

Summary
Systemic lupus erythematosus (SLE) patients are susceptible to the development of posterior reversible encephalopathy syndrome (PRES). The main theory concerning the physiopathology of PRES suggests that there is brain–blood barrier damage, which is associated with endothelial dysfunction, and characterized by vasogenic oedema. However, current evidence regarding its physiopathogenic mechanisms is quite scant. The aim of this study was to analyse the expression of different serum cytokines, as well as vascular endothelial growth factor (VEGF) and soluble CD40 ligand (sCD40L), in patients with PRES/systemic lupus erythematosus (SLE) and to compare them with levels in SLE patients without PRES and in healthy controls. We performed a transversal study in a tertiary care centre in México City. We included 32 subjects (healthy controls, n = 6; remission SLE, n = 6; active SLE, n = 6 and PRES/SLE patients, n = 14). PRES was defined as reversible neurological manifestations (seizures, visual abnormalities, acute confusional state), associated with compatible changes by magnetic resonance imaging (MRI). Serum samples were obtained during the first 36 h after the PRES episode and were analysed by cytometric bead array, Luminex multiplex assay or enzyme‐linked immunosorbent assay (ELISA). Interleukin (IL)‐6 and IL‐10 levels were significantly higher in PRES/SLE patients (P = 0·013 and 0·025, respectively) when compared to the other groups. Furthermore, IL‐6 and IL‐10 levels displayed a positive correlation (r = 0·686, P = 0·007). There were no differences among groups regarding other cytokines, sCD40L or VEGF levels. A differential serum cytokine profile was found in PRES/SLE patients, with increased IL‐6 and IL‐10 levels. Our findings, which are similar to those described in other neurological manifestations of SLE, support the fact that PRES should be considered among the SLE‐associated neuropsychiatric syndromes.
Keywords: IL‐6, IL‐10, posterior reversible encephalopathy, systemic lupus erythematosus
Introduction
Posterior reversible encephalopathy syndrome (PRES) is a clinical–radiological entity, described in 1996 1, characterized by seizures, headache, confusional state and transient visual deficits, associated generally with high blood pressure and specific magnetic resonance imaging (MRI) findings [bilateral and asymmetrical isointensities or hypointensities in T1, hyperintensities in T2 and fluid attenuation inversion recovery (FLAIR) sequences in the parieto‐temporo‐occipital regions] 2. Even though PRES is associated with multiple diseases, its clinical prognosis and radiological outcome is good in both the short and long term, and recurrences are rare 3; however, its potential implications in cognitive function or other clinical outcomes have not been addressed. Different entities have been associated with PRES; among autoimmune diseases, systemic lupus erythematosus (SLE) is the most common, although it is important to emphasize that its prevalence is low (< 1%) 4. However, it should also be considered that PRES could be underdiagnosed in these patients. Up to 50% of SLE patients develop neuropsychiatric manifestations throughout their disease 5, but currently, PRES is not considered as one of the 19 SLE‐associated neuropsychiatric syndromes.
There are different hypotheses regarding the physiopathogenic mechanisms that lead to PRES [i.e. blood–brain barrier disruption, loss of central nervous system (CNS) autoregulation mechanisms, endothelial dysfunction] 2, 6, 7, 8. Regarding the latter, our group recently described the main independent risk factors associated with the development of PRES in SLE patients, including lymphopenia and dyslipidaemia, which could contribute to endothelial dysfunction 9. Also, it has been proposed that, in SLE subjects, PRES can be associated either with blood pressure dysregulation or with an immune‐mediated aetiology 10. However, evidence supporting the potential mechanisms associated with PRES is quite scarce. Therefore, we decided to analyse immunological abnormalities that could be part of the physiopathogenic scheme of PRES in SLE patients. The aim of this study was to address whether PRES/SLE patients show a particular serum cytokine profile. We also specifically measured molecules related to systemic endothelial damage [soluble CD40L and vascular endothelial growth factor (VEGF)], as PRES‐associated vasogenic oedema may cause hypoxia, which is a strong stimulus for their synthesis 11.
Methods
We performed a transversal study between January 2012 and March 2015 in a third‐level hospital in Mexico City (Instituto Nacional de Ciencias Médicas y Nutrición, Salvador Zubirán). We included 32 subjects, divided into the following groups: healthy controls (n = 6), SLE in remission (n = 6), active SLE (n = 6) and PRES with SLE (n = 14). All SLE patients fulfilled ≥ 4 American College of Rheumatology (ACR) criteria 12. Remission was considered when the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score was zero and patients had no immunosuppressive treatment during the 12‐month period before the blood draw; active SLE patients had SLEDAI scores ≥ 6. We excluded subjects with active infections or chronic infections (HIV, viral hepatitis, etc.), pregnancy, SLE‐associated CNS activity and the diagnosis of other concomitant autoimmune diseases. We also excluded patients with other altered laboratory parameters or image studies that could explain their neurological symptoms [i.e. serum electrolytes, thyroid function tests, computerized tomography (CT) with findings different from PRES, electroencephalograph (EEG) with epileptogenic focus, cerebrospinal fluid suggestive of meningitis] or with a previous history of SLE‐associated CNS activity. PRES diagnosis was considered in patients with the appropriate clinical features (seizures, visual deficit, confusional state or headache) and MRI findings of vasogenic oedema (iso or hypointensity in T1 and hyperintensity in T2/FLAIR), as evaluated by a blinded neuroradiologist. PRES remission was considered when PRES‐associated symptoms resolved, regardless of treatment, according to the attending neurologist.
We obtained serum samples from all subjects. All samples were stored frozen until analysis. In patients with PRES, the sample was obtained ≤ 36 h from the beginning of symptoms. We analysed cytokine levels using different methods: cytometric bead array [T helper type 1 (Th1), Th2, Th17; Becton, Dickinson and Company, San Jose, CA, USA], which included interleukin (IL)‐2, IL‐4, IL‐6, IL‐10, tumour necrosis factor (TNF)‐α, interferon (IFN)‐γ and IL‐17A; Bio‐Plex Pro human cytokine 21‐Plex Panel (Bio‐Rad Laboratories, Hercules, CA, USA), which included IL‐1α, IL‐2Rα, IL‐3, IL‐12, IL‐16, IL‐18, cutaneous T cell‐attracting chemokine (CTACK), growth‐related oncogene (GRO‐α), hepatocyte growth factor (HGF), IFN‐2α, leucocyte inhibitory factor (LIF), monocyte chemotactic protein‐3 (MCP‐3), macrophage colony‐stimulating factor (M‐CSF), macrophage inhibitory factor (MIF), monokine induced by IFN‐γ (MIG), β‐nerve growth factor (β‐NGF), stem cell factor (SCF), SCFG‐β, stromal cell‐derived factor‐1α (SDF‐1α), TGF‐β and TNF‐related apoptosis‐inducing ligand (TRAIL); and enzyme‐linked immunosorbent assay (ELISA) for sCD40L (R&D Systems, Inc., Minneapolis, MN, USA) and VEGF (Life Technologies Corporation, Carlsbad, CA, USA). Variables were described as mean, standard error of the mean (s.e.m.) and proportions. For comparison between groups, we used the one‐way analysis of variance (anova) test. Association between variables was assessed by χ2 test. Pearson's test was performed for correlation between variables; P‐values were considered significant at ≤ 0·05. Data were analysed with the support of spss software version 21. The study was approved by the Research and Ethics Committee from the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Results
All included patients were female. Mean age in the PRES/SLE group was 33·3 ± 2·96 years, and did not differ among groups (P = 0·29). Mean SLEDAI score was lower in PRES/SLE patients in comparison with patients from the active SLE group (7·8 ± 1·4 versus 17 ± 3·07, P < 0·0001). Other demographic, clinical and general laboratory features of SLE patients and healthy controls are shown in Table 1.
Table 1.
Demographic, clinical and laboratory characteristics in the different study groups
| Variable | Healthy controls (n = 6) | Remission SLE (n = 6) | Active SLE (n = 6) | PRES/SLE (n = 14) |
|---|---|---|---|---|
| Age (years ± s.e.m.) | 36 ± 4·1 | 34·1 ± 4·8 | 34·6 ± 4·2 | 33·3 ± 2·9 |
| SLEDAI score (points ± s.e.m.) | n.a. | 0 | 17 ± 3·07 | 7·8 ± 1·4 |
| Leucocytes (cells/µl × 103 ± s.e.m.) | n.d. | 4·95 ± 0·49 | 7·05 ± 1·25 | 10·07 ± 2·15 |
| Total lymphocytes (k/μl ± s.e.m.) | n.d. | 1250 ± 185 | 950 ± 140 | 1020 ± 188 |
| Platelets (cells/µl × 103 ± s.e.m.) | n.d. | 224 ± 19·3 | 233 ± 68·9 | 190 ± 31·4 |
| Haemoglobin (g/dl ± s.e.m.) | n.d. | 14·7 ± 0·26 | 9·88 ± 0·61 | 9·80 ± 0·65 |
Values in bold type represent P < 0·05. PRES = posterior reversible encephalopathy syndrome; s.e.m. = standard error of the mean; SLE = systemic lupus erythematosus; SLEDAI = Systemic Lupus Erythematosus Disease Activity Index; n.a. = not applicable; n.d. = not determined.
Among patients with PRES/SLE, 35% had lupus nephritis (confirmed by biopsy), with classes IV + V (ISN/RPS classification) 13 being the most prevalent; other comorbidities are displayed in Table 2. Eleven patients (78%) had elevated blood pressure (> 150/90 mmHg) at the time of the event and, in most cases (78%), the main clinical feature was seizures. Mean time to PRES remission was 2·19 ± 0·58 days. Three patients were classified as ‘immunological’ PRES 10, as they were normotensive and received immunosuppressive treatment specifically for the neurological manifestations. Conversely, 11 patients had hypertensive PRES; they were treated with anti‐hypertensives with clinical improvement. When patients in the latter group received immunosuppressive therapy, it was directed against other features of disease activity (mainly renal and haematological). Most MRI findings in subjects with PRES (64%) were considered typical (unilateral or bilateral occipital abnormalities). As shown in Table 2, there were no statistically significant differences among the SLE groups.
Table 2.
Clinical and laboratory variables in patients with SLE
| Variable | PRES/SLE (n = 14) (n, %) | Remission SLE (n = 6) (n, %) | Active SLE (n = 6) (n, %) |
|---|---|---|---|
| Comorbidities (prior to event of PRES or venipuncture) | |||
| Type 2 diabetes | 1 (7) | 0 (0) | 0 (0) |
| Hypertension | 5 (35) | 0 (0) | 1 (16) |
| Dyslipidaemia * | 2 (14) | 1 (16) | 1 (16) |
| Smoking † | 2 (14) | 0 (0) | 0 (0) |
| Lupus nephritis ‡ | 5 (35) | 0 (0) | 1 (16) |
| sAPS | 1 (7) | 0 (0) | 2 (33) |
| Positive APS serology § | 4 (28) | 1 (16) | 4 (66) |
| At PRES event (or venipuncture) | |||
| Immunosuppressive treatment | 12 (85) | 0 (0) | 4 (66) |
| Prednisone | 7 (50) | – | 3 (50) |
| Prednisone dose (mean ± s.e.m.) (mg) | 23·7 ± 6·9 | – | 26 ± 11·6 |
| Cyclophosphamide | 2 (14) | – | 1 (16) |
| Azathioprine | 2 (14) | – | 3 (50) |
| Mycophenolate Mofetil | 0 (0) | – | 0 (0) |
| High blood pressure ¶ | 11 (78) | 0 (0) | 1 (16) |
| High serum creatinine** | 5 (35) | 0 (0) | 0 (0) |
| Low C3 levels †† | 8 (57) | – | 3 (50) |
| Low C4 levels ‡‡ | 9 (64) | – | 6 (100) |
| Positive anti‐dsDNA §§ | 6 (42) | – | 6 (100) |
*Defined as hypercholesterolaemia ≥ 200 mg/dl and or hypertriglyceridaemia ≥ 150 mg/dl. †Current or active in the past 5 years. ‡Proved by biopsy. §Positive antibodies regardless of clinical manifestations. ¶Defined as blood pressure ≥ 150/90 mmHg. **Defined as serum creatinine ≥ 2·5 mg/dl. ††Defined as C3 levels ≤ 87 g/l. ‡‡Defined as C4 levels ≤ 19 g/l. §§Defined as anti‐dsDNA two times above the upper limit (9·6 UI). Anti‐dsDNA = anti‐double‐stranded DNA antibodies; sAPS = secondary anti‐phospholipid syndrome; s.e.m. = standard error of the mean; PRES = posterior reversible encephalopathy syndrome; SLE = systemic lupus erythematosus.
Regarding cytokine levels, PRES/SLE patients had higher serum IL‐6 [23·70 ± 9·37 versus 2·55 ± 0·65 (healthy controls) versus 2·04 ± 0·62 (remission SLE) versus 8·35 ± 2·43 pg/ml (active SLE), P = 0·043], as well as IL‐10 levels [4·56 ± 1·40 versus 0·83 ± 0·09 (healthy controls) versus 1·00 ± 0·14 (remission SLE) versus 2·26 ± 0·65 pg/ml (active SLE), P = 0·031] when compared to the other groups, as displayed in Fig. 1. Furthermore, in this group, there was a positive correlation between both cytokines (Fig. 2; r = 0·686, P = 0·007). As shown in Table 3, there were no differences in the other cytokines measured (IL‐2, IL‐3, IL‐4, IL‐16, IL‐17A, IL‐18, IL‐12p40, IFN‐γ and TNF‐α) or in soluble CD40L [3923 ± 1200 (healthy controls), 2724 ± 1296 (remission SLE), 1445 ± 1252 (active SLE), 2620 ± 773 pg/ml (PRES/SLE), P = 0·80] and VEGF levels [653 ± 275 (healthy controls), 153 ± 62 (remission SLE), 223 ± 116 (active SLE), 173 ± 66 pg/ml (PRES/SLE), P = 0·77] among the groups (Table 3). There was no association between IL‐6 and IL‐10 levels and the type of MRI abnormalities (IL‐6, χ2 = 0·062, P = 1; IL‐10, χ2 = 4·32, P = 0·086). We also did not find a correlation between those cytokine levels and time to PRES remission (IL‐6 −0·089, P = 0·761; IL‐10 −0·234, P = 0·421).
Figure 1.
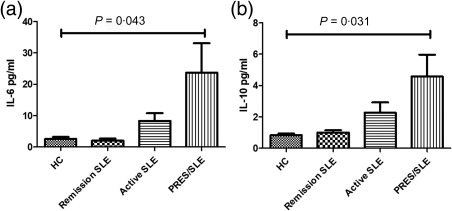
Differential interleukin (IL)‐6 and IL‐10 serum concentrations among different study groups. Serum IL‐6 (a) and IL‐10 (b) levels in healthy controls (n = 6), remission systemic lupus erythematosus (SLE) (n = 6), active SLE (n = 6) and posterior reversible encephalopathy syndrome (PRES)/SLE (n = 14) were measured by cytometric bead array. Bars represent mean ± standard error of the mean (s.e.m.) of each cytokine concentration in each group. P‐values were considered significant at ≤ 0·05.
Figure 2.
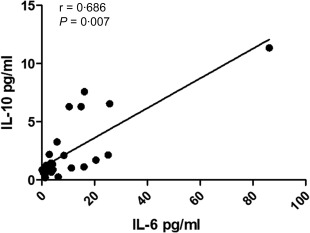
Correlation between serum interleukin (IL)‐6 and IL‐10 among patients with posterior reversible encephalopathy syndrome/systemic lupus erythematosus (PRES/SLE).
Table 3.
Serum cytokines and endothelial damage markers in the different study groups
| Serum cytokine (pg/ml) (mean ± s.e.m.) | Healthy controls (n = 6) | Remission SLE (n = 6) | Active SLE (n = 6) | PRES/SLE (n = 14) |
|---|---|---|---|---|
| IL‐2 | 0·07 ± 0·04 | 0·08 ± 0·05 | 0·35 ± 0·27 | 0·43 ± 0·31 |
| IL‐4 | 0·17 ± 0·15 | 0·23 ± 0·11 | 0·32 ± 0·27 | 0·36 ± 0·27 |
| TNF‐α | 0·45 ± 0·37 | n.d. | 0·32 ± 0·32 | 0·35 ± 0·32 |
| IFN‐γ | 0·08 ± 0·08 | 0·16 ± 0·15 | n.d. | 0·26 ± 0·15 |
| VEGF | 653 ± 275 | 153 ± 62 | 223 ± 116 | 173 ± 66 |
| Soluble CD40L | 3923 ± 1200 | 2724 ± 1296 | 1445 ± 1252 | 2620 ± 773 |
IL = interleukin; IFN = interferon; PRES = posterior reversible encephalopathy syndrome; s.e.m. = standard error of the mean; SLE = systemic lupus erythematosus; TNF = tumour necrosis factor; n.d. = not detected; VEGF = vascular endothelial growth factor.
Discussion
Even though PRES is remarkably associated with SLE 4, 14, 15, and has even been proposed as one of its neuropsychiatric syndromes 16, its pathogenesis in these patients is largely unknown. We found a differential serum cytokine profile in patients with PRES and SLE, characterized by high serum IL‐6 and IL‐10 levels, which could explain some of the disease mechanisms involved.
Endothelial dysfunction, with a resultant blood–brain barrier disruption, has been proposed as one of the physiopathogenic mechanisms in PRES 7. This would be expected in SLE, as endothelial dysfunction and endothelial progenitor cell deficiency have been described in these patients 17 and have been considered to play a role in different complications of the disease, such as accelerated atherosclerosis and thrombotic microangiopathy 18. Also, our group recently described risk factors associated with the development of PRES in SLE patients, including lymphopenia and dyslipidaemia, which could contribute to endothelial dysfunction 9. Interestingly, this is not the first association found between low lymphocyte counts and neuropsychiatric disorders in SLE patients 19.
IL‐6, which we found increased significantly in PRES/SLE patients, could contribute to endothelial damage by inducing the expression of adhesion molecules, which are also considered markers of endothelial activation 7, 20. The high IL‐6 levels we found also agree with previous reports in which IL‐6 was increased in cerebrospinal fluid (CSF) from SLE patients with diverse neuropsychiatric syndromes 21, 22. Furthermore, Asano et al. 23 recently described blood–brain barrier disruption and increased CSF IL‐6 levels in SLE patients with diffuse neuropsychiatric symptoms, suggesting that the source of IL‐6 is not only intrathecal production, but that it may also enter into the CSF from the systemic circulation through the injured blood–brain barrier. Therefore, increased serum IL‐6 could reflect CNS activity. It is noteworthy that we found no correlation between SLEDAI score and IL‐6 levels, so it could be hypothesized that this cytokine has certain CNS specificity in SLE patients.
Conversely, IL‐10 has been considered classically as an anti‐inflammatory cytokine, and has been found recently to exert its anti‐inflammatory and neuroprotective actions in the brain 24, 25, 26. Particularly, it has been shown to inhibit IL‐6 and IL‐6 receptor expression in murine microglia 25, 27. Interestingly, both cytokines have signalling pathways involving signal transducer and activator of transcription‐3 (STAT‐3) activation, but with different temporal patterns, with IL‐10 causing a more sustained phosphorylation and nuclear translocation, leading to the transcription of a specific gene profile and a predominantly anti‐inflammatory response 28. Finally, IL‐6 produced by human umbilical cord multipotent stromal cells was found to induce IL‐10 expression in monocytes, hence promoting a suppressive phenotype 29. Therefore, the fact that both these cytokines were increased, and that there was a positive correlation between their levels, may relate to their joint actions over STAT‐3 and the induction of IL‐10 by IL‐6, as well as a potential counter‐regulatory action mediated by IL‐10 to prevent further damage to endothelial cells 20, 30, 31.
Finally, VEGF levels were not different among groups, which was a somewhat unexpected finding. VEGF has also been considered to be associated with the development of PRES by increasing adhesion molecule expression and inducing blood–brain barrier permeability 11, leading to oedema. We did not find higher serum levels in PRES/SLE patients, but it is possible that VEGF synthesis is only increased locally 32, with direct effects on the brain vasculature, without affecting serum levels.
The main limitation of this study is the small sample size, but it is important to consider that SLE‐associated PRES is an extremely rare entity 4, 15, 33. As cytokine levels were measured at the time of PRES diagnosis, we could not assess whether this profile changed over time and according to clinical features. Also, CSF cytokine levels, which could be more representative of the inflammatory brain scenario, were not measured. However, serum levels might be more feasible to use in routine clinical practice. Finally, we cannot assert that this profile is exclusive to SLE patients with PRES, so prospective studies including other types of neuropsychiatric activity, as well as seizures with other aetiologies, should be performed in order to clarify these issues. Furthermore, studies addressing cytokine levels in patients with non‐autoimmune PRES should be performed, in other to determine whether this profile is specific for PRES associated with SLE or if it can be found in patients with PRES from different aetiologies.
In summary, we found a differential serum cytokine profile in PRES/SLE patients. This could partly explain PRES pathogenesis in this group of patients and, by coinciding with findings in other neuropsychiatric manifestations, it also reinforces the fact that PRES could be considered a new neuropsychiatric syndrome in SLE patients. Additionally, serum IL‐6 and IL‐10 could be used as potential diagnostic markers for this entity. This measurement would be less invasive than CSF cytokine levels. Prospective studies are needed to corroborate our findings and to assess whether this profile is specific for SLE‐associated PRES, as well as its longitudinal behaviour.
Disclosure
The authors declare no financial or other conflicts of interest.
Acknowledgements
This study was supported by a grant from the Consejo Nacional de Ciencia y Tecnología (CONACYT grant 261473 (FOSSIS 2015‐2). We also would like to thank the Flow Cytometry Unit from the Red de Apoyo a la Investigación CIC‐UNAM for the valuable technical support provided.
References
- 1. Hinchey J, Chaves C, Appignani B et al A reversible posterior leukoencephalopathy syndrome. N Engl J Med 1996; 334:494–500. [DOI] [PubMed] [Google Scholar]
- 2. Fischer M, Schmutzhard E. Posterior reversible encephalopathy syndrome. J Neurol 2017; 264:1608–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roth C, Ferbert A. Posterior reversible encephalopathy syndrome: long‐term follow‐up. J Neurol Neurosurg Psychiatry 2010; 81:773–7. [DOI] [PubMed] [Google Scholar]
- 4. Barber CE, Leclerc R, Gladman DD, Urowitz MB, Fortin PR. Posterior reversible encephalopathy syndrome: an emerging disease manifestation in systemic lupus erythematosus. Semin Arthritis Rheum 2011; 41:353–63. [DOI] [PubMed] [Google Scholar]
- 5. West SG. Neuropsychiatric lupus. Rheum Dis Clin North Am 1994; 20:129–58. [PubMed] [Google Scholar]
- 6. Feske SK. Posterior reversible encephalopathy syndrome: a review. Semin Neurol 2011; 31:202–15. [DOI] [PubMed] [Google Scholar]
- 7. Marra A, Vargas M, Striano P, Del Guercio L, Buonanno P, Servillo G. Posterior reversible encephalopathy syndrome: the endothelial hypotheses. Med Hypotheses 2014; 82:619–22. [DOI] [PubMed] [Google Scholar]
- 8. Chen Z, Shen GQ, Lerner A, Gao B. Immune system activation in the pathogenesis of posterior reversible encephalopathy syndrome. Brain Res Bull 2017; 131:93–9. [DOI] [PubMed] [Google Scholar]
- 9. Merayo‐Chalico J, Apodaca E, Barrera‐Vargas A et al Clinical outcomes and risk factors for posterior reversible encephalopathy syndrome in systemic lupus erythematosus: a multicentric case‐control study. J Neurol Neurosurg Psychiatry 2016; 87:287–94. [DOI] [PubMed] [Google Scholar]
- 10. Fujieda Y, Kataoka H, Odani T et al Clinical features of reversible posterior leukoencephalopathy syndrome in patients with systemic lupus erythematosus. Mod Rheumatol 2011; 21:276–81. [DOI] [PubMed] [Google Scholar]
- 11. Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia‐initiated angiogenesis. Nature 1992; 359:843–5. [DOI] [PubMed] [Google Scholar]
- 12. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40:1725. [DOI] [PubMed] [Google Scholar]
- 13. Weening JJ, D'Agati VD, Schwartz MM et al The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004; 15:241–50. [DOI] [PubMed] [Google Scholar]
- 14. Mak A, Chan BP, Yeh IB et al Neuropsychiatric lupus and reversible posterior leucoencephalopathy syndrome: a challenging clinical dilemma. Rheumatology (Oxf) 2008; 47:256–62. [DOI] [PubMed] [Google Scholar]
- 15. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome–an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol 2006; 33:2178–83. [PubMed] [Google Scholar]
- 16. Ishimori ML, Pressman BD, Wallace DJ, Weisman MH. Posterior reversible encephalopathy syndrome: another manifestation of CNS SLE? Lupus 2007; 16:436–43. [DOI] [PubMed] [Google Scholar]
- 17. Lee PY, Li Y, Richards HB et al Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum 2007; 56:3759–69. [DOI] [PubMed] [Google Scholar]
- 18. Castejon R, Jimenez‐Ortiz C, Valero‐Gonzalez S, Rosado S, Mellor S, Yebra‐Bango M. Decreased circulating endothelial progenitor cells as an early risk factor of subclinical atherosclerosis in systemic lupus erythematosus. Rheumatology (Oxf) 2014; 53:631–8. [DOI] [PubMed] [Google Scholar]
- 19. Silva LM, Garcia AB, Donadi EA. Increased lymphocyte death by neglect‐apoptosis is associated with lymphopenia and autoantibodies in lupus patients presenting with neuropsychiatric manifestations. J Neurol 2002; 249:1048–54. [DOI] [PubMed] [Google Scholar]
- 20. Wassmann S, Stumpf M, Strehlow K et al Interleukin‐6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res 2004; 94:534–41. [DOI] [PubMed] [Google Scholar]
- 21. Alcocer‐Varela J, Aleman‐Hoey D, Alarcon‐Segovia D. Interleukin‐1 and interleukin‐6 activities are increased in the cerebrospinal fluid of patients with CNS lupus erythematosus and correlate with local late T‐cell activation markers. Lupus 1992; 1:111–7. [DOI] [PubMed] [Google Scholar]
- 22. Fragoso‐Loyo H, Atisha‐Fregoso Y, Llorente L, Sanchez‐Guerrero J. Inflammatory profile in cerebrospinal fluid of patients with headache as a manifestation of neuropsychiatric systemic lupus erythematosus. Rheumatology (Oxford) 2013; 52:2218–22. [DOI] [PubMed] [Google Scholar]
- 23. Asano T, Ito H, Kariya Y et al Evaluation of blood–brain barrier function by quotient alpha2 macroglobulin and its relationship with interleukin‐6 and complement component 3 levels in neuropsychiatric systemic lupus erythematosus. PLOS ONE 2017; 12:e0186414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strle K, Zhou JH, Shen WH et al Interleukin‐10 in the brain. Crit Rev Immunol 2001; 21:427–49. [PubMed] [Google Scholar]
- 25. Sawada M, Suzumura A, Hosoya H, Marunouchi T, Nagatsu T. Interleukin‐10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J Neurochem 1999; 72:1466–71. [DOI] [PubMed] [Google Scholar]
- 26. Garcia JM, Stillings SA, Leclerc JL et al Role of interleukin‐10 in acute brain injuries. Front Neurol 2017; 8:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heyen JR, Ye S, Finck BN, Johnson RW. Interleukin (IL)‐10 inhibits IL‐6 production in microglia by preventing activation of NF‐kappaB. Brain Res Mol Brain Res 2000; 77:138–47. [DOI] [PubMed] [Google Scholar]
- 28. Braun DA, Fribourg M, Sealfon SC. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J Biol Chem 2013; 288:2986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng Y, Zhang Y, Ye L et al Umbilical cord‐derived mesenchymal stem cells instruct monocytes towards an IL10‐producing phenotype by secreting IL6 and HGF. Sci Rep 2016; 6:37566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saraiva M, O'Garra A. The regulation of IL‐10 production by immune cells. Nat Rev Immunol 2010; 10:170–81. [DOI] [PubMed] [Google Scholar]
- 31. Oshima T, Laroux FS, Coe LL et al Interferon‐gamma and interleukin‐10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res 2001; 61:130–43. [DOI] [PubMed] [Google Scholar]
- 32. Schiff D, Lopes MB. Neuropathological correlates of reversible posterior leukoencephalopathy. Neurocrit Care 2005; 2:303–5. [DOI] [PubMed] [Google Scholar]
- 33. Ferreira TS, Reis F, Appenzeller S. Posterior reversible encephalopathy syndrome and association with systemic lupus erythematosus. Lupus 2016; 25:1369–76. [DOI] [PubMed] [Google Scholar]