

Summary
This study examined the expression of the inhibitory receptor, leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) in fibroblast‐like synoviocytes (FLS) in rheumatoid arthritis (RA) patients to investigate its potential role in the modulation of inflammatory cytokines, matrix metalloproteinases (MMPs) and invasiveness of synoviocytes. LAIR‐1 expression in synovial tissues from RA patients, osteoarthritis patients and healthy donors was analysed by immunohistochemistry. The membrane‐bound form (mLAIR‐1) was detected by flow cytometry. Factors involved in inflammation and MMP activity in FLS were analysed by quantitative polymerase chain reaction (qPCR). LAIR‐1 expression was higher in the synovia of the RA patients than those of the osteoarthritis patients. Co‐immunostaining of vimentin/LAIR‐1 demonstrated that LAIR‐1 was localized mainly in FLS in the RA patients. Surprisingly, primary FLS isolated from the RA patients had low levels of mLAIR‐1 expression, with cytoplasmic distribution. The extracellular domain of LAIR‐1 was shed from the cell surface in response to tumour necrosis factor (TNF)‐α, and this process could be blocked by serine protease inhibitors. Additional experiments indicated that LAIR‐1 over‐expression reduced FLS invasion considerably, which reduced simultaneously the mRNA levels of interleukin (IL)‐6, IL‐8 and MMP‐13 in the presence of TNF‐α. Our study demonstrated that LAIR‐1 is an anti‐inflammatory molecule, and was up‐regulated in FLS in the RA patients; however, cell‐surface LAIR‐1 could be shed from cells in the inflammatory microenvironment in RA. This may weaken the interaction of LAIR‐1 with its ligand, thus reducing the anti‐inflammatory effects of LAIR‐1. These findings suggested that LAIR‐1 may be an important factor involved in the mediation of the progressive joint destruction in RA.
Keywords: fibroblast‐like synoviocytes, inflammation, LAIR‐1, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is an immune‐related disease characterized by inflammatory infiltration of the synovium, leading eventually to cartilage and bone destruction 1. RA affects approximately 0·5–1% of the general population 2, 3. Fibroblast‐like synoviocytes (FLS) are highly active cells that contribute to the development of RA. They localize close to the articular cartilage, which enables them to invade it 4.
Leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) (also known as CD305) is expressed widely by most immune cells, including natural killer (NK) cells, T cells, B cells, monocytes and CD34+ haematopoietic progenitors 5. Similar to other inhibitory receptors, LAIR‐1 contributes to the regulation of the immune system by delivering inhibitory signals via a short cytoplasmic tail that contains two immunoreceptor tyrosine‐based inhibitory motifs (ITIMs) 6. In recent years, the finding that collagen is a functional ligand for LAIR‐1 revealed a novel role of collagen in the regulation of immune responses 7.
LAIR‐1 is an inhibitory receptor presented mainly in haematopoietic cells; it regulates the activities of the immune system and protects against tissue damage and autoimmune dysfunction resulting from hyperactive immune responses. In our previous study, we observed that LAIR‐1 is expressed in synovial CD68+ macrophages in patients with active RA 8. Both soluble human LAIR‐1 and its secreted homologue, LAIR‐2 (CD306) were detectable in the plasma and urine of healthy individuals and RA patients. It has been demonstrated that soluble LAIR‐1 is a useful biomarker for analysing synovial fluid for the presence of RA 9. However, the role of LAIR‐1 in RA pathogenesis is still unclear. The objectives of this study were to investigate the possible functions of LAIR‐1 in FLS in the RA patients.
Herein, we demonstrated that the collagen receptor LAIR‐1 was expressed highly in FLS in the RA patients. However, membrane‐bound LAIR‐1 (mLAIR‐1) was shed from FLS cell membranes in the presence of the inflammatory cytokine, tumour necrosis factor (TNF)‐α. Activated FLS may contribute to the development of inflammation and joint damage. LAIR‐1 over‐expression reduced the expression of the interleukins (ILs)‐6 and IL‐8 and matrix metalloproteinase‐13 (MMP‐13) in the FLS of RA patients.
Materials and methods
Reagents
TNF‐α was obtained from R&D Systems (Minneapolis, MN, USA). Dulbecco's modified Eagle's medium (DMEM), RPMI‐1640, fetal calf serum (FCS), antibiotics, trypsin–ethylenediamine tetraacetic acid (EDTA), phosphate‐buffered saline (PBS) and other products for cell culture were purchased from Invitrogen (Carlsbad, CA, USA). Protease inhibitor panels were purchased from Sigma‐Aldrich (St Louis, MO, USA).
Patients and synovial tissues
Biopsy samples from RA and osteoarthritis (OA) patients were obtained from the Department of Orthopedics at Tangdu Hospital. Human FLS were isolated from the synovial tissues of RA patients during arthroscopic synovectomy of the knee joint. All the RA patients fulfilled the American Rheumatism Association 1987 revised criteria 10. Primary FLS were isolated from synovial tissues via arthroscopic biopsy. Normal human peripheral blood mononuclear cells (PBMC) were isolated in our laboratory using the conventional method. All healthy donors and patients provided written informed consent prior to the donation of synovial tissues and blood samples. The study design was approved by the ethics committee of the Fourth Military Medical University.
Isolation and culture of human FLS
Human primary FLS were isolated and cultured as described previously 11. After careful removal of the adipose and fibrous tissue, fresh synovial tissue was minced and then digested overnight with 1·0 mg/ml collagenase in serum‐free DMEM at 37°C on a horizontal shaker. The cell suspensions were filtered through a stainless steel filter to remove the undigested tissue. The filtrate was centrifuged and the cell precipitate was washed twice with PBS. After that, the cells were resuspended in complete media containing 20% FCS, 100 U/ml penicillin and 100 mg/ml streptomycin. The cells were counted, seeded into culture flasks and cultured overnight in a humidified, 5% CO2 atmosphere at 37°C. The non‐adherent cells were then washed out. The adherent cells were cultured in complete medium, and the culture medium was replaced every week. Upon confluence, cells were trypsinized and then transferred to new plastic dishes in a split ratio of 1 : 3. Passages three to eight of the FLS were used in subsequent experiments. The human FLS cell line MH7A was purchased from Jennio biological technology (Guangzhou, China) and cultured in RPMI‐1640 media supplemented with 10% FCS at 37°C.
Lentiviral packaging and infection
Commercially available lentiviral LV‐LAIR‐1 constructs (Shanghai GenePharma Co., Ltd, Shanghai, China) were modified to over‐express LAIR‐1. The ITIM mutants of LAIR‐1 (Tyr253 of ITIM1 to Phe, Tyr283 of ITIM2 to Phe) were generated using a QuikChange site‐directed mutagenesis kit (Stratagene, La Jolla, CA, USA). The human MH7A rheumatoid FLS cells or primary FLS were infected with LV‐control, LV‐LAIR‐1 or LV‐LAIR‐1‐mut in the presence of 5 μg/ml polybrene (Sigma‐Aldrich). The lentiviral vectors expressed GFP, and the infection efficiency was evaluated by fluorescent microscopy to test for GFP expression.
Haematoxylin and eosin (H&E) and immunohistochemical staining
Fresh synovial tissues were fixed in 4% paraformaldehyde for 48 h, embedded in paraffin, and then serial sections (4 μm thick) were stained with haematoxylin and eosin (H&E). For immunohistochemical staining, sections were treated for 10 min in 0·3% H2O2/methanol to block endogenous peroxidase activity. After three washes in PBS, the sections were incubated with 10% normal goat serum to reduce non‐specific protein binding. Next, the sections were incubated overnight at 4°C with anti‐human LAIR‐1 monoclonal antibody (clone 9.1C3) 12 and anti‐human vimentin antibody (Cell Signaling Technology, Danvers, MA, USA). Bound primary antibodies were detected using a streptavidin‐conjugated horseradish peroxidase (HRP) kit (Immunocruz Staining System, Santa Cruz, Dallas, TX, USA), according to the manufacturer's instructions. Antibody complexes were then visualized by incubation with the 3,3'‐diaminobenzidine (DAB) chromogen. Sections were counterstained with Mayer's haematoxylin for 30 s, dehydrated through an ethanol series, cleaned, mounted and examined using light microscopy. In negative controls, the primary antibody was replaced with normal mouse immunoglobulin (Ig)G (mIgG) at the same concentration.
For primary FLS, cells were fixed and incubated overnight at 4°C with anti‐human LAIR‐1 monoclonal antibody and anti‐human vimentin antibody. Antibodies were then detected using an HRP/DAB system. Stained sections were photographed with a CKX41 inverted microscope (Olympus, Tokyo, Japan).
For immunofluorescence staining, sections were fixed and blocked using the same procedure as that for immunohistochemical (IHC) staining. Sections were then co‐stained with Cy3‐conjugated anti‐LAIR‐1 antibody (clone 9.1C3) and Alexa Fluor 488‐conjugated vimentin or 488‐conjugated CD45 antibodies (Cell Signaling Technology) overnight at 4°C. After three PBS washes, each sample was mounted and observed with an Olympus Fluoview 1000 confocal microscope. Images were processed and analysed with FV10‐ASW 1.4 viewer software (Olympus).
Flow cytometric analysis
After incubation with Fc‐blocker (2·4 G2; BD Biosciences Pharmingen, San Diego, CA, USA), cell‐surface antigens were detected using phycoerythrin (PE)‐labelled anti‐LAIR‐1 monoclonal antibody (clone 9.1C3) or PE‐labelled mouse IgG1 isotype antibody at 4°C for 30 min, and then washed twice.
For intracellular staining, FLS isolated from the RA patients were incubated in culture medium in the presence of 10 μg/ml Brefeldin A (Sigma‐Aldrich) for 5 h. The cells were washed twice with PBS, fixed in 4% paraformaldehyde (PFA) and permeabilized with 0·5% saponin (Sigma‐Aldrich) in PBS with 0·1% BSA. Then, the cells were stained intracellularly with the PE‐labelled anti‐LAIR‐1 antibody or PE‐labelled mouse IgG1 isotype antibody for 30 min at room temperature (RT) and washed. All the prepared cells were analysed using a fluorescence activated cell sorter (FACS)Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). Data were processed using FlowJo version 7.6.1 software (Tree Star, Inc., Ashland, OR, USA).
Invasion assay
A chemotactic cell invasion assay was performed using 24‐well Transwell units (Costar, Cambridge, NY, USA). Cells were plated in the upper chamber, which consisted of polycarbonate filter inserts (8‐µm pore size) coated with Matrigel matrix (BD Biosciences, Franklin Lakes, NJ, USA). The chemoattractant in the lower chamber was supplemented with medium containing 10% FCS. The cells were cultured for 24 h at 37°C in a CO2 incubator. The cells on the upper surface were removed, and those attached to the lower side of the membrane were fixed and stained with crystal violet. Cells were then counted under a microscope in five randomly selected fields.
Proliferation assay with 5‐ethynyl‐2'‐deoxyuridine (EdU)
The impact of LAIR‐1 on the FLS proliferation was measured by EdU incorporation assay using an EdU assay kit (Ribobio, Guangzhou, China), according to the manufacturer's instructions. Briefly, FLS cells at a density of 5 × 103 cells per well were cultured in triplicate in 96‐well plates and infected with lentivirus vector (LV‐control; negative control) or LAIR‐1 lentivirus vector (LV‐LAIR‐1) for 48 h, and then exposed to 50 μM of EdU for an additional 4 h at 37°C. The cells were fixed with 4% formaldehyde for 15 min at room temperature and permeabilized with 0·5% Triton X‐100 for 20 min at room temperature. After washing thrice with PBS, cells in each well were reacted with 100 μl of ×1 Apollo reaction cocktail for 30 min. Subsequently, the DNA content in each well was stained with 100 μl of Hoechst 33342 (5 μg/ml) for 30 min and visualized under a fluorescent microscope (Olympus).
Quantitative real‐time polymerase chain reaction (qPCR) assay
RNA was extracted from the cells using Trizol Reagent. For qPCR analysis, we used the commercial kit from Roche Biosystems (Pleasanton, CA, USA). PCR was performed using SYBR 2× Universal PCR Master Mix (ABI, Foster City, CA, USA). All‐in‐OneTM qPCR primers for detecting mRNA for human LAIR‐1, IL‐6, IL‐8, MMP‐1, MMP‐2, MMP‐3, MMP‐9, MMP‐13, cyclooxygenase‐2 (COX‐2) [prostaglandin‐endoperoxide synthase 2 (PTGS2)], prostaglandin E2 (PGE2) (PTGES2), tissue inhibitor of metalloproteinases 1 (TIMP‐1) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) were synthesized by GeneCopoeia Biotechnologies (Guangzhou, China). GAPDH was used as an internal control. Relative gene expression was quantified by using the comparative Ct method (2–ΔΔCT), as described by the manufacturer.
Statistical analysis
All data were expressed as means ± standard error of the mean (s.e.m.), and statistical significance was determined using one‐way analysis of variance for comparisons among the groups. The least significant difference t‐test was used for multiple comparisons. P‐values < 0·05 were considered statistically significant.
Results
Increased LAIR‐1 expression in the RA synovial tissues
All the RA, OA and healthy individual biopsy samples expressed LAIR‐1 based on immunohistochemical analysis. The levels of LAIR‐1 expression among the RA patients were variable. Staining was intense and widespread in certain patients, while others exhibited moderate expression levels restricted chiefly to the synovial membrane (Fig. 1). Additionally, LAIR‐1 was expressed at much lower levels in the healthy synovial tissues and synovia of the OA patients. H&E staining for samples from each patient and healthy donor was also performed.
Figure 1.
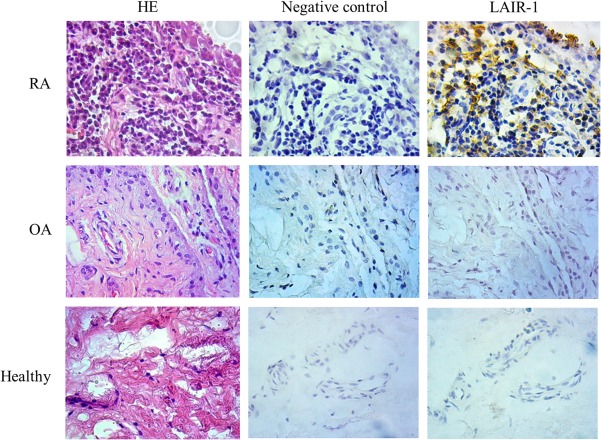
Expression of leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) in inflamed and healthy synovial tissues. Immunohistochemical staining revealed LAIR‐1 expression (brown staining with haematoxylin counterstain) in the synovial tissues from rheumatoid arthritis (RA) patients, osteoarthritis patients and healthy individuals. IgG isotype served as the negative control in the staining; ×400 magnification.
Co‐localization of LAIR‐1 with the fibroblast marker, vimentin
As FLS play a crucial role in synovial pathophysiology in RA, we investigated the expression of LAIR‐1 on the surfaces of synovial FLS in situ in patients with active RA. Consecutive sections were used to detect LAIR‐1 and vimentin by immunohistochemistry. Vimentin is a marker of rheumatoid synovial fibroblasts. LAIR‐1 was expressed highly in the synovial lining and moderately in the underlying layer adjacent to the lining (Fig. 2a). Furthermore, using immunofluorescence staining, we observed a high correlation between the co‐localization of vimentin and LAIR‐1 in the samples from the RA patients (Fig. 2b). The expression of the collagenase, MMP‐13 is acknowledged widely to be also localized in the RA lining, and this is highly relevant for studying synovial invasion and cartilage degradation. Furthermore, double immunofluorescence staining of CD45 and LAIR‐1 was performed, and the results demonstrated that LAIR‐1 was also present in a few leucocytes that infiltrated into the synovial tissues. Taken together, the elevated expression of LAIR‐1 in the synovial lining supports the potential contribution of LAIR‐1 to the progression of RA.
Figure 2.
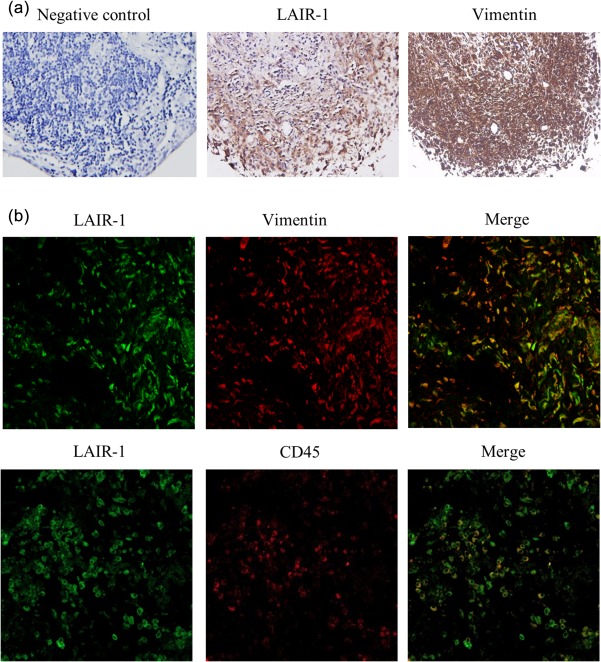
Immunolabelling of leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) and vimentin in human rheumatoid arthritis (RA) synovial tissue. (a) Consecutive synovial sections from the RA patients were used to detect LAIR‐1 and vimentin. Levels of LAIR‐1 expression were higher in the synovial lining than the sublining layer. IgG isotype served as the negative control in the staining; ×400 magnification. (b) Co‐localization of LAIR‐1/vimentin and LAIR‐1/CD45 in the synovial lining was detected by immunofluorescence staining and confocal microscopy; ×400 magnification. These results were confirmed by examining synovial membrane specimens from six RA patients. Immunolabelling of a representative sample is shown.
LAIR‐1 was expressed mainly in the cytoplasm of FLS in the RA tissues
We isolated primary FLS from the RA patients to evaluate LAIR‐1 expression. As shown in Fig. 3a, compared with the PBMC, the mRNA level of LAIR‐1 in FLS was significantly low, indicating that the expression level of LAIR‐1 mRNA was higher in the haematopoietic cells than the cells of mesenchymal origin. There was variation in the surface expression of LAIR‐1 among donors, as analysed by flow cytometry. Among the 11 primary FLS samples isolated from RA patients, two had a positive cell subpopulation (5·9 and 6·1%), whereas the other nine patients had a peak shift rather than a positive subpopulation. The percentage of FLS that stained positive for LAIR‐1 (5·2 ± 2·5%) was not high (Fig. 3b left). Higher levels of LAIR‐1 expression were observed in the cells of the RA synovial tissues; however, the levels of cell‐surface LAIR‐1 were low. This result is not completely consistent with that of the IHC analysis of the RA synovial tissues. Therefore, intracellular staining for LAIR‐1 was performed. A significantly higher percentage of LAIR‐1 positive FLS, calculated as 17·7 ± 3·1%, was observed (Fig. 3b, right).
Figure 3.
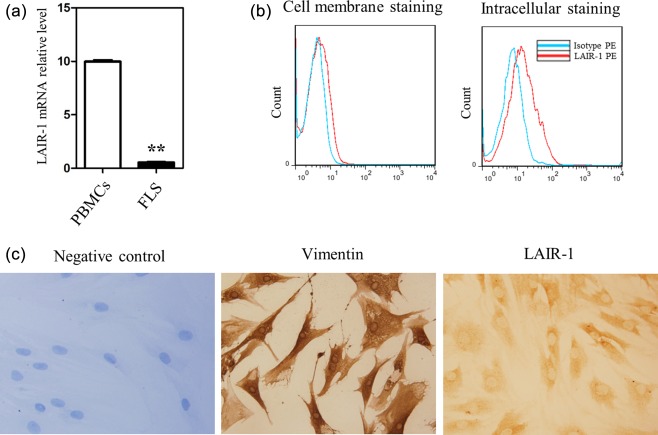
Expression of leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) in primary fibroblast‐like synoviocytes (FLS) isolated from the rheumatoid arthritis (RA) patients. (a) Levels of LAIR‐1 mRNA in normal human peripheral peripheral blood mononuclear cells (PBMC) (n = 3) and FLS (n = 5) as shown by quantitative polymerase chain reaction (qPCR). Reaction without template cDNA served as the negative control, and showed no melting curve. **P < 0·01 versus the PBMC group. (b) Flow cytometric analysis of the LAIR‐1‐positive FLS populations from the RA patients (n = 11). Intracellular staining and flow cytometric analysis of LAIR‐1 were also performed with phycoerythrin (PE)‐labelled monoclonal antibodies (mAbs) (n = 5). Representative flow cytometry graphs are shown. (c) Immunohistochemical images show the expression of LAIR‐1 and vimentin in the primary FLS isolated from the RA patients. IgG isotype‐stained sections did not stain positively; ×400 magnification. Data are representative of FLS from six different RA patients.
Furthermore, we performed IHC staining for LAIR‐1 in the primary FLS isolated from the RA patients. The cells were also stained with anti‐vimentin antibodies, which were enriched in isolated FLS (> 99%). Interestingly, LAIR‐1 was localized mainly in the cytoplasm of FLS from the RA patients, suggesting that LAIR‐1 was translated and retained in these cells (Fig. 3b).
Effect of LAIR‐1 on the proliferation and invasion of FLS from the RA patients
MH7A is a transformed human synovial cell line derived from the intra‐articular soft tissue of the knee joint of an RA patient 13. Few MH7A cells expressed LAIR‐1 (0·6 ± 0·2%). To study its function in MH7A cells, we over‐expressed LAIR‐1 using lentiviral vectors (LV‐LAIR‐1). The efficiency of the lentiviral infection was evaluated by counting GFP‐positive cells under a fluorescence microscope at 48 h post‐infection (Supporting information, Fig. S1a). Next, qPCR was performed to evaluate the levels of LAIR‐1 mRNA in the MH7A cells (Supporting information, Fig. S1b). EdU assays showed that enhanced LAIR‐1 expression did not inhibit MH7A cell proliferation (Fig. 4a,b). Next, we next examined the effect of LAIR‐1 on the invasive activity of FLS. After lentiviral LAIR‐1 over‐expression and TNF‐α stimulation (10 ng/ml), the number of cells invading the basal membrane decreased by 60% compared with the LV control group (Fig. 4c,d). These results indicated that, induced by TNF‐α, LAIR‐1 might contribute to the invasive behaviour of the cells.
Figure 4.
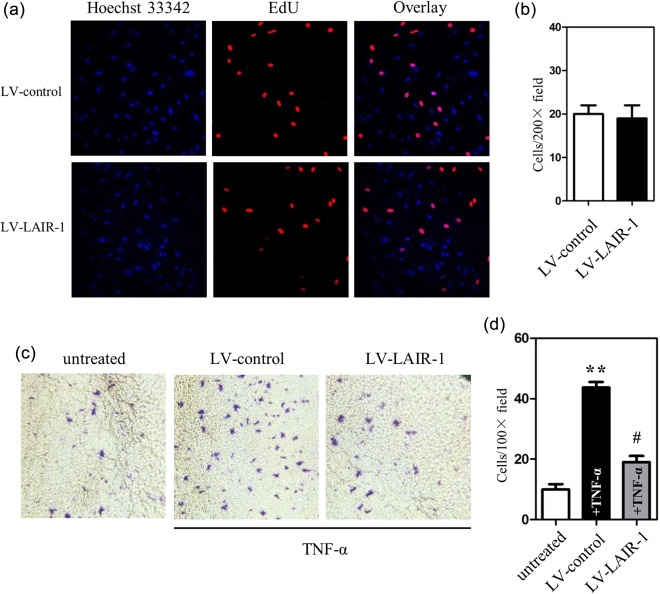
Role of leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) in the modulation of the cell proliferation and invasion of fibroblast‐like synoviocytes (FLS). (a) MH7A cells at a density of 5 × 103 cells per well were cultured in 96‐well plates and transfected with either a negative control lentiviral vector (LV‐control) or the LAIR‐1 overexpression lentiviral vector (LV‐LAIR‐1) for 48 h. Cells were then exposed to 5‐ethynyl‐2'‐deoxyuridine (EdU) (50 μM) for 4 h, followed by staining with the Apollo reaction cocktail for 30 min. EdU‐labelled proliferating cells were examined by fluorescence microscopy. Data shown are representative images of individual groups (n = 6 per group) from three independent experiments; ×200 magnification. (b) Cell numbers per microscopic field were plotted in the histogram. (c) LV‐LAIR‐1 was used to study the role of LAIR‐1 in tumour necrosis factor (TNF)‐α‐induced FLS invasion. Photomicrographs show cells that have passed through the membrane. MH7A cells at a density of 4 × 104 cells per well were cultured in a 24‐well Transwell insert. After infection with LV‐LAIR‐1 or LV‐control for 24 h, cells were treated with TNF‐α. FLS were allowed to invade for 24 h; ×200 magnification. (d) Cell number per microscopic field was plotted in the histogram. **P < 0·01 versus the untreated control group. #P < 0·05 versus TNF‐α‐treated LV control cells.
Down‐regulated expression of cell‐surface LAIR‐1 in the presence of TNF‐α
LAIR‐1 and LAIR‐2 are biomarkers of inflammation 14. The levels of soluble LAIR‐1/CD305 and LAIR‐2/CD306 were higher in the RA patients than the healthy subjects 9, 15. Quantification of the levels of soluble LAIR‐1 and LAIR‐2 in RA patients may help to monitor the disease progression. However, the origin of soluble LAIR‐1 is still unclear.
A key inflammatory event in RA is the TNF‐α overproduction 16. This pathway drives both synovial inflammation and joint destruction. In Fig. 5a, the flow cytometric analysis of the MH7A cells infected with LV‐LAIR‐1 revealed that 35·0 ± 1·3% of the cells were positive for LAIR‐1 expression without any other treatment (blank group, mean fluorescence intensity = 109·5 ± 8·5). TNF‐α treatment (10 ng/ml) decreased the proportion of the cells expressing LAIR‐1 significantly (22·7 ± 1·1%, mean fluorescence intensity = 95·6 ± 4·8); however, IL‐6 (10 ng/ml) did not have any effect (34·8 ± 1·0%, mean fluorescence intensity = 106·6 ± 5·5). A time–course analysis revealed that cell‐surface LAIR‐1 in the MH7A cells could be shed to approximately 30% at 6 h after TNF‐α‐stimulation (Supporting information, Fig. S2). Using cultures of primary FLS isolated from RA patients infected with LV‐LAIR‐1, we observed that these cells also exhibited a significant decrease in mLAIR‐1 levels after treatment with 10 ng/ml TNF‐α under the same conditions (Fig. 5c,d). TNF‐α treatment decreased the proportion of cells expressing LAIR‐1 significantly from 57·2 ± 3·8% (mean fluorescence intensity = 44·2 ± 1·6) to 17·8 ± 1·5% (mean fluorescence intensity = 20·5 ± 1·8).
Figure 5.
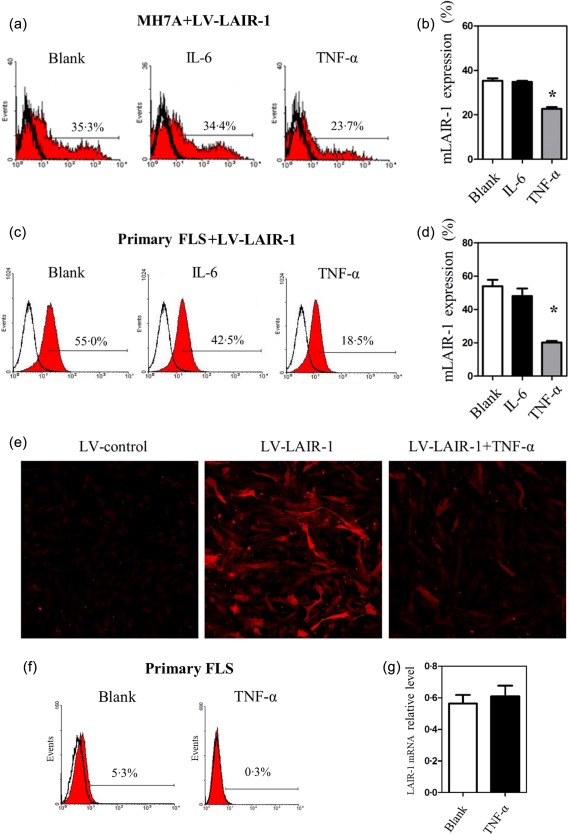
Tumour necrosis factor (TNF) decreased leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) expression significantly at the cell surface. (a,b) Flow cytometric analysis and statistics of the MH7A cells infected with lentiviral vector (LV)‐LAIR‐1 for 24 h. MH7A cells were then treated with IL‐6 or TNF‐α (10 ng/ml) for another 24 h. A blank group served as the control. *P < 0·05 versus the blank group and data were obtained from three independent experiments. (c,d) Flow cytometric analysis and statistics of primary fibroblast‐like synoviocytes (FLS) from rheumatoid arthritis (RA) patients infected with LV‐LAIR‐1 (n = 3). FLS were treated with the same conditions as the MH7A cells. (e) Images obtained by laser scanning confocal microscopy showing LAIR‐1 expression (red). MH7A cells were infected with the LV‐control or LV‐LAIR‐1 lentiviral vector. LV‐LAIR‐1‐infected cells were then stimulated in the presence or absence of TNF‐α (10 ng/ml) for 24 h; ×100 magnification. (f) Flow cytometric analysis of cell‐surface LAIR‐1 expression in primary FLS from RA patients (n = 3). Cells were treated with TNF‐α (10 ng/ml) for 24 h, and a blank group was included as the control. (g) Primary FLS from RA patients treated with TNF‐α for 24 h were collected, and then RNA was extracted for qPCR analysis, and a blank group was included as the control (n = 3).
LAIR‐1 expression in the presence or absence of TNF‐α was examined further in the LV‐LAIR‐1‐transfected MH7A cells by laser scanning confocal microscopy. Compared with the LV‐control lentiviral vector‐transfected cells, significantly increased LAIR‐1 expression was observed in the LV‐LAIR‐1‐transfected MH7A cells. However, LAIR‐1expression in the LV‐LAIR‐1‐transfected MH7A cells was down‐regulated significantly by the treatment with 10 ng/ml TNF‐α for 24 h (Fig. 5e).
The expression levels of cell surface LAIR‐1 in primary FLS was determined by flow cytometry analysis with or without TNF‐α stimulation, and qPCR analysis was used to assess the mRNA levels of LAIR‐1 in primary FLS. The flow cytometry results showed that although the level of cell surface LAIR‐1 was low, it could still be shed following TNF‐α treatment (Fig. 5f). The qPCR results revealed that the LAIR‐1 mRNA levels in primary FLS with and without TNF‐α treatment did not differ significantly (Fig. 5g). This suggested that although TNF‐α had no effect on the mRNA level or endogenous expression of LAIR‐1, it can be shed from the cell membrane, releasing a soluble form, under TNF‐α stimulation in primary FLS.
Shedding of the LAIR‐1 receptor is inhibited by a serine protease inhibitor
To identify enzymes that are involved in the production of soluble LAIR‐1, we tested a collection of protease inhibitors for their ability to block the shedding of the membrane‐bound form selectively (mLAIR‐1) following TNF‐α stimulation. In these inhibitors, group 1 included 6‐aminocaproic acid (5 mg/ml), which is reported to inhibit Factor VIIa, chymotrypsin, lysine carboxypeptidase, plasmin and plasminogen activator. Soybean trypsin inhibitor (1 : 1 stoichiometric binding) inhibits trypsin; E‐64 (10 μM) is a highly selective cysteine protease inhibitor. Bestatin (40 μM) is a specific inhibitor of aminopeptidase B, leucine aminopeptidase and triaminopeptidase. Group 2 included phosphoramidon (10 μM), which is a strong inhibitor of many bacterial metalloendoproteinases, thermolysin and elastase; antipain hydrochloride (10 μM) is an inhibitor of serine/cysteine proteases; aprotinin (500 nM) is a competitive serine protease inhibitor. Group 3 included leupeptin (50 μM), which is a reversible inhibitor of serine and thiol proteases. Pepstatin A (1 μg/ml) is an inhibitor of acid proteases. Group 4 included chymostatin (50 μM), which is an inhibitor of several proteases.
Of these four groups, group 1 protease inhibitors had no effect on TNF‐α‐induced mLAIR‐1 shedding. Group 2 protease inhibitors partly reversed TNF‐α‐induced mLAIR‐1 shedding, which included the serine protease inhibitors aprotinin and antipain. Group 3 protease inhibitors blocked mLAIR‐1 shedding from FLS totally, including the serine protease inhibitor leupeptin. Finally, group 4 protease inhibitor, chymostatin, increased mLAIR‐1 expression (Fig. 6). This is because chymostatin is a strong inhibitor for many proteases, including chymotrypsin‐like serine proteases. Cumulatively, these results suggest that serine and acid proteases may contribute to inflammation‐induced mLAIR‐1 shedding.
Figure 6.
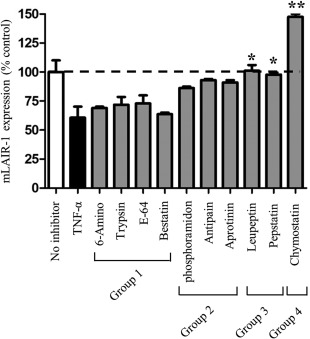
Effects of protease inhibitors on the shedding of the tumour necrosis factor (TNF)‐induced membrane‐bound form of leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (mLAIR‐1). MH7A cells pretreated with the lentiviral vector (LV)‐LAR‐1 lentiviral vector were stimulated with TNF‐α alone or in combination with various protease inhibitors for 24 h. The membrane‐bound form of LAIR‐1 was quantified by flow cytometry. Data are shown as the percentage of mLAIR‐1 expression compared to control cells (no inhibitor). Group 1: no effect; group 2: partly inhibited; group 3: totally inhibited; group 4: increased expression. Data are representative of three independent experiments. Statistical differences in mLAIR‐1 expression between groups treated with TNF‐α only and those treated in combination with the indicated inhibitors are indicated. *P < 0·05; **P < 0·01.
LAIR‐1 inhibited TNF‐α‐induced expression of IL‐6 and IL‐8 in FLS
TNF‐α plays an important role in transforming FLS in RA into cells with an invasive phenotype. Therefore, we examined the expression of inflammation‐related cytokines in MH7A cells in the presence or absence of TNF‐α by qPCR. Untreated MH7A cells expressed only low levels of IL‐6, IL‐8 and COX‐2. Inflammatory cytokine levels were not affected by the LV‐LAIR‐1 transfection. Compared with the untreated controls, the TNF‐α treatment increased the mRNA levels of IL‐6, IL‐8 and COX‐2 significantly, while no significant changes in the expression levels of tissue inhibitor of metallopeptidase‐1 (TIMP‐1) or PTGES2 were observed after the TNF‐α treatment.
IL‐6 and IL‐8 drive persistent inflammation and joint destruction 17. Transfection with the LV‐LAIR‐1 lentiviral vector reduced TNF‐α‐induced IL‐6 expression significantly (2·33 ± 0·71 versus· 0·89 ± 0·49; P < 0·05). As with IL‐6, IL‐8 expression also decreased significantly owing to the LV‐LAIR‐1 lentiviral vector transfection (2·00 ± 0·49 versus 1·14 ± 0·24; P < 0·05).
LAIR‐1 inhibited TNF‐α‐induced MMP‐13 expression in FLS
FLS play critical roles in RA by synthesizing proteinases, such as MMPs, that cause joint destruction. Among these MMPs, gelatinases (MMP‐2 and MMP‐9) play a leading role in the cleavage of proteins in the ECM 18. MMP‐13 is the precursor of MMP‐3. It is noteworthy that MMP‐13 regulation by the collagen matrix in FLS in the RA patients is a critical event in progressive joint destruction 11. The untreated MH7A cells produced negligible MMP‐1 quantities, apart from small quantities of MMP‐13, MMP‐2, MMP‐9 and MMP‐3. Compared with the untreated controls, TNF‐α increased the mRNA levels of MMP‐3, the gelatinase MMP‐9 and the collagenases MMP‐1 and MMP‐13 significantly (Fig. 7). Interestingly, LV‐LAIR‐1 lentiviral‐vector transfection reduced TNF‐α‐induced MMP‐13 expression significantly (2·90 ± 0·58 versus·1·14 ± 0·24; P < 0·05).
Figure 7.
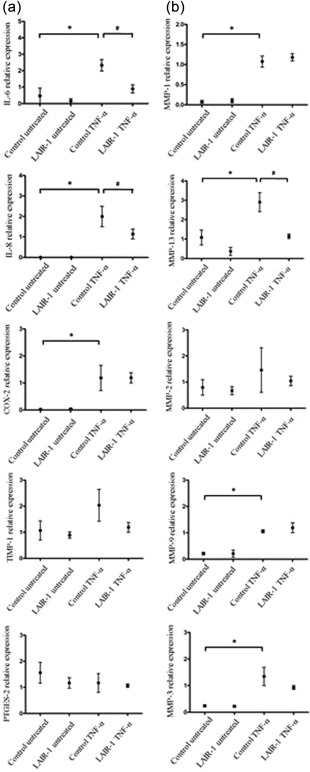
Effects of lentiviral leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) expression on the tumour necrosis factor (TNF)‐induced inflammatory factors and matrix metalloproteinases (MMPs) in fibroblast‐like synoviocytes (FLS). MH7A cells were subjected to quantitative polymerase chain reaction (qPCR) for determining mRNA levels of the inflammatory factors (a) and MMPs (b). Control cells were incubated with the media alone (untreated control and untreated LAIR‐1 groups). Lentiviral vector (LV)‐control or LV‐LAIR‐1 lentiviral vector‐infected MH7A cells were incubated with 10 ng/ml TNF‐α in 1% serum‐free media for 24 h (control TNF‐α and TNF‐α‐induced LAIR‐1 groups). Relative mRNA levels were normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) *P < 0·05 versus untreated control cells, #P < 0·05 versus control TNF‐α cells. Data were obtained from three independent experiments.
In the microenvironment of synovial fluid and tissues in rheumatoid arthritis, excess inflammatory factors and over‐activated proteases may cause the shedding of cell‐membrane LAIR‐1, and thus give rise to low LAIR‐1 level, decrease the interaction of LAIR‐1 with its ligand and inhibit the inhibitory cellular signal transduction of LAIR‐1. As we have mentioned that collagen is a functional, high‐affinity ligand for LAIR‐1, we consider that it is reasonable that LAIR‐1 may be expressed on FLS, and function as a modulator in the extracellular matrix‐FLS interaction in RA. Anti‐inflammatory and anti‐protease drugs may activate the anti‐inflammatory signal by maintaining normal LAIR‐1 function.
Furthermore, we introduced lentiviral LV‐LAIR‐1 constructs with mutations in both the ITIMs into MH7A cells, and observed the effect of ITIMs on anti‐inflammation. We observed that the loss of ITIMs in the cytoplasmic region diminished the anti‐inflammatory effect of LAIR‐1 on FLS, and the results are illustrated in Supporting information, Fig. S3.
Discussion
LAIR‐1 is an inhibitory receptor that is expressed in the majority of mononuclear leucocytes. Recent studies have suggested that LAIR‐1 has anti‐inflammatory effects. However, it is unclear whether LAIR‐1 is expressed in FLS in RA patients. In this study, we demonstrated LAIR‐1 expression in synovial tissues and FLS using immunohistochemistry and flow cytometry. LAIR‐1 was up‐regulated in the synovial tissues of the RA patients, and localized mainly in FLS. However, mLAIR‐1 levels in the primary FLS isolated from RA patients were low. We also observed intracellular LAIR‐1 retention by immunohistochemical staining, suggesting that mLAIR‐1 may be shed from the cell surface under pathological conditions. In cultured FLS from the RA patients, we observed that the levels of LAIR‐1 expression were down‐regulated by TNF‐α. Furthermore, LAIR‐1 over‐expression regulated the TNF‐α‐induced invasive behaviour of FLS negatively.
LAIR‐1 is a useful biomarker for RA in synovial fluid. LAIR‐1 is found as both the membrane‐bound form (mLAIR‐1) and the soluble form (sLAIR‐1). Olde Nordkamp et al. showed that urinary levels of sLAIR‐1 were significantly higher in RA patients compared to healthy controls 9. Additionally, sLAIR‐1 and LAIR‐2 levels in urine correlated significantly with markers of inflammation. We also found that mLAIR‐1 expression was decreased significantly in circulating CD4+ T cells in RA patients compared to both OA patients and healthy individuals. In contrast, mLAIR‐1 was elevated in CD14+ monocytes and local CD68+ macrophages in synovial tissues from RA patients compared to healthy patients. Consistent with previous studies, we found that mLAIR‐1 was shed from cells following stimulation with TNF‐α. This may provide the source of the increased soluble LAIR‐1 present in RA patients. This study is the first, to our knowledge, to show a mechanism for mLAIR‐1 shedding from FLS under the inflammatory conditions associated with RA.
The regulatory roles of the soluble form of the receptor have been researched extensively in recent years 19. Soluble forms of receptors of cytokines and growth factors are generated either by differential mRNA splicing or ectodomain shedding 20. Soluble forms of receptors can function as antagonists or agonists 21. Soluble LAIR‐1 may be released from cells, bind to a ligand and thereby prevent signalling via full‐length receptors. However, it is still unknown how membrane‐bound interacts with the ligand collagen compared to sLAIR‐1, and how this affects cellular responses. Future studies should explore the function of sLAIR‐1 in collagen‐activating signalling and determine whether it is dysregulated in patients with RA.
An increase in TNF‐α levels have been observed in the synovial fluid and synovium of patients with RA, and is produced mainly by macrophage‐like synoviocytes and FLS in RA synovitis joints 22. Accumulating evidence demonstrates that proinflammatory cytokines play pivotal roles in the physiopathology of RA. Furthermore, TNF inhibitors and an anti‐IL‐6 receptor monoclonal antibody have shown significant efficacy in RA treatments 23, 24. RA joints also display activation of the PGE2 pathway, and there are high levels of this mediator in the synovial fluid. The enzymes that synthesize PGE2 (PTGES, COX‐1 and COX‐2) are also present in the synovium. COX‐1 is a constitutively expressed enzyme present under normal conditions, while COX‐2 is induced by inflammation and co‐localizes with PTGES in synovial tissue 25. In the present study, the in‐vitro effect of LAIR‐1 on proinflammatory cytokine production was investigated in FLS of RA patients. Over‐expression of LAIR‐1 decreased expression of IL‐6 and IL‐8 mRNA in FLS significantly in the presence of TNF‐α, but this had no effect on COX‐2 and PTGES2.
In RA, the MMP family promotes the invasiveness of FLS and the erosion of cartilage 11, 26. MMP‐13 is of special interest with regard to cartilage degradation due to its preference for type II collagen in articular cartilage 27. Here, we demonstrated that MMP‐13 mRNA in synovial fibroblasts of RA patients was induced by the inflammatory factor, TNF‐α. Furthermore, over‐expression of wild‐type LAIR‐1 in human synoviocytes decreased the levels of MMP‐13 dramatically. Thus, LAIR‐1 may inhibit TNF‐α‐induced rheumatoid FLS invasion through suppression of IL‐6, IL‐8 and MMP‐13 expression.
Collagen is a key component of the extracellular matrix. It provides essential structural support for connective tissues, and it is also involved intimately in controlling cell growth, differentiation and morphogenesis 28. Transmembrane receptors include integrins, glycoprotein VI, discoidin domain receptors (DDRs), and LAIR‐1 mediate the recognition of the collagen triple helix. A collagen receptor, DDR2, has been linked to both OA and RA 11, 29. Another receptor bearing an immunoglobulin‐like motif, osteoclast‐associated receptor (OSCAR), serves not only an essential role in osteoclastogenesis, but it has also been demonstrated recently that collagen type I (ColI) and collagen type II (ColII) are ligands for OSCAR. An OSCAR–collagen interaction in monocytes plays a proinflammatory role in monocyte‐derived cells and may contribute on multiple levels to RA pathogenesis 30. Collagens function as inhibitory ligands of LAIR‐1 in in‐vitro assays. LAIR‐1 binds to several different collagen types, including fibrillary collagens I–III, collagen IV and certain transmembrane collagens 7. Most notably, the LAIR‐1‐collagen interaction may play a pivotal role in pathological processes in RA patients.
Altogether, TNF‐α mediated the shedding of mLAIR‐1 from the cell surface, and down‐regulated mLAIR‐1 expression. This increased sLAIR‐1 levels in the RA patients. The in‐vitro experiments also demonstrated that high LAIR‐1 expression suppressed the production of proinflammatory cytokines and MMPs in FLS. Future studies should explore thoroughly the cellular events downstream of LAIR‐1.
Disclosure
None.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Analysis of the effect of lentiviral vector (LV)‐leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) on MH7A cells. (a) Bright‐field and fluorescence micrographs (green) showing the efficiency of LV‐LAIR‐1 transfection. (b) LAIR‐1 overexpression in the MH7A cells was confirmed by quantitative polymerase chain reaction (qPCR). LAIR‐1 levels increased significantly in the MH7A cells. *P < 0·05.
Fig. S2. Time–course of levels of membrane‐bound leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) upon tumour necrosis factor (TNF)‐α induction (10 ng/ml) in MH7A cells over‐expressing LV‐LAIR‐1. Data are representative of three independent experiments.
Fig. S3. Effect of lentiviral vector (LV)‐leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) mutation in cytoplasmic immunoreceptor tyrosine‐based inhibitory motif (ITIM) on the production of the inflammatory factors, interleukin (IL)‐6 and matrix metalloproteinase (MMP)‐13, in MH7A cells. (a) LV‐LAIR‐1‐Mut infection in the MH7A cells was confirmed by quantitative polymerase chain reaction (qPCR). LAIR‐1 levels increased significantly in the MH7A cells. *P < 0·05 versus LV‐control. (b) LAIR‐1‐ITIM mutant exerted diminished anti‐inflammatory effects on MH7A cells in the presence of tumour necrosis factor (TNF)‐α. *P < 0·05 versus control. Data were obtained from three independent experiments.
Acknowledgements
We are grateful to Professor Boquan Jin (Department of Immunology, the Fourth Military Medical University) for providing useful critical comments. This work was supported by the Major Research Plan of National Natural Science Foundation of China (no. 91442105) and the National Natural Science Foundation of China (no. 81371926, no. 81370388, no. 81100002 and no. 81671575).
Contributor Information
R. Zhuang, Email: fmmuzhr@fmmu.edu.cn
Y. Ding, Email: dinyonza@fmmu.edu.cn
References
- 1. Gui H, Liu X, Wang ZW, He DY, Su DF, Dai SM. Expression of cannabinoid receptor 2 and its inhibitory effects on synovial fibroblasts in rheumatoid arthritis. Rheumatology (Oxf) 2014; 53:802–9. [DOI] [PubMed] [Google Scholar]
- 2. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010; 376:1094–108. [DOI] [PubMed] [Google Scholar]
- 3. Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov 2003; 2:473–88. [DOI] [PubMed] [Google Scholar]
- 4. Leech MT, Morand E. Fibroblasts and synovial immunity. Curr Opin Pharmacol 2013; 13:565–9. [DOI] [PubMed] [Google Scholar]
- 5. Meyaard L. The inhibitory collagen receptor LAIR‐1 (CD305). J Leukoc Biol 2008; 83:799–803. [DOI] [PubMed] [Google Scholar]
- 6. Meyaard L, Adema GJ, Chang C, Woollatt E, Sutherland GR. LAIR‐1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity 1997; 7:283–90. [DOI] [PubMed] [Google Scholar]
- 7. Lebbink RJ, de Ruiter T, Adelmeijer J et al Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR‐1. J Exp Med 2006; 203:1419–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y, Lv K, Zhang CM, Jin BQ, Zhuang R, Ding Y. The role of LAIR‐1 (CD305) in T cells and monocytes/macrophages in patients with rheumatoid arthritis. Cell Immunol 2014; 287:46–52. [DOI] [PubMed] [Google Scholar]
- 9. Olde Nordkamp MJ, van Roon JA, Douwes M, de Ruiter T, Urbanus RT, Meyaard L. Enhanced secretion of leukocyte‐associated immunoglobulin‐like receptor 2 (LAIR‐2) and soluble LAIR‐1 in rheumatoid arthritis: LAIR‐2 is a more efficient antagonist of the LAIR‐1‐collagen inhibitory interaction than is soluble LAIR‐1. Arthritis Rheum 2011; 63:3749–57. [DOI] [PubMed] [Google Scholar]
- 10. Arnett FC, Edworthy SM, Bloch DA et al The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988; 31:315–24. [DOI] [PubMed] [Google Scholar]
- 11. Su J, Yu J, Ren T et al Discoidin domain receptor 2 is associated with the increased expression of matrix metalloproteinase‐13 in synovial fibroblasts of rheumatoid arthritis. Mol Cell Biochem 2009; 330:141–52. [DOI] [PubMed] [Google Scholar]
- 12. Ouyang W, Ma D, Lin D et al 9.1C3 is identical to LAIR‐1, which is expressed on hematopoietic progenitors. Biochem Biophys Res Commun 2003; 310:1236–40. [DOI] [PubMed] [Google Scholar]
- 13. Miyazawa K, Mori A, Okudaira H. Establishment and characterization of a novel human rheumatoid fibroblast‐like synoviocyte line, MH7A, immortalized with SV40 T antigen. J Biochem 1998; 124:1153–62. [DOI] [PubMed] [Google Scholar]
- 14. Lebbink RJ, van den Berg MC, de Ruiter T et al The soluble leukocyte‐associated Ig‐like receptor (LAIR)‐2 antagonizes the collagen/LAIR‐1 inhibitory immune interaction. J Immunol 2008; 180:1662–9. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y, Ding Y, Huang Y, Zhang C, Boquan J, Ran Z. Expression of leukocyte‐associated immunoglobulin‐like receptor‐1 (LAIR‐1) on osteoclasts and its potential role in rheumatoid arthritis. Clinics (São Paulo) 2013; 68:475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feldmann M, Brennan F, Maini RN. Rheumatoid arthritis. Cell 1996; 85:307–10. [DOI] [PubMed] [Google Scholar]
- 17. Choy EH, Isenberg D, Garrood T et al Therapeutic benefit of blocking interleukin‐6 activity with an anti‐interleukin‐6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double‐blind, placebo‐controlled, dose‐escalation trial. Arthritis Rheum 2002; 46:3143–50. [DOI] [PubMed] [Google Scholar]
- 18. Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol 2008; 40:1362–78. [DOI] [PubMed] [Google Scholar]
- 19. Chockalingam A, Cameron J, Brooks JC, Leifer CA. Negative regulation of signaling by a soluble form of toll‐like receptor 9. Eur J Immunol 2011; 41:2176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sugaya S, Sakimoto T, Shoji J, Sawa M. Regulation of soluble interleukin‐6 (IL‐6) receptor release from corneal epithelial cells and its role in the ocular surface. Jpn J Ophthalmol 2011; 55:277–82. [DOI] [PubMed] [Google Scholar]
- 21. Monti P, Brigatti C, Krasmann M, Ziegler AG, Bonifacio E. Concentration and activity of the soluble form of the interleukin‐7 receptor α in type 1 diabetes identifies an interplay between hyperglycemia and immune function. Diabetes 2013; 62:2500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di Giovine FS, Nuki G, Duff GW. Tumour necrosis factor in synovial exudates. Ann Rheum Dis 1988; 47:768–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ogata A, Hirano T, Hishitani Y, Tanaka T. Safety and efficacy of tocilizumab for the treatment of rheumatoid arthritis. Clin Med Insights Arthritis Musculoskelet Disord 2012; 5:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti‐TNF therapy: past, present and future. Int Immunol 2015; 27:55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gheorghe KR, Sadique S, Leclerc P et al Limited effect of anti‐rheumatic treatment on 15‐prostaglandin dehydrogenase in rheumatoid arthritis synovial tissue. Arthritis Res Ther 2012; 14:R121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tolboom TC, Pieterman E, van der Laan WH et al Invasive properties of fibroblast‐like synoviocytes: correlation with growth characteristics and expression of MMP‐1, MMP‐3, and MMP‐10. Ann Rheum Dis 2002; 61:975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitchell PG, Magna H, Reeves LM et al Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase‐13 from human osteoarthritic cartilage. J Clin Invest 1996; 97:761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leitinger B. Transmembrane collagen receptors. Annu Rev Cell Dev Biol 2011; 27:265–90. [DOI] [PubMed] [Google Scholar]
- 29. Xu L, Peng H, Glasson S et al Increased expression of the collagen receptor discoidin domain receptor 2 in articular cartilage as a key event in the pathogenesis of osteoarthritis. Arthritis Rheum 2007; 56:2663–73. [DOI] [PubMed] [Google Scholar]
- 30. Schultz HS, Guo L, Keller P et al OSCAR‐collagen signaling in monocytes plays a proinflammatory role and may contribute to the pathogenesis of rheumatoid arthritis. Eur J Immunol 2016; 46:952–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Analysis of the effect of lentiviral vector (LV)‐leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) on MH7A cells. (a) Bright‐field and fluorescence micrographs (green) showing the efficiency of LV‐LAIR‐1 transfection. (b) LAIR‐1 overexpression in the MH7A cells was confirmed by quantitative polymerase chain reaction (qPCR). LAIR‐1 levels increased significantly in the MH7A cells. *P < 0·05.
Fig. S2. Time–course of levels of membrane‐bound leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) upon tumour necrosis factor (TNF)‐α induction (10 ng/ml) in MH7A cells over‐expressing LV‐LAIR‐1. Data are representative of three independent experiments.
Fig. S3. Effect of lentiviral vector (LV)‐leucocyte‐associated immunoglobulin (Ig)‐like receptor‐1 (LAIR‐1) mutation in cytoplasmic immunoreceptor tyrosine‐based inhibitory motif (ITIM) on the production of the inflammatory factors, interleukin (IL)‐6 and matrix metalloproteinase (MMP)‐13, in MH7A cells. (a) LV‐LAIR‐1‐Mut infection in the MH7A cells was confirmed by quantitative polymerase chain reaction (qPCR). LAIR‐1 levels increased significantly in the MH7A cells. *P < 0·05 versus LV‐control. (b) LAIR‐1‐ITIM mutant exerted diminished anti‐inflammatory effects on MH7A cells in the presence of tumour necrosis factor (TNF)‐α. *P < 0·05 versus control. Data were obtained from three independent experiments.