

Abstract
Background
The ataxia telangiectasia mutated (ATM) gene is a moderate-risk breast cancer susceptibility gene; germline loss-of-function variants are found in up to 3% of hereditary breast and ovarian cancer (HBOC) families who undergo genetic testing. So far, no clear histopathological and molecular features of breast tumours occurring in ATM deleterious variant carriers have been described, but identification of an ATM-associated tumour signature may help in patient management.
Methods
To characterise hallmarks of ATM-associated tumours, we performed systematic pathology review of tumours from 21 participants from ataxia-telangiectasia families and 18 participants from HBOC families, as well as copy number profiling on a subset of 23 tumours. Morphology of ATM-associated tumours was compared with that of 599 patients with no BRCA1 and BRCA2 mutations from a hospital-based series, as well as with data from The Cancer Genome Atlas. Absolute copy number and loss of heterozygosity (LOH) profiles were obtained from the OncoScan SNP array. In addition, we performed whole-genome sequencing on four tumours from ATM loss-of-function variant carriers with available frozen material.
Results
We found that ATM-associated tumours belong mostly to the luminal B subtype, are tetraploid and show LOH at the ATM locus at 11q22–23. Unlike tumours in which BRCA1 or BRCA2 is inactivated, tumours arising in ATM deleterious variant carriers are not associated with increased large-scale genomic instability as measured by the large-scale state transitions signature. Losses at 13q14.11-q14.3, 17p13.2-p12, 21p11.2-p11.1 and 22q11.23 were observed. Somatic alterations at these loci may therefore represent biomarkers for ATM testing and harbour driver mutations in potentially ‘druggable’ genes that would allow patients to be directed towards tailored therapeutic strategies.
Conclusions
Although ATM is involved in the DNA damage response, ATM-associated tumours are distinct from BRCA1-associated tumours in terms of morphological characteristics and genomic alterations, and they are also distinguishable from sporadic breast tumours, thus opening up the possibility to identify ATM variant carriers outside the ataxia-telangiectasia disorder and direct them towards effective cancer risk management and therapeutic strategies.
Electronic supplementary material
The online version of this article (10.1186/s13058-018-0951-9) contains supplementary material, which is available to authorized users.
Keywords: ATM, Breast tumour, Pathology, Genetic instability, OncoScan array, Copy number, Loss of heterozygosity, Genomic signature
Background
Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder caused by biallelic inactivating variants in the ataxia-telangiectasia mutated (ATM) gene. The phenotype is characterised by progressive neuronal degeneration, immunological deficiency, genetic instability, hypersensitivity to ionising radiation and agents that cause DNA double-strand breaks, and a predisposition to malignancies, particularly lymphoid tumours [1–3]. Epidemiological studies on A-T families showed that heterozygous ATM deleterious variant carriers (hereafter referred to as HetAT) are also at increased risk of other cancer types [4–6], notably of breast cancer (BC) in female relatives [7, 8]. It is estimated that 0.5% to 1% of the general population are HetAT, and studies conducted in hereditary breast and ovarian cancer (HBOC) families or early-onset BC cases showed that deleterious ATM alleles confer a two- to four-fold increase in BC risk for carriers as compared with non-carriers [9, 10]. Therefore, most published case-control studies or family-based studies described such ATM alleles as moderate-risk BC susceptibility alleles, although this risk may differ according to the type of variant [9, 10]. Consequently, ATM is now included in nearly all multigene panels used for HBOC genetic testing that include, in addition to BRCA1 and BRCA2, other moderate- to high-risk genes coding for tumour suppressor proteins acting in critical processes of DNA repair pathways [11]. However, results of ATM testing are usually not issued to patients, owing to the imprecise absolute risk estimates and a lack of management recommendations for ATM variant carriers and their relatives [11]. Nevertheless, National Comprehensive Cancer Network guidelines recommend an annual screening mammogram and annual MRI with contrast enhancement beginning at age 40, or earlier based on family BC history for HetAT women [12]. Moreover, Australia has national best practice guidelines addressing the variant c.7271T>G; these guidelines are based on those that apply to the management of BC risk in BRCA2 deleterious variant carriers [13, 14]. If a consensus was made to define ATM as a gene with clinical utility, specific pathological and genomic features associated with ATM inactivation in tumours could help identify subjects with no strong personal or family history of BC. A genetic test for ATM may thus be offered to them and their relatives and thereby direct those individuals towards effective cancer risk management and therapeutic strategies.
So far, no clear histopathological and molecular features have been described for ATM-associated breast tumours (i.e., tumours developed by subjects carrying one or two mutated copies of ATM), and well-documented ATM tumour series are very limited. Researchers in three Australian studies on familial BC investigated breast tumours developed by HetAT participants carrying either a truncating variant (TV) or a missense variant (essentially the c.7271T>G; p.Val2424Gly variant) [13, 15]. The first two studies, carried out on 21 and 35 tumours, respectively, suggested that histologically, breast tumours from HetAT subjects do not resemble the tumours from BRCA1 mutation carriers [15], and no difference was observed between the histological grade of ATM-associated tumours and a series of age-matched control tumours [13]. The third study, focusing on six tumours from carriers of the c.7271T>G variant, revealed that all tumours presented with the luminal A or B molecular subtype [16]. Finally, no consistent pattern of loss of the normal allele was reported in an Australian series (N = 17) or in a French series (N = 16) of tumours from carriers of putative BC-associated ATM variants [13, 17].
The purpose of the present study was to determine whether breast tumours developed by ATM variant carriers show distinctive histopathological and genomic features as compared with ‘sporadic’ tumours, and also whether they resemble the breast tumours described in carriers of a deleterious variant of other known BC susceptibility genes, in particular the BRCA1-associated phenotype [18]. To this end, we conducted a pathology review of a series of tumours composed of 3 breast tumours from 3 A-T subjects (who were therefore homozygous or compound heterozygous ATM variant carriers), 20 tumours from 18 HetAT subjects from A-T families, and 18 tumours from 18 HetAT subjects from HBOC families who were non-carriers of other known high-risk variants. We also performed single-nucleotide polymorphism (SNP) array genomic profiling to assess somatic loss of heterozygosity (LOH) at the ATM locus and to investigate absolute copy number and LOH profiles at the genome-wide level. Finally, to complete the repertoire of somatic alterations of ATM-associated breast tumours, we performed whole-genome sequencing (WGS) on four tumours from HetAT participants with available frozen material.
Methods
Study participants and tumour material
Breast tumour samples were selected from carriers of a deleterious ATM variant from four different research resources: the French retrospective study on A-T families (Retro-AT) [19], the French prospective cohort on women related to an A-T child (CoF-AT) [8], the GENESIS study [20] and the Kathleen Cuningham Foundation Consortium for research into Familial Breast Cancer (kConFab) study [21]. Briefly, Retro-AT [19] was carried out between 1994 and 1997 to assess cancer risk in A-T families living in France. Thirty-four A-T families were identified during this period, and 27 of them were subsequently included in the CoF-AT prospective study. CoF-AT is an ongoing national prospective cohort study of A-T families which was initiated in 2003 to investigate environmental and genetic risk factors for BC in HetAT and non-HetAT (i.e., non-carriers of an ATM variant) women. All women aged 18 and over were eligible to participate in the study. At inclusion, participants provided a blood sample to determine whether they carried one of the ATM-inactivating variants identified in the A-T child of the family. As of June 2017, 415 women (213 HetAT and 202 non-HetAT) belonging to 105 A-T families had been enrolled in CoF-AT, and 37 study participants from Retro-AT or CoF-AT had developed BC, including 23 HetAT women, 11 non-HetAT women and 3 A-T subjects having inherited two inactivated copies of ATM (2 females and 1 male). Breast tumour material from the 3 A-T subjects and from 18 HetAT subjects could be retrieved for the present study (Table 1).
Table 1.
Clinical characteristics of ATM mutation carriers, and available tumour material used for analyses
| Study | Patient ID | Sex | Nucleotide change | Effect on protein | Variant type* | Tumour ID | Age at diagnosis | Stade | Neoadjuvant treatment | Conservation | OncoScan Analysis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A-T families (Retro-AT + CoF-AT) | AT1 | M | c.2839-580_577del4(-/-) | cryptic splice site | TV (1) | T0072-L | 28 | Invasive | Unknown | FFPE | Yes |
| AT2 | F | c.8585-2A>C | frameshift | TV | T0075-L | 42 | Invasive | Unknown | Bouin | No | |
| c.5189G>T | p.Arg1730Leu | MS (C0) | |||||||||
| AT3 | F | c.2413C>T | p.Arg805X | TV | T0249-L | 30 | Invasive | Unknown | FFPE | Yes | |
| c.7517_7520delGAGA | p.Arg2506ThrfsX3 | TV | |||||||||
| 1 | F | c.3576G>A | p.Ser1135_Lys1192del58 | TV | T0001-L | 62 | Invasive | No | FFPE+Frozen | No | |
| 2 | F | c.2839-580_577del4 | cryptic splice site | TV | T0002-L | 60 | Invasive | No | Bouin | No | |
| 3 | F | c.5644C>T | p.Arg1882X | TV | T0003-L | 45 | Invasive | No | FFPE | No | |
| 4 | F | c.3802delG | p.Val1268X | TV | T0005-L | 65 | Invasive | No | FFPE | Yes | |
| 5 | F | c.6007-2A>T | frameshift | TV | T0007-R | 74 | Invasive | No | FFPE | No | |
| 6 | F | c.3085dupA | p.Thr1029AsnfsX19 | TV | T0008-R | 51 | Invasive | Unknown | FFPE | No | |
| 7 | F | c.3894dupT | p.Ala1299CysfsX3 | TV | T0009-L | 36 | Invasive | No | FFPE | Yes | |
| 8 | F | c.6007-2A>T | frameshift | TV | T0010-L | 30 | Invasive | Unknown | Bouin | No | |
| 9 | F | c.6404_6405insTT | p.Arg2136X | TV | T0015-L | 40 | Invasive | No | FFPE+Frozen | Yes | |
| 10 | F | c.2466fs | del exon19-65 | TV | T0016-R | 62 | Invasive | No | Bouin | No | |
| T0016-L | 72 | Invasive | No | FFPE | Yes | ||||||
| 11 | F | c.3153+1G>A | frameshift | TV | T0073-L | 77 | Invasive | Unknown | Bouin | No | |
| 12 | F | c.8489T>G | p.Val2830Gly | MS (C65) | T0074-L | 62 | Invasive | Unknown | Bouin | No | |
| 13 | F | c.73-2A>G | frameshift | TV | T0076-R | 47 | Invasive | No | FFPE | Yes | |
| 14 | F | c.3754_3756delTATinsCA | p.Met2918IlefsX21 | TV | T0077-L | 66 | Invasive | No | FFPE+Frozen | Yes | |
| T0077-R | 66 | Invasive | No | FFPE+Frozen | Yes | ||||||
| 15 | F | c.8140C>T | p.Gln2714X | TV | T0078-L | 55 | Invasive | No | FFPE | Yes | |
| 16 | F | c.5644C>T | p.Arg1882X | TV | T0181-R | 39 | In situ | Unknown | FFPE | No | |
| 17 | F | c.8083G>A | p.Gly2695Ser | MS (C55) | T0247-R | 48 | Invasive | No | FFPE | Yes | |
| 18 | F | c.7928-2A>C | frameshift | TV | T0248-L | 35 | Invasive | Unknown | FFPE | Yes | |
| GENESIS | 19 | F | c.2413C>T | p.Arg805X | TV | T0045-R | 31 | Invasive | No | No material | - |
| T0045-R | 67 | In situ | No | FFPE | Yes | ||||||
| 20 | F | c.8584+1G>A | frameshift | TV | T0091-L | 51 | In situ | No | FFPE | No | |
| 21 | F | c.3058dupA | p.Leu1019fs | TV | T0099-L | 39 | Invasive | No | FFPE | Yes | |
| 22 | F | c.5497-2A>C | p.Val1833IlefsX | TV (2) | T0111-L | 32 | Invasive | No | FFPE | No | |
| 23 | F | c.9008A>T | p.Asn3003Ile | MS (C65) | T0118-L | 64 | Invasive | No | FFPE | No | |
| 24 | F | c.1464G>T | p.Trp488Cys | MS (C65) | T0120-R | 65 | Invasive | No | FFPE | Yes | |
| 25 | F | c.5527delC | p.Phe1843fs | TV | T0123-R | 74 | Invasive | No | FFPE | Yes | |
| 26 | F | c.5750G>C | p.Arg1917Thr | MS (C65) | T0191-L | 45 | Invasive | Unknown | FFPE | No | |
| 27 | F | c.1236-2A>T | p.Trp412X | TV | T0192-R | 42 | In situ | Unknown | FFPE | No | |
| 28 | F | c.8614C>A | p.His2872Asn | MS (C65) | T0218-R | 54 | Invasive | Unknown | FFPE | No | |
| 29 | F | c.8494C>T | p.Arg2832Cys | MS (3) (C45) | T0220-R | 42 | Invasive | No | FFPE | Yes | |
| kConFab | 30 | F | c.3801delG | p.Glu1267fs | TV | T0173-L | 44 | Invasive | Unknown | No material | - |
| T0173-R | 49 | Invasive | Unknown | FFPE | Yes | ||||||
| 31 | M | c.6820G>A | p.Ala2274Thr | MS (C55) | T0174-R | 45 | In situ | Unknown | FFPE | Yes | |
| 32 | F | c.4909+1G>A | frameshift | TV (4) | T0175-R | 42 | Invasive | Unknown | FFPE | Yes | |
| 33 | F | c.8266A>T | p.Lys2756X | TV (5) | T0176-R | 60 | Invasive | Unknown | FFPE | Yes | |
| 34 | F | c.8158G>C | p.Asp2720His | MS (C65) | T0177-L | 59 | Invasive | Unknown | FFPE | No | |
| 35 | F | c.8266A>T | p.Lys2756X | TV (5) | T0179-R | 41 | Invasive | Unknown | No material | - | |
| T0179-L | 50 | Invasive | Unknown | FFPE | Yes | ||||||
| 36 | F | c.7176_7177insT | p.Ser2394PhefsX9 | TV | T0180-R | 43 | Invasive | Unknown | FFPE | Yes |
AT ataxia-telangiectasia, F female, M male, TV truncating variant, MS missense substitution. L left, R right, FFPE formalin-fixed, paraffin-embedded tissue sample, Bouin Bouin-fixed, paraffin-embedded tissue sample
(1,2,3,4,5) Reported as pathogenic for A-T in ClinVar
*Align-GVGD grades are indicated in brackets for MS variants
GENESIS is a national study on HBOC families identified through French family cancer clinics [20]. Index cases were women diagnosed with invasive mammary carcinoma or in situ ductal carcinoma, having at least one sister affected with BC, and with a negative test result for a pathogenic variant in BRCA1 and BRCA2. ATM carriers of a TV or of a rare likely deleterious missense substitution (MS) were identified during the course of a large-scale case-control mutation-screening study (F. Lesueur, PhD, unpublished data, March 2018). Tumour material from 11 of them was assessed in the present study (Table 1). In addition, we investigated tumours from seven HetAT subjects enrolled in the Australian kConFab study [21] (Table 1).
Selection of ATM variant carriers
Individuals included in the study were either homozygous, compound heterozygous or heterozygous carriers of a variant considered pathogenic for A-T disorder. We also selected HetAT BC participants from HBOC families carrying a TV that had not necessarily been reported in A-T families, as well as carriers of a rare likely deleterious MS classified as C65, C55 or C45 according to the Align-GVGD tool as previously described [10, 22]. Carriers of the p.Val2424Gly variant identified in kConFab were not included in this study, because tumour characteristics of carriers of this variant had been already investigated [13, 15, 16]. In total, 41 tumour tissues were available from 3 A-T and 36 HetAT subjects for histopathological review (Table 1).
Pathology review
The Hematoxylin and Eosin Stained (HES) breast tumour tissue was reviewed and scored for morphology features and graded by two pathologists (AVS and GB) using the modified system of Elston et al. [23]. The World Health Organisation classification of tumours of the breast was used to determine histological subtype of ATM-associated tumours, and TNM stage according to tumour size, nodal infiltration and metastasis status [24]. Oestrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) status, as well as the expression of proliferating marker Ki-67, was obtained from histopathology reports held by diagnostic laboratories. When incomplete, hormonal status was determined by immunohistochemistry (IHC) staining at Institut Curie. Tumours were considered HER2+ if they were scored 3+ by IHC or for tumours scored 2+ by IHC if fluorescence in situ hybridisation showed an HER2 gene amplification. Tumours were classified using IHC data according to the St. Gallen molecular subtypes as follows: triple-negative (ER−, PR− and HER2−), HER2-overexpressing (ER−, PR− and HER2+), luminal A (ER+, PR+/−, HER2− and Ki-67 < 20%), luminal B (ER+, PR+/−, HER2− and Ki-67 ≥ 20%), and luminal B/HER2+ (ER+, PR+/−, HER2+ and Ki-67 ≥ 20%) [25].
Morphological features of ATM-associated breast tumours were compared with the series of breast tumours from patients who had surgery at Institut Curie between 2005 and 2006, named the PICBIM series (from the programme incitatif et collaboratif - Cancer du sein: invasion et motilité). None of the PICBIM patients received neoadjuvant treatment. Patients who had developed a previous cancer at any site were excluded, as were known BRCA1 or BRCA2 mutation carriers. In this series, ATM mutation status of participants had not been determined. In total, 516 patients diagnosed with invasive carcinoma and 83 patients diagnosed with in situ carcinoma served as control subjects.
DNA preparation and confirmation of the familial ATM deleterious variant
Tumour DNA was extracted from tumour-enriched areas (with ≥ 50% tumour content when possible) delimited from the most representative HES-stained slides for the 35 tumours for which formalin-fixed, paraffin-embedded (FFPE) material was available (Table 1). The relevant areas were macrodissected from four 10-μm sections, and DNA was purified using the NucleoSpin Tissue protocol according to the manufacturer’s instructions (Macherey-Nagel, Düren, Germany). DNA quantity and quality were assessed using a Qubit fluorometer (Life Technologies/Thermo Fisher Scientific, Carlsbad, CA, USA) and SYBR Green-based qPCR assay (Promega, Madison, WI, USA). Of the 35 available FFPE tumour DNA samples, sufficient quantity and quality to perform subsequent molecular analyses were obtained for 23 of them. Matched blood DNA was extracted with the QuickGene-610L automated system (AutoGen, Holliston, MA, USA) according to the manufacturer’s instructions. The presence of the familial ATM deleterious variant was confirmed in all analysable blood and tumour DNA samples by Sanger targeted sequencing on the Applied Biosystems ABI 3500xL DNA analyser (Thermo Fisher Scientific, Forest City, CA, USA).
Copy number variation analysis
Copy number variation (CNV) analysis using the Affymetrix OncoScan SNP array (Thermo Fisher Scientific, Santa Clara, CA, USA) was performed on the 23 FFPE ATM-associated tumours, including 2 tumours from 2 A-T participants (Table 1). Data were analysed with the Genome Alteration Print (GAP) method, which takes into account both ploidy and large-scale genomic rearrangements [18, 26]. Copy number ranged from zero to eight copies, and all segments exceeding eight copies were ascribed eight-copy status. Chromosome number was estimated by the sum of the copy numbers detected at the peri-centric regions. Output processing files derived from the GAP tool were analysed using VAMP in-house software [27] to define the boundaries of regions recurrently altered in ATM-mutated tumours. Copy loss and gain for near-diploid tumours were called for the segments with zero or one copy and four or more copies, respectively. Copy loss and gain for near-tetraploid tumours were called for the segments with less than or equal to two and six or more copies, respectively. LOH status was ascribed to regions having monoallelic content, regardless of copy number. LOH associated with copy loss was referred as LOH/loss. Breakpoints (changes in the copy number or major allele counts within chromosomes) in each genomic profile were characterised on the basis of resulting absolute copy number profile and after filtering for regions with < 50 SNP variations. Recurrent alterations (CNV, LOH) among the cohort were obtained using the CNTools R package (version 1.24.0; R Foundation for Statistical Computing, Vienna, Austria) and a homemade script. In order to find inherent grouping structure, hierarchical clustering was performed using the alteration status (absence/presence of an alteration) per segment using the Jaccard distance and the Ward linkage function, available in the vegan R package (version 2.3-3).
Validation of LOH status at ATM locus using microsatellites
Tumour and matched blood DNA were evaluated on a subset of participants by using a PCR-based LOH assay with four fluorescence-labelled microsatellite markers (namely D11S1113, D11S1819, D11S2179 and D11S1778) spanning a 14.4-Mbp region encompassing the MRE11A and ATM genes. Capillary electrophoresis was performed on the ABI 3500xL DNA analyser. Raw electrophoretic data were analysed with GeneMarker software version 1.3 (SoftGenetics, LLC, State College, PA, USA) to assess allele ratios. We considered LOH at the ATM locus when the allele ratio fell below 50% in the tumour DNA sample.
Whole-genome sequencing
WGS was performed on four tumour-normal DNA pairs from three participants for whom frozen tumour tissue was available (Table 1). Paired-end libraries were prepared from 2 μg of DNA using the TruSeq DNA PCR-Free Low-Throughput Sample Preparation Kit (Illumina, San Diego, CA, USA) and were sequenced on the HiSeq 2500 instrument (Illumina). Tumour DNA was sequenced at a higher depth of coverage (100×) than the germline counterpart (30×). Sequencing reads were mapped to the reference genome (assembly hg19) using Burroughs-Wheeler Aligner (version 0.7.5a) [28]. Regions of CNV and LOH were identified using the FACETS algorithm (version 0.5.6) [29], and single-nucleotide variations (SNVs) and indels were called using VarScan 2 [30]. Somatic variants were filtered and annotated using an in-house pipeline.
Statistical analysis
Statistical analyses were performed using STATA version 14.1 software (StataCorp, College Station, TX, USA). Two-tailed tests with a 5% significance level were used throughout. Logistic regressions were used to assess the level of association between the presence of an ATM variant and various features of interest when comparing the ATM series with the PICBIM series. Fisher’s exact test (FET) was used to assess molecular subtype differences between ATM-associated tumours and breast tumours from sporadic cases from The Cancer Genome Atlas (TCGA) [31] and from the Norwegian series [32].
Results
Histopathological features associated with ATM variant status
Clinicopathological and IHC features were evaluated on 3 breast tumours from 3 A-T participants and 38 tumours from 36 HetAT participants. This tumour series was compared with BC cases enrolled in the PICBIM program of Institut Curie. An overview of the features examined in both series is presented in Table 2.
Table 2.
Clinical and histological features of ATM-associated invasive breast carcinomas compared with those of sporadic cases
| Clinicopathological variable | PICBIM series (N = 516) | ATM series (all tumours, TV + MS) (N = 36) | P value a | ATM series (1st primary tumours only) (N = 32) | P value a | ATM series (excluding MS in HBOC families) (N = 31) | P value a |
|---|---|---|---|---|---|---|---|
| Histological subtype | |||||||
| Ductal carcinoma | 434 | 30 | Reference | 27 | Reference | 25 | Reference |
| Lobular carcinoma | 54 | 2 | 0.55 | 2 | 0.69 | 2 | 0.78 |
| Others | 28 | 3 | 0.43 | 2 | 0.78 | 3 | 0.25 |
| Unknown | 0 | 1 | – | 1 | – | 1 | – |
| Histological grade | |||||||
| I | 88 | 4 | Reference | 3 | Reference | 4 | Reference |
| II | 191 | 16 | 0.34 | 15 | 0.24 | 13 | 0.58 |
| III | 236 | 15 | 0.64 | 13 | 0.55 | 14 | 0.75 |
| Unknown | 1 | 1 | – | 1 | – | – | – |
| Architecture | |||||||
| 1–2 | 126 | 0 | Reference | 7 | Reference | 6 | Reference |
| 3 | 390 | 30 | 0.97 | 21 | 0.86 | 20 | 0.96 |
| Unknown | 0 | 6 | – | 4 | – | 5 | – |
| Mitosis | |||||||
| 0–1 | 221 | 15 | Reference | 14 | Reference | 12 | Reference |
| 2 | 103 | 9 | 0.58 | 8 | 0.70 | 9 | 0.31 |
| 3 | 191 | 6 | 0.10 | 6 | 0.13 | 5 | 0.15 |
| Unknown | 1 | 6 | – | 4 | – | 5 | – |
| Nuclear grade | |||||||
| 1 | 30 | 2 | Reference | 2 | Reference | 2 | Reference |
| 2 | 225 | 7 | 0.25 | 7 | 0.22 | 5 | 0.12 |
| 3 | 261 | 21 | 0.96 | 19 | 0.89 | 19 | 0.9 |
| Unknown | 0 | 6 | – | 4 | – | 5 | – |
| Tumour size (cm) | |||||||
| pT1 (< 2) | 329 | 23 | Reference | 20 | Reference | 19 | Reference |
| pT2 (2–5) | 166 | 8 | 0.37 | 7 | 0.41 | 7 | 0.48 |
| pT3 (> 5) | 14 | 2 | 0.37 | 2 | 0.30 | 2 | 0.26 |
| pT4 | 7 | 0 | – | 0 | – | 0 | – |
| Unknown | 0 | 3 | – | 3 | – | 3 | – |
| Pushing margins | |||||||
| Absent | 461 | 19 | Reference | 17 | Reference | 15 | Reference |
| Present | 49 | 2 | 0.98 | 2 | 0.93 | 2 | 0.78 |
| Unknown | 6 | 15 | – | 13 | – | 14 | – |
| Emboli | |||||||
| Absent | 315 | 13 | Reference | 11 | Reference | 13 | Reference |
| Present | 199 | 14 | 0.08 | 14 | 0.04 | 11 | 0.32 |
| Unknown | 2 | 9 | – | 7 | – | 7 | – |
| N stage | |||||||
| pN0 | 286 | 18 | Reference | 15 | Reference | 15 | Reference |
| pN1 | 151 | 10 | 0.80 | 9 | 0.66 | 9 | 0.55 |
| pN2 | 59 | 1 | 0.23 | 1 | 0.32 | 1 | 0.35 |
| pN3 | 16 | 1 | 0.96 | 1 | 0.81 | 1 | 0.76 |
| pNx | 5 | 6 | – | 6 | – | 5 | – |
| Oestrogen receptor | |||||||
| Positive | 307 | 34 | Reference | 31 | Reference | 29 | Reference |
| Negative | 209 | 1 | 0.003 | 1 | 0.004 | 1 | 0.005 |
| Unknown | 0 | 1 | – | – | – | 1 | – |
| Progesterone receptor | |||||||
| Positive | 278 | 26 | Reference | 23 | Reference | 23 | Reference |
| Negative | 236 | 8 | 0.03 | 8 | 0.07 | 6 | 0.02 |
| Unknown | 2 | 2 | – | 1 | – | 2 | – |
| HER2 | |||||||
| Negative | 433 | 25 | Reference | 23 | Reference | 21 | Reference |
| Positive | 83 | 6 | 0.77 | 5 | 0.97 | 6 | 0.53 |
| Unknown | 0 | 5 | – | 4 | – | 4 | – |
| Ki-67 | |||||||
| < 20% | 173 | 11 | Reference | 10 | Reference | 8 | Reference |
| ≥ 20% | 336 | 18 | 0.55 | 17 | 0.59 | 17 | 0.97 |
| Unknown | 7 | 7 | – | 5 | – | 6 | – |
| Molecular subtype | |||||||
| TNBC | 142 | 1 | Reference | 1 | Reference | 1 | Reference |
| HER2 | 66 | 0 | N/A | 0 | N/A | 0 | N/A |
| Luminal A | 180 | 10 | 0.06 | 9 | 0.08 | 7 | 0.13 |
| Luminal B | 111 | 13 | 0.009 | 12 | 0.010 | 12 | 0.010 |
| Luminal B/HER2 | 17 | 4 | 0.005 | 4 | 0.005 | 4 | 0.005 |
| Unknown | 0 | 8 | – | 6 | – | 7 | – |
Abbreviations: ATM, Ataxia-telangiectasia mutated, HBOC Hereditary breast and ovarian cancer, HER2 Human epidermal growth factor receptor 2, MS Missense substitution, PICBIM Programme incitatif et collaboratif - Cancer du sein: invasion et motilité series, TV Truncating variant, TNBC Triple-negative breast cancer
a P value adjusted for sex and for age at diagnosis
Among the 41 reviewed ATM-associated breast tumours, 36 were invasive carcinomas and 5 were in situ carcinomas. Overall, subjects with invasive carcinoma and subjects with in situ carcinoma from the ATM series tend to be diagnosed at a younger age than subjects from the PICBIM series (mean age 52.4 vs. 56.2 years, P = 0.08 for invasive carcinomas; 45.5 vs. 54.1 years, P = 0.07 for in situ carcinomas). This can be explained by the fact that women related to an A-T child or belonging to an HBOC family are more likely to benefit from early detection of the disease owing to their higher risk of developing BC than the general population. We also compared mean age at diagnosis in participants who developed invasive breast carcinoma between the studies, and we observed no difference (CoF-AT/Retro-AT 51.9, GENESIS 55.9, kConFab 50.6, P = 0.90).
Invasive breast carcinomas developed by HetAT and A-T participants were mostly ductal carcinomas (86%) with an intermediate to high grade (II–III), which was the same as the distribution of histological types and grades found in the invasive tumours from the PICBIM series. With respect to the IHC of tumours arising in ATM variant carriers, 97% of ATM-associated tumours were ER+, which was significantly higher than the proportion of ER+ tumours in the PICBIM series (59%, P = 0.004). Low to moderate lymphocytic infiltration was observed in ATM-associated tumours (data not shown), but this information was not available in the PICBIM, so no comparison could be performed.
Molecular subtypes could be determined for 28 of 36 ATM-associated invasive breast tumours. ATM-associated breast tumours were mostly luminal B (46%) and luminal A (36%), and the distribution of the molecular subtypes differed significantly from that of the PICBIM series. In particular the luminal B and luminal B/HER2+ subtypes were over-represented among tumours developed by HetAT and A-T participants (P = 0.009 and P = 0.005, respectively) (Table 2). Because the PICBIM series might not reflect the distribution of the molecular subtypes of invasive breast tumours in the general population, we also compared the ATM series with a series of 1423 primary breast tumours from a Norwegian population-based survey of women born between 1886 and 1977 [32], as well as with 501 invasive breast tumours characterised with the PAM50 test [33, 34] available in TCGA, after exclusion of carriers of a TV in BRCA1, BRCA2 and ATM [31]. We found that the proportion of luminal B tumours was also significantly higher in ATM-associated tumours than in the Norwegian study (PFET = 0.03) and in the TCGA series (PFET = 0.02), whereas the prevalence rates of luminal B/HER2+ tumours and of triple-negative breast tumours in the two latter series were similar to those observed in ATM-associated tumours (Fig. 1).
Fig. 1.
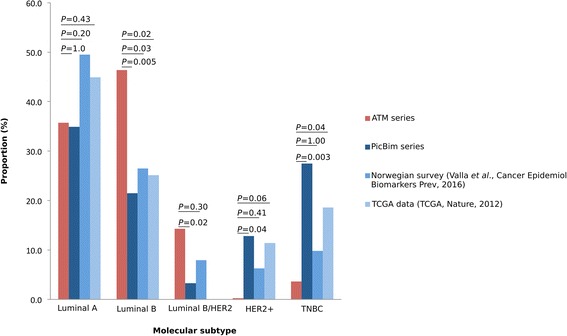
Distribution of molecular subtypes in the ataxia-telangiectasia mutated (ATM) series and in the three control series. PAM50 classification was used for The Cancer Genome Atlas (TCGA) data, which explains the absence of luminal B/Human epidermal growth factor receptor 2-positive (HER2+) tumours in this series. Fisher’s exact test was used to assess difference between ATM series and control series. PICBIM Programme incitatif et collaboratif - Cancer du sein: invasion et motilité series
We next performed two sensitivity tests. First, analyses comparing clinical and histological features of ATM-associated tumours with those of the PICBIM series were repeated after exclusion of four ATM-associated invasive breast tumours, three of which were second primary tumours (T0016-L, T0173-R and T0179-L) whose morphology and histology might have been affected by treatment of the first primary BC. The fourth tumour (T0077-L) was from a patient with synchronous bilateral tumours (T0077-R and T0077-L); we randomly excluded one of the two tumours to take into account only one tumour per patient in the analysis. The results remained unchanged (Table 2).
Second, we repeated the analyses after exclusion of five invasive tumours developed by carriers of a missense variant not reported so far as pathogenic for A-T, namely c.1464G>T (p.Trp488Cys), c.5750G>C (p.Arg1917Thr), c.8158G>C (p.Asp2720His), c.8614C>A (p.His2872Asn) and c.9008A>T (p.Asn3003Ile), to avoid possible misclassification of the deleterious effect of the variant based on in silico prediction only. Again, the results remained unchanged (Table 2).
We also compared features of the five in situ carcinomas (two ER+, two ER− and one with undetermined ER status) of the ATM series with those of the 83 in situ carcinomas from the PICBIM series, and we observed no difference in nuclear grade, tumour size and hormonal status between the two groups of tumours. However, owing to the low number of in situ carcinomas observed in this series, it was not possible at this stage to draw any conclusions about the characteristics of the in situ tumours developed by HetAT participants.
Genome-wide copy number and LOH profiles of ATM breast tumours
High-quality SNP array data were obtained for 23 FFPE breast tumours; 2 tumours were from A-T subjects, and 21 tumours were from HetAT subjects. Tumour ploidy inferred from the absolute segmental copy number profiles and genotype status by the GAP method [26] identified 16 of 23 (70%) near-tetraploid tumours and 7 of 23 (30%) near-diploid tumours. CNVs and regions of LOH were subsequently determined by taking into account the ploidy of each tumour. No evidence of the homologous recombination deficiency (HRD) signature as measured by large-scale state transition genomic signature [18, 35] was observed among the ATM-associated tumours (Fig. 2a).
Fig. 2.
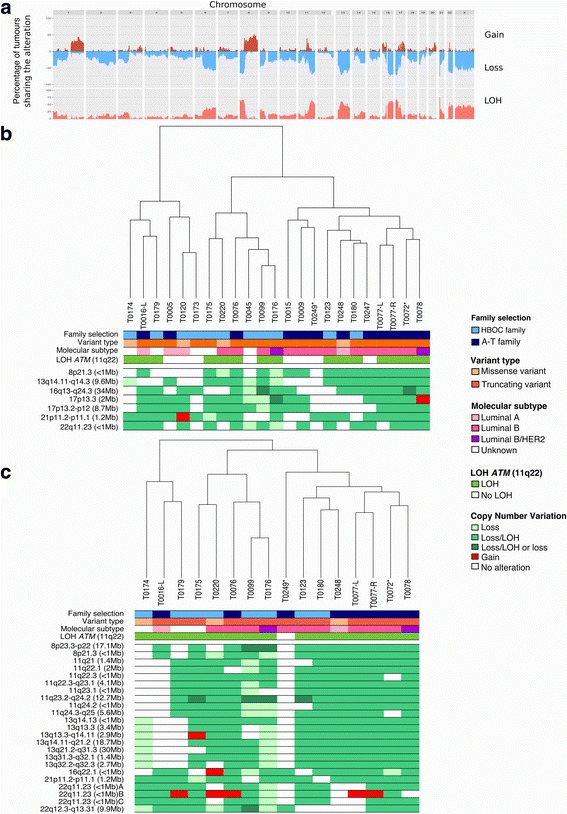
Copy number variation profiles of ataxia-telangiectasia mutated (ATM)-associated tumours analysed with the OncoScan array. a Genome-wide view of cumulative copy number variations present in the 23 ATM-associated tumours. Gains are indicated in red, losses in blue, and loss of heterozygosity (LOH) in orange. b Cluster dendrogram and genomic regions altered in ≥ 70% of the 23 analysed tumours. Tumours from Ataxia-telangiectasia (A-T) children are indicated by asterisks. c Cluster dendrogram and genomic regions altered in ≥ 70% of the 16 tumours with confirmed biallelic inactivation of ATM. Tumours from A-T children are indicated by asterisks. Loss The two alleles are present in the tumour, Loss/LOH Only one allele is present in the tumour, Loss/LOH or Loss Consecutive segmental regions characterised as either ‘Loss/LOH’ or ‘Loss’, HBOC Hereditary breast and ovarian cancer, HER2 Human epidermal growth factor receptor 2
Because ‘two-hit’ inactivation of the causative gene is regarded as a principal feature of molecular pathogenesis of most hereditary tumours, we next examined the LOH status of ATM-associated tumours at 11q22–23 in the tumours from HetAT participants. LOH was found in 14 of the 21 tumours (67%) developed by HetAT participants. Microsatellite analysis performed on a subset of 14 tumours confirmed the OncoScan results, except for 1 tumour (T0005). The one exception may be due to differences in sensitivity of the two methods. In the subsequent analyses, OncoScan results were not considered for this tumour. Sanger sequencing of the tumour-blood DNA pairs suggested that the ATM wild-type allele was lost in all tumours that underwent LOH at this locus (data not shown).
Genome-wide profiling of the 23 ATM-associated tumours revealed multiple copy number aberrations, including those previously reported in breast tumours, such as losses at 8p and gains at 8q [31], occurring in 50% and 70% of ATM-associated tumours, respectively (Fig. 2a and b). Copy number losses at 16q, 17p and 22q, which are known features of breast tumours of the luminal A and B subtypes [31, 36], were also seen in 70% of ATM-associated tumours (Fig. 2b). In addition, 70% of ATM-associated tumours showed copy number losses at 13q14.11-q14.3, 17p13.2-p12 and 21p11.2-p11.1 (Fig. 2b). The 13q14.11-q14.3 locus is 9.6 Mbp long and contains 90 genes, including LCP1 (lymphocyte cytosolic protein 1) and RB1 (RB transcriptional co-repressor 1) (Table 3). This region is included in the 13q12.3-q21 locus identified in high-grade luminal BRCA2-associated tumours as described by Pecuchet et al. [37]. The 17p13.2-p12 locus contains 166 cancer-related genes, including TP53 and MAP2K4 (Table 3). The 21p11.2-p11.1 locus is 1.2 Mbp long and contains only the PTEN-related tyrosine phosphatase gene TPTE, the pseudogene TEKT4P2 and four microRNAs (MIR3648-1, MIR3648-2, MIR3687-1 and MIR3687-2) (Fig. 2b and Table 3). A complete list of genes located in segment losses observed in ≥ 70% of ATM-associated breast tumours is provided in Additional file 1: Table S1.
Table 3.
Copy number losses recurrently observed in the 23 ATM-associated breast tumours
| Locus | Associated morphology | All ATM tumours (N = 23) | ATM tumours with proven biallelic inactivation of ATM (n = 16) | ATM tumours sequenced by WGS (n = 4) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cytogenetic band | Number of genes | Cancer genesa | Cytogenetic band | Number of genes | Cancer genesa | Cytogenetic band | Number of genes | Cancer genesa | ||
| 6q | 20% of ATM tumours | 6q23.3-q27 | 190 | ARID1B, ECT2L, ESR1, EZR, FGFR10P, QKI, TNFAIP3 | ||||||
| 8p | Breast cancer | 8p21.3 | 6b | – | 8p23.3-p12 | 216 | WRN, NRG1 | 8p21.3 | 6b | – |
| 11q | ATM tumours | 11q22 | 2 | ATM, ZBTB16 | 11q21-q25 | 276 | ATM, ZBTB16, DDX10, POU2AF1, SDHD, ZBTB16, PAFAH1B2, PDCK7, KMT2A, MAML2, DDX6, BCL9L, CBL, ARHGEF12, KCNJ5 | 11q21-q24.2 | 236 | ATM, ZBTB16, DDX10, POU2AF1, SDHD, ZBTB16, PAFAH1B2, PDCK7, KMT2A, MAML2, DDX6, BCL9L, CBL |
| 13q | 70% ATM tumours | 13q14.11-q14.3 | 90 | LCP1, RB1 | 13q13.3-q32.3 | 320 | LHFP, FOXO1, LCP1, RB1 | 13q13.3-q32.3 | 320 | LHFP, FOXO1, LCP1, RB1 |
| 16q | Luminal tumours | 16q13-q24.3 | 364 | HERPUD1, CDH11, CBFB, CTCF, CDH1, ZFHX3, MAF, CBFA2T3, FANCA | 16q22.1 | 1c | – | 16q22.1 | 1c | – |
| 17p | Luminal tumours | 17p13.3 | 46 | YWHAE | – | – | 17p13.3 | 46 | YWHAE | |
| 17p | Luminal tumours | 17p13.2-p12 | 166 | USP6, RABEP1, TP53, GAS7, MAP2K4 | – | – | ||||
| 19p | 40% of ATM tumours | 19p13.3-p13.2 | 256 | FSTL3, GNA11, MAP2K2, MLLT1, SH3GL1, STK11, TCF3 | ||||||
| 21p | 70% of ATM tumours | 21p11.2-p11.1 | 2a | – | 21p11.2-p11.1 | 2d | – | |||
| 22q | Luminal B tumours, 70% ATM tumours | 22q11.23 | 2b | 22q11.23 | 3e | GSTT1 | ||||
| Luminal B tumours, 70% ATM tumours | – | – | – | 22q12.3 | 199 | APOBEC3B, MKL1, EP300 | ||||
aAs reported in the COSMIC database
bThis region contains PPP3CC, SORBS3, PDLIM2, BIN3, BIN3-IT1 and EGR3
cThis region contains WWP2 and the microRNA MIR140
dThis region contains TEKT4P2, TPTE and four miRNAs (MIR3648-1, MIR3648-2, MIR3687-1, MIR3687-2)
eThis region contains the pseudogenes GSTTP1 and GSTTP2
When we restricted the analysis to the 16 tumours in which biallelic inactivation of ATM was demonstrated (i.e., breast tumours from A-T participants and breast tumours from HetAT participants showing LOH at the ATM locus), we found that copy number losses at 8p, 11q, 13q and 22q corresponded to longer chromosome segments than the ones described in the 23 ATM-associated tumours. The segment loss at 21p11 was the same as the one initially described (Fig. 2c and Table 3).
We also performed unsupervised hierarchical clustering analyses of CNV data. This analysis did not allow separation of ATM-associated tumours according to molecular subtype, LOH status at 11q22 (ATM locus), type of inherited variant (TV vs. MS) or origin of HetAT participants (A-T families or HBOC families) (Fig. 2b). Interestingly, the synchronous bilateral tumours from HetAT patient T0077 showed similar CNV profiles, whereas tumours from the two A-T participants showed quite distinctive features (Fig. 2b and c).
Finally, although the hierarchical clustering of the CNV data did not separate the tumours according to the variant type (TV vs. MS), we performed a sensitivity analysis excluding tumours of the four HetAT participants carrying an MS. This analysis confirmed that loci 8p21, 13q14, 16q13-q24, 17p13-p12 and 21p11 were sites of recurrent alterations found in ≥ 70% of ATM-associated tumours (Additional file 2: Figure S1). However, after exclusion of these 4 tumours, the boundaries of altered loci were extended, and locus 22q11 was lost in only 12 of the 19 analysed tumours.
Comparison with publicly available data
We used the publicly available data from TCGA [31] accessible through cBioportal [38] to investigate whether the genes listed in Table 3 were specifically lost in ATM-associated tumours. A total of 745 TCGA tumour samples with available CNV data were used. Those tumours were from individuals who developed invasive primary breast tumours and did not carry a deleterious variant in ATM, BRCA1 or BRCA2. In addition to LOH at the ATM locus, which was observed more frequently in ATM-associated tumours (67%) than in the TCGA ‘sporadic’ tumours (40.1%) (P = 0.02), several genes at other loci appeared more frequently lost in ATM-associated tumours, including TPTE (21p11.2-p11.1), GSTT1, GSTTP1 and GSTTP2 (22q11.23), as well as LCP1, RB1 (13q14), YWHAE, USP6, RABEP1 and MAP2K4 (17p13.3-p12) (Additional file 3: Table S2).
Deep whole-genome sequencing of ATM-associated tumours
Whole-genome massively parallel sequencing of four ATM-associated breast tumours (T0001-L, T0015-L, T0077-L and T0077-R) and their respective germline DNA was used to characterise the genetic landscape of ATM-associated tumours at base pair resolution. Tumour DNA was sequenced at a mean depth of coverage of 97× (range 82×–104×), and paired blood DNA was sequenced at a mean depth of coverage of 36× (range 35×–37×). CNV patterns obtained from WGS data for frozen tumours T0015-L, T0077-L and T0077-R were compared with CNV patterns obtained in the OncoScan analysis in the corresponding FFPE tumours. Loss/LOH was confirmed by whole-genome analysis for loci 8p21.3, 11q21-q24.2 (containing ATM), 13q13.3-q32.3, 16q22.1 and 17p13.3, whereas discordant results were obtained at locus 21p11.2-p11.1 for one tumour and at locus 22p11.23 for two tumours (Fig. 3a). Divergent ploidy estimations between the WGS analysis (ploidy 3.5) and the OncoScan analysis (ploidy 4) or the use of different tumour sections to prepare tumour DNA may explain these discrepancies. No OncoScan data were available for tumour T0001, but the CNV profile obtained from the WGS data showed LOH at 11q21-q24.2 (containing ATM) and also loss/LOH at 13q13.3-q32.3, 17p13.3 and 22q12.3-q13.31.
Fig. 3.
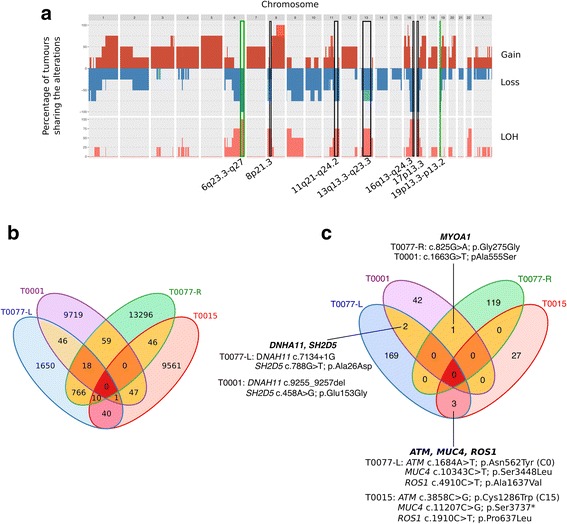
Copy number variation and single-nucleotide variant (SNV) profiles of ataxia-telangiectasia mutated (ATM)-associated tumours obtained by deep whole-genome sequencing (WGS). a Cumulative profiles of copy number gains, losses and of loss of heterozygosity (LOH) regions obtained from WGS of four ATM-associated tumours. Black boxes indicate the genomic regions identified in the OncoScan analysis; green boxes indicate the new genomic regions identified by WGS. b Venn diagram representing the number of somatic SNVs and indels shared between the four tumours. c Venn diagram representing the number of genes altered and shared between the four tumours
In addition we found that the four tumour genomes shared a region of copy number loss/LOH at 6q23.3-q27, which contains ESR1 encoding the ER, as well as a region of copy number loss at 19p13.3-p13.2 measuring 7.9 Mbp (Fig. 3a) and containing 256 genes (Table 3). Going back to the OncoScan data, we found that these two latter regions were indeed altered but in < 40% of the analysed FFPE tumour genomes.
On the basis of our analysis of high-confidence SNVs identified in each ATM-associated tumour genome, we next looked for potential driver mutations. Post-filtering, 51,161 SNVs were identified, 1004 of which were shared by 2 tumours and 29 of which were shared by 3 tumours (Fig. 3b). Only 794 SNVs were shared by the synchronous bilateral tumours T0077-L and T0077-R (Fig. 3b). When analyses were restricted to the coding part of the genome (exome), no genes were found to be altered either in all four tumours or in the two tumours from patient T0077 (Fig. 3c). Six genes were found to be altered in two tumours: MYO1A, DNAH11, SH2D5, ATM, MUC4 and ROS1 (Fig. 3c). However, only DNAH11 variants (c.7134+1G>A and c.9255_9257del) and the nonsense variant in MUC4 (c.11207C>G) are likely to have a deleterious impact on the gene product function and therefore might represent candidate driver mutations. The two ATM somatic variants identified in tumours T0015 and T0077-L were predicted as benign variants according to the Align-GVGD prediction tool (Fig. 3c).
Discussion
This exploratory study in which we investigated both the histological and molecular features of breast tumours developed by subjects who inherited one or two mutated copies of ATM describes, to our knowledge, the largest series of ATM-associated tumours reported to date. One asset of the study design is that the vast majority of participants included in the study carried a loss-of-function or missense variant that had been identified in an A-T family, hence avoiding introduction of noise into the analysis that would be due to misclassification of an ATM variant based on the impact on the protein function. Moreover, all ATM-associated breast tumours and the control series were blindly reviewed by trained reference pathologists of Institut Curie (AVS and GB), thus ensuring unbiased scoring of the morphological features.
The study revealed that most ATM-associated breast tumours are luminal B or luminal B/HER2+ tumours, which is consistent with a recent case-control study showing that ATM TV carriers are at increased risk of developing ER+ breast tumours [39]. Moreover, tetraploidy, loss of the wild-type allele at the ATM locus, and copy number loss/LOH at loci 13q14.11-q14.3, 17p13.2-p12, 21p11.2-p11.1 and 22q11.23 are hallmarks of breast tumours developed by ATM variant carriers.
In comparison with breast tumours associated with other BC susceptibility genes, we thus confirm previous observations showing that ATM-associated tumours do not resemble BRCA1-associated tumours [13, 15] or PALB2-associated tumours, which are also predominantly triple-negative tumours [40, 41]. Like BRCA2- and CHEK2-associated tumours, ATM-associated tumours are mostly luminal tumours [42–44] but they do not show a particular histological subtype as observed in BRCA1- (medullary) [45], BRCA2- (lobular) [45], CDH1- (lobular) [46], and PTEN-associated tumours (apocrine) [47].
The absence of histological resemblance between BRCA1- and ATM-associated tumours was reflected at the molecular level. Indeed, ATM-associated tumours do not show the HRD signature characterised by large-scale state transitions [18, 48], suggesting that tumorigenesis in BRCA1 variant carriers and ATM variant carriers occurs by different mechanisms. ATM-associated tumours also differ from luminal BRCA2-associated tumours, which can also display the HRD signature [35]. Our results are consistent with recently published results on tumour-derived genome sequences from seven BCs from TCGA carrying an ATM TV [49]. However, the absence of HRD in ATM-associated tumours does not exclude the possibility that HetAT subjects who develop BC may be sensitive to cisplatin and/or poly(ADP)-ribose polymerase (PARP) inhibitors, as reported by others for HetAT subjects who developed prostate cancer [50]. Furthermore, it was shown that olaparib induces significant killing of ATM-deficient lymphoid tumour cells from patients with chronic lymphocytic leukaemia [51].
Interestingly, tumour genomic profiling revealed that ~ 70% of ATM-associated breast tumours are tetraploid. Polyploidy can be triggered by cell fusion, endoreplication or abortive cell cycle [52]. ATM is required for three cell cycle checkpoints- G1/S border, S phase and G2/M- after DNA double-strand breaks, so the emergence of polyploidy could be due to cell cycle checkpoint defects linked to inactivation of ATM in breast tumours. Of note, tetraploidy was also reported in BRCA2-associated tumours associated with the luminal molecular subtype and loss of the normal allele [53], although this result was not confirmed when BRCA2 CNV profiles were investigated with SNP array and GAP methods [37].
We found that LOH at the ATM locus was more frequent in tumours from HetAT subjects (67%) than in ‘sporadic’ tumours from TCGA (40%) and from previous studies investigating LOH in tumours from sporadic BC cases (20–40%) [54, 55]. This observation is consistent with Knudson’s ‘two-hit’ model in which the second allele of the tumour suppressor gene would be an early event in the oncogenic process of hereditary BC. Similar results were found in BRCA1- and BRCA2-associated tumours [48]. With regard to ATM-associated tumours, one cannot exclude the possibility that biallelic inactivation of ATM occurs through promoter methylation of the gene in the seven tumours not showing LOH at the ATM locus or through point or small-size sequence variation (Fig. 1c). Another possible explanation for carriers of a deleterious missense variant would be that such alterations might have a dominant negative effect and therefore do not require inactivation of the second allele to impact gene product function. Finally, as previously reported, we did not observe a clear pattern of the biallelic inactivation of ATM according to variant type (TVs vs. deleterious or likely deleterious MS) [13].
In ATM-associated tumours, the cumulative profile of copy number losses, gains and regions in LOH revealed several genomic regions frequently altered in breast tumours, and in particular in luminal tumours, which was consistent with the molecular subtypes defined by IHC staining in our ATM series. Nevertheless, when comparing to TCGA sporadic tumours, the following copy number losses or regions of LOH appeared to be specific to ATM-associated tumours: 13q14.11-q14.3 (LHFP, FOXO1, LCP1, RB1), 21p11.2-p11.1 (TPTE, TEKT4P2, MIR3648-1, MIR3648-2, MIR3687-1, MIR3687-2) and 22q11.23 (GSTT1, GSTTP1, GSTTP2). Interestingly, loss of RB1 has been associated with a poor response to hormone therapy [56], and expression of LCP1 has been proposed as a biomarker of advanced tumour stage and tumour severity [57]. Unfortunately, in our study protein expression analysis could not be performed to validate the diminution of expression of these genes in ATM-associated tumours, owing to limited material.
To extend the repertoire of somatic alterations in ATM-associated tumours, we performed WGS on the four frozen tumours available. Only four deleterious variants located in the two genes DNAH11 and MUC4 were identified in two tumours. Little is known about the role of these two genes in tumourigenesis. However, the diminution of MUC4 expression has been associated with tumour progression and with an increase infiltration of immune CD8+ T and natural killer cells [58]. Remarkably, no mutation in TP53 and PIK3CA was detected in any of the four tumours, although these two genes are frequently mutated in luminal B tumours [59]. Despite the very limited sample size, we found that at the somatic level, ATM-associated tumours were more homogeneous in terms of CNV than in terms of SNV. In particular the two primary tumours from patient T0077 shared only 2.1% of SNVs. Moreover, no specific mutation signature as defined by Alexandrov et al. [60] could be identified using the SNV patterns of these four tumours only, and a larger tumour series should be sequenced to determine whether such signatures can discriminate ATM-associated tumours.
Conclusions
Altogether, ATM-associated tumours do not show the hormone receptor deficiency profile, and it is not clear whether breast tumours developed by HetAT patients could be targeted by alkylating agents or PARP inhibitors [50, 51]. Nonetheless, hallmarks of ATM-associated tumours were found and could help to identify ATM variant carriers outside an A-T context or an HBOC family context. More studies are needed to investigate whether genes located at loci 13q, 21p and 22q could harbour potential new therapeutic targets and whether RB1 deficiency could be a predictive biomarker for hormone therapy response for patients with BC carrying one or two mutated copies of ATM.
Additional files
Table S1. Genomic regions showing copy number loss or loss/LOH in at least 70% of ATM-associated tumours. (XLSX 70 kb)
{kind=link}
Figure S1. Copy number variation profiles of ATM-associated tumours analysed with the OncoScan array. a Genome-wide view of cumulative CNVs present in the 19 ATM-associated tumours from participants carrying one or two copies of a TV. Gains are indicated in red, losses in blue, and LOH in orange. b Cluster dendrogram and genomic regions altered in at least 70% of the 19 analysed tumours. AT children are indicated by asterisks. Loss: the two alleles are present in the tumour; Loss/LOH: only one allele is present in the tumour; Loss/LOH or Loss: consecutive segmental regions characterized as either ‘Loss/LOH’ or ‘Loss’. (PNG 510 kb)
Table S2. Comparison of genes altered at the copy number level between ATM-associated tumours and tumours from TCGA. (XLSX 13 kb)
Acknowledgements
We are most grateful to the families who so willingly participated in the study. We acknowledge David Gentien and Cécile Reyes for technical expertise. We thank all the CoF-AT collaborating cancer clinics: Institut Curie, Paris: Dominique Stoppa-Lyonnet, Bruno Buecher, Antoine de Pauw and Sophie Lejeune-Dumoulin; Hôpital Arnaud de Villeneuve, Montpellier: Isabelle Coupier; ICM Val d’Aurelle, Montpellier: Isabelle Coupier; CHU de Nîmes: Audrey Combès; Centre François Baclesse, Caen: Pascaline Berthet; Hôpital de la Timone, Marseille: Hélène Zattara; Clinique Universitaire Saint-Luc, Bruxelles: Nicolas Janin and Karin Dahan; Centre Oscar Lambret, Lille: Philippe Vennin† and Claude Adenis; Institut Bergonié, Bordeaux: Michel Longy; Centre Jean Perrin, Clermont-Ferrand: Jacques-Olivier Bay; Centre Jean-Paul Strauss, Strasbourg: Jean-Pierre Fricker; Centre Catherine de Sienne, Nantes: Alain Lortholary; CHU de Poitiers, Poitiers: Brigitte Gilbert-Dussardier; Hôpital d’Enfants, Dijon: Laurence Faivre and Caroline Jacquot; Centre Antoine Lacassagne, Nice: Marc Frenay; Institut Claudius Regaud, IUCT Oncopole, Toulouse: Laurence Gladieff; CHU de Grenoble, Grenoble: Dominique Leroux; CHU de Lyon, Lyon: Gaétan Lesca; Gustave Roussy, Villejuif: Agnès Chompret†; Centre Léon Bérard, Lyon: Christine Lasset; Hôtel-Dieu, Chambéry: James Lespinasse; CHI Toulon La Seyne-sur-Mer, Toulon: Xavier Tchiknavorian; Centre Eugène Marquis, Rennes: Catherine Dugast†; CHU du Mans, Le Mans: Dominique Martin-Coignard; CHRU Hôpital Caremeau, Nîmes: Jean Chiesa; Hôpital Universitaire Dupuytren, Limoges: Laurence Venat-Bouvet; Centre René Gauducheau, Nantes: Capucine Delnatte; Hôpital Porte-Madeleine, Orléans: Sonia Nizard; CHU de Nancy, Nancy: Bruno Leheup; Hôpital Sainte-Musse, Toulon: Patrick Collignon; Polyclinique Courlancy, Reims: Liliane Demange†; CHU de Besançon-Hôpital Jean Minjoz, Besançon: Pierre Rohlrich; CHU Vaudois, Lausanne: Florence Fellmann; CHU d’Angers, Angers: Isabelle Pellier; Hôpitaux de Rouen, Rouen: Julie Tinat. We thank more specifically our late colleague Josué Feingold† who was instrumental in the setup of the CoF-AT study. We are also thankful to the GENESIS study collaborators and more specifically to Sylvie Mazoyer, Francesca Damiola and Laure Barjhoux, who managed the GENESIS biological resource until December 2015. In addition, we thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the family cancer clinics, and the clinical follow-up study (which has received funding from the National Health and Medical Research Council [NHMRC], the National Breast Cancer Foundation, Cancer Australia and the National Institutes of Health [USA]) for their contributions to this resource, as well as the many families who contribute to kConFab.
† Deceased
Funding
This work was supported by Inserm and Ministère de la Recherche (grants 01P0751, 01P0752, 01P0753, 01P0754 and 01P0755), Electricité de France (conseil scientifique de Radioprotection d’EDF; grants EP 2002-03, EP 2004-03 and RB 2016-22), Fondation de France (grants 2001009761 and 2005011201), La Ligue (grants PRE04/NA, PRE07/NA and PRE2015 LNCC/NA), La Ligue Comité du Maine et Loire, La Ligue Comité de Paris (grant RS16/75-72), MGEN Union, ITMO Santé Publique d’AVIESAN (grant AAP12-COH-110), Institut National du Cancer (grant INCa-9578), the comprehensive cancer centre SiRIC (Site de Recherche Intégrée sur le Cancer; grant INCa-DGOS-4654), Fondation ARC pour la recherche sur le cancer (grant PJA 20151203365), and the Agence Nationale de la Recherche, program investissements d’avenir (grant ANR-10-EQPX-03). ALR was the recipient of a doctoral fellowship from the Fondation ARC pour la recherche sur le cancer. kConFab is supported by a grant from the National Breast Cancer Foundation and previously by the National Health and Medical Research Council (NHMRC); the Queensland Cancer Fund; the cancer councils of New South Wales, Victoria, Tasmania and South Australia; and the Cancer Foundation of Western Australia.
Availability of data and materials
The datasets generated during and/or analysed during the present study are not publicly available, owing to confidentiality reasons, but anonymised versions may be available from the corresponding author on reasonable request. Anonymised OncoScan data are available in the GEO database under the accession series number GSE111711.
Abbreviations
- A-T
Ataxia-telangiectasia
- ATM
Ataxia-telangiectasia mutated
- BC
Breast cancer
- Bouin
Bouin-fixed, paraffin-embedded tissue sample
- CNV
Copy number variation
- CoF-AT
French prospective cohort on women related to an A-T child
- ER
Oestrogen receptor
- F
Female
- FET
Fisher’s exact test
- FFPE
Formalin-fixed, paraffin-embedded tissue sample
- GAP
Genome Alteration Print tool
- HBOC
Hereditary breast and ovarian cancer
- HER2
Human epidermal growth factor receptor 2
- HES
Hematocylin and Eosin Stained
- HetAT
Heterozygous ATM deleterious variant carrier
- HRD
Homologous recombination deficiency
- IHC
Immunohistochemistry
- kConFab
Kathleen Cuningham Foundation Consortium for research into Familial Breast Cancer study
- L
Left
- LOH
Loss of heterozygosity
- M
Male
- MS
Missense substitution
- Non-HetAT
Non-carrier of an ATM variant
- PARP
Poly(ADP)-ribose polymerase
- PICBIM
Programme incitatif et collaboratif - Cancer du sein series: invasion et motilité
- PR
Progesterone receptor
- R
Right
- Retro-AT
French retrospective study on A-T families
- SNP
Single-nucleotide polymorphism
- SNV
Single-nucleotide variant
- TCGA
The Cancer Genome Atlas
- TNBC
Triple-negative breast cancer
- TV
Truncating variant
- WGS
Whole-genome sequencing
Authors’ contributions
ALR, JH, AVS, DSL, NA and FL conceived of and designed the study. EC, DLG, JB, SEM, ML, IB, SB, JOB, PB, OC, BB, LF, MF, MGV, PG, NJ, SL, CM, SM, LVB, HZ, JPF, LG, IC, GCT, AVS and DSL acquired data (e.g., invited and managed patients, provided facilities). ALR, NM, LF, TP, EG, GB, CDd’E, JH, DSL, NA and FL analysed and interpreted data (molecular analysis, statistical analysis, bioinformatics). ALR, LF, EG, JH, GCT, DSL, NA and FL wrote, reviewed and/or revised the manuscript. ALR, NM, PLR, MGD, AL, WC, VR and ML provided administrative, technical or material support (e.g., reporting or organizing data, constructing databases). FL supervised the study. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent for genetic studies and use of medical records for the present analyses was obtained from all participants enrolled in the Retro-AT, CoF-AT, GENESIS and kConFab research programs. The appropriate local ethics committee (Comité de Protection des Personnes [CCP] Ile-de-France III 2002/2006) and the French data protection authority (Commission Nationale de l’Informatique et des Libertés [CNIL]) approved the individual resource collections; Retro-AT, CoF-AT, and GENESIS study protocols; and the specific study on tumour material of ATM carriers. The kConFab resource collection and the specific study on tumour material of ATM carriers were approved by the Peter MacCallum Cancer Centre Ethics Committee and the Queensland Institute of Medical Research Human Research Ethics Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13058-018-0951-9) contains supplementary material, which is available to authorized users.
Contributor Information
Anne-Laure Renault, Email: anne-laure.renault@curie.fr.
Noura Mebirouk, Email: noura.mebirouk@curie.fr.
Laetitia Fuhrmann, Email: laetitia.fuhrmann@curie.fr.
Guillaume Bataillon, Email: guillaume.bataillon@curie.fr.
Eve Cavaciuti, Email: eve.cavaciuti@curie.fr.
Dorothée Le Gal, Email: dorothee.legal@curie.fr.
Elodie Girard, Email: elodie.girard@curie.fr.
Tatiana Popova, Email: tatiana.popova@curie.fr.
Philippe La Rosa, Email: philippe.la-rosa@curie.fr.
Juana Beauvallet, Email: juana.beauvallet@curie.fr.
Séverine Eon-Marchais, Email: Severine.Eon-Marchais@curie.fr.
Marie-Gabrielle Dondon, Email: marie-gabrielle.dondon@curie.fr.
Catherine Dubois d’Enghien, Email: catherine.dubois-denghien@curie.fr.
Anthony Laugé, Email: anthony.lauge@curie.fr.
Walid Chemlali, Email: walid.chemlali@curie.fr.
Virginie Raynal, Email: virginie.raynal@curie.fr.
Martine Labbé, Email: martine.labbe@gustaveroussy.fr.
Ivan Bièche, Email: ivan.bieche@curie.fr.
Sylvain Baulande, Email: sylvain.baulande@curie.fr.
Jacques-Olivier Bay, Email: jobay@chu-clermontferrand.fr.
Pascaline Berthet, Email: pberthet@baclesse.unicancer.fr.
Olivier Caron, Email: olivier.caron@gustaveroussy.fr.
Bruno Buecher, Email: bruno.buecher@curie.fr.
Laurence Faivre, Email: laurence.faivre@chu-dijon.fr.
Marc Fresnay, Email: marc.frenay@nice.unicancer.fr.
Marion Gauthier-Villars, Email: marion.gauthier-villars@curie.fr.
Paul Gesta, Email: paul.gesta@ch-niort.fr.
Nicolas Janin, Email: nicolas.janin@uclouvain.be.
Sophie Lejeune, Email: sophie.lejeune@chru.lille.fr.
Christine Maugard, Email: christine.maugard@chru-strasbourg.fr.
Sébastien Moutton, Email: sebastien.moutton@chu-bordeaux.fr.
Laurence Venat-Bouvet, Email: laurence.venat@chu-limoges.fr.
Hélène Zattara, Email: helene.zattara@ap-hm.fr.
Jean-Pierre Fricker, Email: jfricker@strasbourg.fnclcc.fr.
Laurence Gladieff, Email: gladieff.laurence@iuct-oncopole.fr.
Isabelle Coupier, Email: i-coupier@chu-montpellier.fr.
Georgia Chenevix-Trench, Email: georgia.trench@qimrberghofer.edu.au.
Janet Hall, Email: janet.hall@inserm.fr.
Anne Vincent-Salomon, Email: anne.salomon@curie.fr.
Dominique Stoppa-Lyonnet, Email: dominique.stoppa-lyonnet@curie.fr.
Nadine Andrieu, Email: nadine.andrieu@curie.fr.
Fabienne Lesueur, Email: fabienne.lesueur@curie.fr.
References
- 1.Micol R, Ben Slama L, Suarez F, Le Mignot L, Beaute J, Mahlaoui N, Dubois d’Enghien C, Lauge A, Hall J, Couturier J, et al. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. J Allergy Clin Immunol. 2011;128(2):382–9.e1. doi: 10.1016/j.jaci.2011.03.052. [DOI] [PubMed] [Google Scholar]
- 2.Suarez F, Mahlaoui N, Canioni D, Andriamanga C, Dubois d’Enghien C, Brousse N, Jais JP, Fischer A, Hermine O, Stoppa-Lyonnet D. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: a report from the French national registry of primary immune deficiencies. J Clin Oncol. 2015;33(2):202–208. doi: 10.1200/JCO.2014.56.5101. [DOI] [PubMed] [Google Scholar]
- 3.Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood. 1996;87(2):423–438. [PubMed] [Google Scholar]
- 4.Borresen AL, Andersen TI, Tretli S, Heiberg A, Moller P. Breast cancer and other cancers in Norwegian families with ataxia-telangiectasia. Genes Chromosomes Cancer. 1990;2(4):339–340. doi: 10.1002/gcc.2870020412. [DOI] [PubMed] [Google Scholar]
- 5.Swift M, Chase CL, Morrell D. Cancer predisposition of ataxia-telangiectasia heterozygotes. Cancer Genet Cytogenet. 1990;46(1):21–27. doi: 10.1016/0165-4608(90)90004-T. [DOI] [PubMed] [Google Scholar]
- 6.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97(11):813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 7.Andrieu N, Cavaciuti E, Laugé A, Ossian K, Janin N, Hall J, Stoppa-Lyonnet D. Ataxia-telangiectasia genes and breast cancer risk in a French family study. J Dairy Res. 2005;72 Spec No:73–80. doi: 10.1017/S0022029905001147. [DOI] [PubMed] [Google Scholar]
- 8.Renault AL, Mebirouk N, Cavaciuti E, Le Gal D, Lecarpentier J, d’Enghien CD, Laugé A, Dondon MG, Labbé M, Lesca G, et al. Telomere length, ATM mutation status and cancer risk in ataxia-telangiectasia families. Carcinogenesis. 2017;38(10):994–1003. doi: 10.1093/carcin/bgx074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 10.Tavtigian SV, Oefner PJ, Babikyan D, Hartmann A, Healey S, Le Calvez-Kelm F, Lesueur F, Byrnes GB, Chuang SC, Forey N, et al. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am J Hum Genet. 2009;85(4):427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, Devilee P, Meindl A, Couch FJ, Southey M, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372(23):2243–2257. doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daly MB, Pilarski R, Berry M, Buys SS, Farmer M, Friedman S, Garber JE, Kauff ND, Khan S, Klein C, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Cancer Netw. 2017;15(1):9–20. doi: 10.6004/jnccn.2017.0003. [DOI] [PubMed] [Google Scholar]
- 13.Goldgar DE, Healey S, Dowty JG, Da Silva L, Chen X, Spurdle AB, Terry MB, Daly MJ, Buys SM, Southey MC, et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. 2011;13(4):R73. doi: 10.1186/bcr2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Os NJ, Roeleveld N, Weemaes CM, Jongmans MC, Janssens GO, Taylor AM, Hoogerbrugge N, Willemsen MA. Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet. 2016;90(2):105–117. doi: 10.1111/cge.12710. [DOI] [PubMed] [Google Scholar]
- 15.Balleine RL, Murali R, Bilous AM, Farshid G, Waring P, Provan P, Byth K, Thorne H. kConFab Investigators, Kirk JA. Histopathological features of breast cancer in carriers of ATM gene variants. Histopathology. 2006;49(5):523–532. doi: 10.1111/j.1365-2559.2006.02538.x. [DOI] [PubMed] [Google Scholar]
- 16.Waddell N, Cocciardi S, Johnson J, Healey S, Marsh A, Riley J, da Silva L, Vargas AC, Reid L. kConFab Investigators, et al. Gene expression profiling of formalin-fixed, paraffin-embedded familial breast tumours using the whole genome-DASL assay. J Pathol. 2010;221(4):452–461. doi: 10.1002/path.2728. [DOI] [PubMed] [Google Scholar]
- 17.Bubien V, Bonnet F, Dupiot-Chiron J, Barouk-Simonet E, Jones N, de Reynies A, MacGrogan G, Sevenet N, Letouzé E, Longy M. Combined tumor genomic profiling and exome sequencing in a breast cancer family implicates ATM in tumorigenesis: a proof of principle study. Genes Chromosomes Cancer. 2017;56(11):788–799. doi: 10.1002/gcc.22482. [DOI] [PubMed] [Google Scholar]
- 18.Popova T, Manie E, Rieunier G, Caux-Moncoutier V, Tirapo C, Dubois T, Delattre O, Sigal-Zafrani B, Bollet M, Longy M, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72(21):5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 19.Janin N, Andrieu N, Ossian K, Lauge A, Croquette MF, Griscelli C, Debre M, Bressac-de-Paillerets B, Aurias A, Stoppa-Lyonnet D. Breast cancer risk in ataxia telangiectasia (AT) heterozygotes: haplotype study in French AT families. Br J Cancer. 1999;80(7):1042–1045. doi: 10.1038/sj.bjc.6690460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinilnikova OM, Dondon MG, Eon-Marchais S, Damiola F, Barjhoux L, Marcou M, Verny-Pierre C, Sornin V, Toulemonde L, Beauvallet J, et al. GENESIS: a French national resource to study the missing heritability of breast cancer. BMC Cancer. 2016;16:13. doi: 10.1186/s12885-015-2028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mann GJ, Thorne H, Balleine RL, Butow PN, Clarke CL, Edkins E, Evans GM, Fereday S, Haan E, Gattas M, et al. Analysis of cancer risk and BRCA1 and BRCA2 mutation prevalence in the kConFab familial breast cancer resource. Breast Cancer Res. 2006;8(1):R12. doi: 10.1186/bcr1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB, Group IUGVW In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat. 2008;29(11):1327–1336. doi: 10.1002/humu.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elston CW, Ellis IO, Pinder SE. Pathological prognostic factors in breast cancer. Crit Rev Oncol Hematol. 1999;31(3):209–223. doi: 10.1016/S1040-8428(99)00034-7. [DOI] [PubMed] [Google Scholar]
- 24.Böcker W. WHO classification of breast tumors and tumors of the female genital organs: pathology and genetics [in German] Verh Dtsch Ges Pathol. 2002;86:116–119. [PubMed] [Google Scholar]
- 25.Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thürlimann B, Senn HJ. Panel members. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013;24(9):2206–2223. doi: 10.1093/annonc/mdt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popova T, Manie E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009;10(11):R128. doi: 10.1186/gb-2009-10-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.La Rosa P, Viara E, Hupe P, Pierron G, Liva S, Neuvial P, Brito I, Lair S, Servant N, Robine N, et al. VAMP: visualization and analysis of array-CGH, transcriptome and other molecular profiles. Bioinformatics. 2006;22(17):2066–2073. doi: 10.1093/bioinformatics/btl359. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44(16):e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valla M, Vatten LJ, Engstrom MJ, Haugen OA, Akslen LA, Bjorngaard JH, Hagen AI, Ytterhus B, Bofin AM, Opdahl S. Molecular subtypes of breast cancer: long-term incidence trends and prognostic differences. Cancer Epidemiol Biomark Prev. 2016;25(12):1625–1634. doi: 10.1158/1055-9965.EPI-16-0427. [DOI] [PubMed] [Google Scholar]
- 33.Nielsen TO, Parker JS, Leung S, Voduc D, Ebbert M, Vickery T, Davies SR, Snider J, Stijleman IJ, Reed J, et al. A comparison of PAM50 intrinsic subtyping with immunohistochemistry and clinical prognostic factors in tamoxifen-treated estrogen receptor-positive breast cancer. Clin Cancer Res. 2010;16(21):5222–5232. doi: 10.1158/1078-0432.CCR-10-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, Cronin M, Baehner FL, Watson D, Bryant J, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol. 2006;24(23):3726–3734. doi: 10.1200/JCO.2005.04.7985. [DOI] [PubMed] [Google Scholar]
- 35.Manie E, Popova T, Battistella A, Tarabeux J, Caux-Moncoutier V, Golmard L, Smith NK, Mueller CR, Mariani O, Sigal-Zafrani B, et al. Genomic hallmarks of homologous recombination deficiency in invasive breast carcinomas. Int J Cancer. 2016;138(4):891–900. doi: 10.1002/ijc.29829. [DOI] [PubMed] [Google Scholar]
- 36.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pecuchet N, Popova T, Manie E, Lucchesi C, Battistella A, Vincent-Salomon A, Caux-Moncoutier V, Bollet M, Sigal-Zafrani B, Sastre-Garau X, et al. Loss of heterozygosity at 13q13 and 14q32 predicts BRCA2 inactivation in luminal breast carcinomas. Int J Cancer. 2013;133(12):2834–2842. doi: 10.1002/ijc.28315. [DOI] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Decker B, Allen J, Luccarini C, Pooley KA, Shah M, Bolla MK, Wang Q, Ahmed S, Baynes C, Conroy DM, et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J Med Genet. 2017;54(11):732–741. doi: 10.1136/jmedgenet-2017-104588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cybulski C, Kluzniak W, Huzarski T, Wokolorczyk D, Kashyap A, Jakubowska A, Szwiec M, Byrski T, Debniak T, Gorski B, et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: a prospective cohort analysis. Lancet Oncol. 2015;16(6):638–644. doi: 10.1016/S1470-2045(15)70142-7. [DOI] [PubMed] [Google Scholar]
- 41.Musolino A, Bella MA, Bortesi B, Michiara M, Naldi N, Zanelli P, Capelletti M, Pezzuolo D, Camisa R, Savi M, et al. BRCA mutations, molecular markers, and clinical variables in early-onset breast cancer: a population-based study. Breast. 2007;16(3):280–292. doi: 10.1016/j.breast.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Cybulski C, Huzarski T, Byrski T, Gronwald J, Debniak T, Jakubowska A, Górski B, Wokołorczyk D, Masojć B, Narod SA, et al. Estrogen receptor status in CHEK2-positive breast cancers: implications for chemoprevention. Clin Genet. 2009;75(1):72–78. doi: 10.1111/j.1399-0004.2008.01111.x. [DOI] [PubMed] [Google Scholar]
- 43.Keeney MG, Couch FJ, Visscher DW, Lindor NM. Non-BRCA familial breast cancer: review of reported pathology and molecular findings. Pathology. 2017;49(4):363–370. doi: 10.1016/j.pathol.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Massink MP, Kooi IE, Martens JW, Waisfisz Q, Meijers-Heijboer H. Genomic profiling of CHEK2*1100delC-mutated breast carcinomas. BMC Cancer. 2015;15:877. doi: 10.1186/s12885-015-1880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mavaddat N, Barrowdale D, Andrulis IL, Domchek SM, Eccles D, Nevanlinna H, Ramus SJ, Spurdle A, Robson M, Sherman M, et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA) Cancer Epidemiol Biomark Prev. 2012;21(1):134–147. doi: 10.1158/1055-9965.EPI-11-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N, Suriano G, Zaor S, Van Manen L, Gilpin C, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297(21):2360–2372. doi: 10.1001/jama.297.21.2360. [DOI] [PubMed] [Google Scholar]
- 47.Banneau G, Guedj M, MacGrogan G, de Mascarel I, Velasco V, Schiappa R, Bonadona V, David A, Dugast C, Gilbert-Dussardier B, et al. Molecular apocrine differentiation is a common feature of breast cancer in patients with germline PTEN mutations. Breast Cancer Res. 2010;12(4):R63. doi: 10.1186/bcr2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, Smith-McCune K, Broaddus R, Lu KH, Chen J, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49(10):1476–1486. doi: 10.1038/ng.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15(8):1781–1791. doi: 10.1158/1535-7163.MCT-15-0945. [DOI] [PubMed] [Google Scholar]
- 52.Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584(12):2652–2661. doi: 10.1016/j.febslet.2010.03.044. [DOI] [PubMed] [Google Scholar]
- 53.Jonsdottir AB, Stefansson OA, Bjornsson J, Jonasson JG, Ogmundsdottir HM, Eyfjord JE. Tetraploidy in BRCA2 breast tumours. Eur J Cancer. 2012;48(3):305–310. doi: 10.1016/j.ejca.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 54.di Iasio MG, Calin G, Tibiletti MG, Vorechovsky I, Benediktsson KP, Taramelli R, Barbanti-Brodano G, Negrini M. Refinement of the LOH region 1 at 11q23.1 deleted in human breast carcinomas and sublocalization of 11 expressed sequence tags within the refined region. Oncogene. 1999;18(8):1635–1638. doi: 10.1038/sj.onc.1202453. [DOI] [PubMed] [Google Scholar]
- 55.Rio PG, Pernin D, Bay JO, Albuisson E, Kwiatkowski F, De Latour M, Bernard-Gallon DJ, Bignon YJ. Loss of heterozygosity of BRCA1, BRCA2 and ATM genes in sporadic invasive ductal breast carcinoma. Int J Oncol. 1998;13(4):849–853. doi: 10.3892/ijo.13.4.849. [DOI] [PubMed] [Google Scholar]
- 56.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8(9):714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koide N, Kasamatsu A, Endo-Sakamoto Y, Ishida S, Shimizu T, Kimura Y, Miyamoto I, Yoshimura S, Shiiba M, Tanzawa H, et al. Evidence for critical role of lymphocyte cytosolic protein 1 in oral cancer. Sci Rep. 2017;7:43379. doi: 10.1038/srep43379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cho JS, Park MH, Lee JS, Yoon JH. Reduced MUC4 expression is a late event in breast carcinogenesis and is correlated with increased infiltration of immune cells as well as promoter hypermethylation in invasive breast carcinoma. Appl Immunohistochem Mol Morphol. 2015;23(1):44–53. doi: 10.1097/PAI.0000000000000041. [DOI] [PubMed] [Google Scholar]
- 59.Marchio C, Geyer FC, Ng CK, Piscuoglio S, De Filippo MR, Cupo M, Schultheis AM, Lim RS, Burke KA, Guerini-Rocco E, et al. The genetic landscape of breast carcinomas with neuroendocrine differentiation. J Pathol. 2017;241(3):405–419. doi: 10.1002/path.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genomic regions showing copy number loss or loss/LOH in at least 70% of ATM-associated tumours. (XLSX 70 kb)
Figure S1. Copy number variation profiles of ATM-associated tumours analysed with the OncoScan array. a Genome-wide view of cumulative CNVs present in the 19 ATM-associated tumours from participants carrying one or two copies of a TV. Gains are indicated in red, losses in blue, and LOH in orange. b Cluster dendrogram and genomic regions altered in at least 70% of the 19 analysed tumours. AT children are indicated by asterisks. Loss: the two alleles are present in the tumour; Loss/LOH: only one allele is present in the tumour; Loss/LOH or Loss: consecutive segmental regions characterized as either ‘Loss/LOH’ or ‘Loss’. (PNG 510 kb)
Table S2. Comparison of genes altered at the copy number level between ATM-associated tumours and tumours from TCGA. (XLSX 13 kb)
Data Availability Statement
The datasets generated during and/or analysed during the present study are not publicly available, owing to confidentiality reasons, but anonymised versions may be available from the corresponding author on reasonable request. Anonymised OncoScan data are available in the GEO database under the accession series number GSE111711.