997; 26(3 Suppl 1):15S–20S. [DOI] [PubMed] [Google Scholar]
- 5. Rehermann B. Chronic infections with hepatotropic viruses: mechanisms of impairment of cellular immune responses. Semin Liver Dis 2007; 27:152–60. [DOI] [PubMed] [Google Scholar]
- 6. Wedemeyer H, He XS, Nascimbeni M, Davis AR, Greenberg HB, Hoofnagle JH et al Impaired effector function of hepatitis C virus‐specific CD8+ T‐cells in chronic hepatitis C virus infection. J Immunol 2002; 169:3447–58. [DOI] [PubMed] [Google Scholar]
- 7. Yao ZQ, King E, Prayther D, Yin D, Moorman J. T cell dysfunction by hepatitis C virus core protein involves PD‐1/PDL‐1 signaling. Viral Immunol 2007; 20:276–87. [DOI] [PubMed] [Google Scholar]
- 8. Zhao B‐B, Zheng S‐J, Gong L‐L, Wang Y, Chen C‐F, Jin W‐J et al T lymphocytes from chronic HCV‐infected patients are primed for activation‐induced apoptosis and express unique pro‐apoptotic gene signature. PLoS One 2013; 8:e77008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lucas M, Vargas‐Cuero AL, Lauer GM, Barnes E, Willberg CB, Semmo N et al Pervasive influence of hepatitis C virus on the phenotype of antiviral CD8+ T‐cells. J Immunol 2004; 172:1744–53. [DOI] [PubMed] [Google Scholar]
- 10. Burke Schinkel SC, Carrasco‐Medina L, Cooper CL, Crawley AM. Generalized liver‐ and blood‐derived CD8+ T‐cell impairment in response to cytokines in chronic hepatitis C virus infection. PLoS One 2016; 11:e0157055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Large MK, Kittlesen DJ, Hahn YS. Suppression of host immune response by the core protein of hepatitis C virus: possible implications for hepatitis C virus persistence. J Immunol 1999; 162:931–8. [PubMed] [Google Scholar]
- 12. Sundstrom S, Ota S, Dimberg LY, Masucci MG, Bergqvist A. Hepatitis C virus core protein induces an anergic state characterized by decreased interleukin‐2 production and perturbation of mitogen‐activated protein kinase responses. J Virol 2005; 79:2230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev 2007; 5:453–63. [DOI] [PubMed] [Google Scholar]
- 14. Polyak SJ, Klein KC, Shoji I, Miyamura T, Lingappa JR. Assemble and interact: pleiotropic functions of the HCV core protein In: Tan SL, ed. Hepatitis C Viruses: Genomes and Molecular Biology. Norfolk, UK: Horizon Bioscience, 2006. [PubMed] [Google Scholar]
- 15. Kanto T, Hayashi N, Takehara T, Hagiwara H, Mita E, Naito M et al Buoyant density of hepatitis C virus recovered from infected hosts: two different features in sucrose equilibrium density‐gradient centrifugation related to degree of liver inflammation. Hepatology 1994; 19:296–302. [PubMed] [Google Scholar]
- 16. Katsarou K, Lavdas AA, Tsitoura P, Serti E, Markoulatos P, Mavromara P et al Endocytosis of hepatitis C virus non‐enveloped capsid‐like particles induces. Cell Mol Life Sci 2010; 67:2491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maillard P, Krawczynski K, Nitkiewicz J, Bronnert C, Sidorkiewicz M, Gounon P et al Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J Virol 2001; 75:8240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kittlesen DJ, Chianese‐Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T‐lymphocyte proliferation. J Clin Invest 2000; 106:1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen A, Gaddipati S, Hong Y, Volkman DJ, Peerschke EI, Ghebrehiwet B. Human T‐cells express specific binding sites for C1q. Role in T cell activation and proliferation. J Immunol 1994; 153:1430–40. [PubMed] [Google Scholar]
- 20. Yao ZQ, Prayther D, Trabue C, Dong ZP, Moorman J. Differential regulation of SOCS‐1 signalling in B and T lymphocytes by hepatitis C virus core protein. Immunology 2008; 125:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao ZQ, Nguyen DT, Hiotellis AI, Hahn YS. Hepatitis C virus core protein inhibits human T lymphocyte responses by a complement‐dependent regulatory pathway. J Immunol [Internet] 2001;167 URL http://www.jimmunol.org/content/167/9/5264.long [accessed on 10 August 2017] [DOI] [PubMed] [Google Scholar]
- 22. Crawley AM, Katz T, Parato K, Angel JB. IL‐2 receptor γ chain cytokines differentially regulate human CD8+CD127+ and CD8+CD127– T cell division and susceptibility to apoptosis. Int Immunol 2009; 21:29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michalek RD, Rathmell JC. The metabolic life and times of a T‐cell. Immunol Rev 2010; 236:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H‐R, Hwang K‐A, Park S‐H, Kang I. IL‐7 and IL‐15: biology and roles in T‐cell immunity in health and disease. Crit Rev Immunol 2008; 28:325–39. [DOI] [PubMed] [Google Scholar]
- 25. Murphy K. Janeway's Immunobiology, 8th edn New York, NY: Garland Science, 2012. [Google Scholar]
- 26. Crawley AM, Vranjkovic A, Faller E, McGuinty M, Busca A, Burke SC et al Jak/STAT and PI3K signaling pathways have both common and distinct roles in IL‐7‐mediated activities in human CD8+ T‐cells. J Leukoc Biol 2014; 95:117–27. [DOI] [PubMed] [Google Scholar]
- 27. Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7‐mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med 2004; 200:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T‐cell receptor proximal signaling via the Src‐family kinases, Lck and Fyn, influences T‐cell activation, differentiation, and tolerance. Immunol Rev 2009; 228:9–22. [DOI] [PubMed] [Google Scholar]
- 29. Tewari K, Walent J, Svaren J, Zamoyska R, Suresh M. Differential requirement for Lck during primary and memory CD8+ T cell responses. Proc Natl Acad Sci USA 2006; 103:16388–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golden‐Mason L, Hahn YS, Strong M, Cheng L, Rosen HR. Extracellular HCV‐core protein induces an immature regulatory phenotype in NK cells: implications for outcome of acute infection. PLoS One 2014; 9:e103219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of pre‐incubation with hepatitis C virus core protein on Granzyme B and interleukin‐10 release from CD8+ T‐cells.