

Abstract
Background
Cardiovascular disease (CVD) describes the pathological conditions of the heart and blood vessels. Despite the large number of studies on CVD and its etiology, its key modulators remain largely unknown. To this end, we performed a comprehensive proteomic analysis of blood plasma, with the scope to identify disease-associated changes after placing them in the context of existing knowledge, and generate a well characterized dataset for further use in CVD multi-omics integrative analysis.
Methods
LC–MS/MS was employed to analyze plasma from 32 subjects (19 cases of various CVD phenotypes and 13 controls) in two steps: discovery (13 cases and 8 controls) and test (6 cases and 5 controls) set analysis. Following label-free quantification, the detected proteins were correlated to existing plasma proteomics datasets (plasma proteome database; PPD) and functionally annotated (Cytoscape, Ingenuity Pathway Analysis). Differential expression was defined based on identification confidence (≥ 2 peptides per protein), statistical significance (Mann–Whitney p value ≤ 0.05) and a minimum of twofold change.
Results
Peptides detected in at least 50% of samples per group were considered, resulting in a total of 3796 identified proteins (838 proteins based on ≥ 2 peptides). Pathway annotation confirmed the functional relevance of the findings (representation of complement cascade, fibrin clot formation, platelet degranulation, etc.). Correlation of the relative abundance of the proteins identified in the discovery set with their reported concentrations in the PPD was significant, confirming the validity of the quantification method. The discovery set analysis revealed 100 differentially expressed proteins between cases and controls, 39 of which were verified (≥ twofold change) in the test set. These included proteins already studied in the context of CVD (such as apolipoprotein B, alpha-2-macroglobulin), as well as novel findings (such as low density lipoprotein receptor related protein 2 [LRP2], protein SZT2) for which a mechanism of action is suggested.
Conclusions
This proteomic study provides a comprehensive dataset to be used for integrative and functional studies in the field. The observed protein changes reflect known CVD-related processes (e.g. lipid uptake, inflammation) but also novel hypotheses for further investigation including a potential pleiotropic role of LPR2 but also links of SZT2 to CVD.
Electronic supplementary material
The online version of this article (10.1186/s12967-018-1476-9) contains supplementary material, which is available to authorized users.
Keywords: Cardiovascular disease, Proteomics, Plasma, LC–MS/MS, Proteomic dataset, Biomarker, Drug discovery
Background
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality worldwide [1]. CVD comprises multiple clinical conditions affecting the heart and blood vessels which may vary in severity. These include coronary artery disease, peripheral arterial disease, rheumatic heart disease, heart failure, ischemic stroke, myocardial infarction, cardiomyopathy, and others [1]. Common molecular features that have been associated to CVD include oxidative stress, inflammation and extracellular matrix remodeling [2, 3]. However, understanding the molecular mechanisms underlining the onset and progression of the CVD remains incomplete, hindering the development of methods for prevention, early diagnosis, prognosis and targeted therapies. Early stages of CVD can be, at least in part, reversible, but since they are clinically asymptomatic, their diagnosis is challenging. Furthermore, the available treatment strategies mainly aim to suppress the symptoms (i.e. hypertension, hypercholesterolemia and heart load) and not the etiologies of the disease [4, 5]. Thus, a need for deeper understanding of the key molecular mechanisms responsible for the observed pathology in CVD clearly still exists.
A consensus that has arisen the last years is that to tackle a multifactorial and complex disease, such as CVD, a global view of the underlying molecular pathophysiology is required [6]. Towards this direction, several genome-wide association studies (GWASs) have been conducted in the recent years that support the existence of a genetic impact even in late-onset CVD. Smith et al. reviewed statistically robust (with regard to multiple testing, p < 5 × 10−8) GWAS on late-onset CVD and relative traits. The analysis highlighted 92 genetic loci being associated with coronary artery disease, carotid artery disease, ischemic stroke, aortic aneurysm, peripheral vascular disease, atrial fibrillation, valvular disease and correlates of vascular and myocardial function. While only a few of these variations have an established underlying mechanism in the disease, the novel molecular players and pathways revealed by these studies provide new insights in the pathophysiology of the disease [7]. However, the biggest part of heritability of cardiovascular traits is not yet explained. This could be due to the low minor allele frequencies and rare variants that are excluded in GWASs, as well as the gene-environment and gene–gene (epistatic) interactions that are usually not taken into account [8]. Along the same lines, multiple studies on gene expression in CVD have been conducted with the employment of transcriptomics approaches. Chen et al. compared 15 studies that yielded in total 706 genes differentially expressed between coronary artery disease and controls, but only 23 of these genes were replicated in 2, maximal 3 different studies [9]. These differentially expressed genes included annexin A3, fibrinogen alpha chain, guanylate kinase 1, haptoglobin and interferon gamma. The low number of reproducible results may be due to small study cohorts and genetic variability [9].
In parallel to the studies on genetic polymorphisms and transcriptome, several studies investigating changes at the protein level in tissue, plaques, plasma and urine associated with CVD have been conducted (reviewed in [10]). Investigation at the proteome level is of special value when studying disease associated mechanisms, as proteins integrate genomic information with environmental impact, thus changes at the protein level are expected to reflect the disease phenotype especially well. Based on the non- (urine) or minimally invasive (plasma) means for their collection, analysis of the urine and plasma proteome in association to CVD has gained special attention. Urinary protein markers that have been suggested in this context include orosomucoid 1 for chronic heart failure [11]; CD14 in stable coronary artery disease [12]; and uromodulin in hypertension [13]. Extensive analysis of the urine peptidome, using capillary electrophoresis–mass spectrometry (CE–MS), has also been described revealing biomarker profiles for coronary artery disease [14]; heart failure [15]; stroke [16]; hypertension with left ventricular diastolic dysfunction [17, 18]; and acute coronary syndromes [19].
Multiple protein changes have been also reported to date in plasma. These studies, involving use of both gel-based and non-gel-based techniques (reviewed in [20]), addressed the identification of features associated with specific CVD phenotypes, including abdominal aneurism progression [21]; fibrin clot-bound proteins in acute myocardial infarction [22]; stable coronary artery disease pathophysiology [23]; prognostic biomarkers in peripheral arterial occlusive disease [24]; venous thromboembolism risk prediction [25]; and extracellular matrix remodeling in hypertrophic cardiomyopathy [26]. Significant changes in multiple proteins included various forms of apoliproteins [23]; thrombospondin 1 [10]; alpha 2 macroglobulin [10]; transthyretin [24]; complement factor B [24]; platelet-derived growth factor β [25]; and fibronectin [26].
Complementing existing studies and to further improve on global understanding of molecular mechanisms of CVD, we investigated plasma proteomic changes common to different clinical etiologies and severity of CVD from patients undergoing vascular surgery or organ donors, where vascular tissue was available for assessment of CVD. The comparison is made to controls of expected high inflammatory profile (donors deceased from accidents) without any cardiovascular symptom/event, and results are analyzed in the context of existing relevant literature (workflow is presented in Fig. 1). Multiple significant findings include changes previously reported at the GWAS and other molecular levels, collectively providing a well characterized and comprehensive protein dataset to support further data integration studies in the field.
Fig. 1.
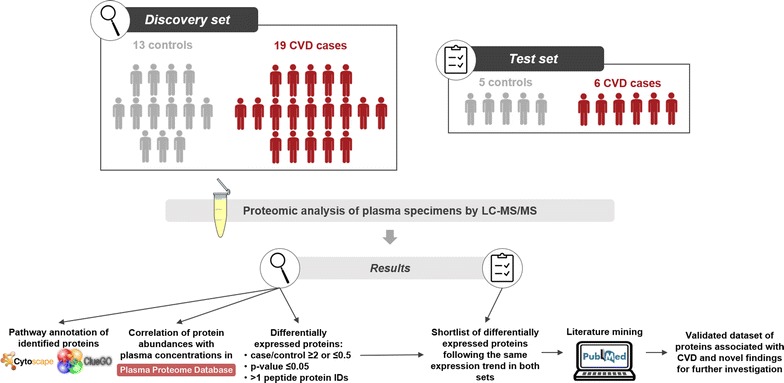
Summary of the workflow followed in the present study. For increased validity, subjects were divided randomly into discovery and test sets. After proteomic analysis of the plasma specimens, pathway annotation was performed for the discovery set proteins and their abundance was correlated with the respective plasma concentrations in the plasma proteome database. Differentially expressed proteins were defined with the following criteria: case/control ratio ≥ 2 or ≤ 0.5; Mann–Whitney p-value ≤ 0.05; protein identifications based on multiple peptides. The differentially expressed proteins with identical expression trend in both sets were shortlisted and extensive literature mining was performed. The resulting validated dataset consists of proteins previously found associated with CVD and novel findings to be further investigated in the context of CVD pathophysiology
Methods
Clinical samples
Blood plasma specimens were collected within the context of a prospective study on CVD (part of the FP7 project SysVASC: Systems Biology to Identify Molecular Targets for Vascular Disease Treatment), from control participants with no cardiovascular background (n = 13) and CVD patients (n = 19) in Medical University of Graz, Graz, Austria. All control and over 40% of the CVD (n = 8) samples were collected from deceased donors. The study was approved by the respective local ethics committee (approval number: 26-355 ex 13/14). CVD subjects were characterized by established vascular disease and had at least one of the following conditions: peripheral artery disease; coronary artery disease; arterial hypertension; cerebral hemorrhage; or carotid stenosis. The characteristics of the participants in this study are presented in Table 1.
Table 1.
Characteristics of the participants
| Variable | Control group (n = 13) | Case group (n = 19) |
|---|---|---|
| Demographics | ||
| Gender [n (%)] | 12 (92.3%) M, 1 (7.7%) F | 10 (52.6%) M, 9 (47.4%) F |
| Age (years) | 42.50 ± 16.8 | 65.8 ± 10.4 |
| Physical characteristics | ||
| Weight (kg) | 82.5 ± 8.7 | 75.1 ± 10.6 |
| Height (cm) | 177.8 ± 9.0 | 169.3 ± 7.3 |
| Body mass index (kg/m2) | 26.1 ± 2.1 | 26.2 ± 3.3 |
| Clinical information [n (%)] | ||
| Hypertension | 2 (15.4%) | 18 (94.7%) |
| Obesity | 1 (7.7%) | 2 (10.5%) |
| Diabetes | 0 (0%) | 5 (26.3%) |
| Dyslipoproteinemia | 0 (0%), 1 (7.7%) NA | 2 (10.5%) |
| Left ventricular hypertrophy | 1 (7.7%) | 6 (31.6%) |
| Angina pectoris | 0 (0%) | 1 (5.3%) |
| Intermittent claudication | 0 (0%), 1 (7.7%) NA | 7 (36.8%) |
| Myocardial infarction | 0 (0%) | 3 (15.8%) |
For gender: M male, F female. Hypertension occurrence corresponds to systolic blood pressure ≥ 140/90 mmHg. Obesity is defined by a body mass index > 30 kg/m2. For dyslipoproteinemia all the following measurements have to occur: total cholesterol ≥ 5.2 mmol/l (≥ 200 mg/dl); low-density lipoprotein (LDL) cholesterol ≥ 3.4 mmol/l (≥ 130 mg/dl); high-density lipoproteins (HDL) cholesterol ≤ 1.0 mmol/l (≤ 40 mg/dl); and triglycerides ≥ 1.7 mmol/l (≥ 150 mg/dl)
NA not available data
Sample preparation
Plasma samples were processed with the filter-aided sample preparation protocol, as previously described by Wisniewski et al. [27]. Specifically, protein concentration of plasma was determined by Bradford assay and 200 µg of protein per sample was mixed with 0.2 ml of 8 M urea in 0.1 M Tris/HCl, pH 8.5 (urea buffer). Reduction was performed with 0.1 M dithioerythritol (DTE) for 20 min (room temperature—RT) and the samples were loaded into 30 kDa Amicon Ultra Centrifugal Filters, Ultracel (Merck Millipore). The filters were then centrifuged at 16,000g for 15 min and washed twice with 0.2 ml of urea buffer. The concentrates were then mixed with 0.1 ml of 50 mM iodoacetamide in urea solution and incubated in the dark, at RT for 20 min (alkylation), followed by centrifugation for 10 min. Then, the filters were washed once with 0.1 ml of urea solution and twice with 0.1 ml of 50 mM NH4HCO3, pH 8.5. Finally, samples were digested with trypsin (proteomics grade) overnight, in the dark at RT (trypsin/protein ratio: 1/100) and the peptides were collected by centrifugation of the filter units for 10 min at 16,000g in clean tubes. The filters were washed once more with 40 µl of 50 mM NH4HCO3 and eluted peptides were collected in the same tube. The eluates were lyophilized and stored at − 20 °C until further processing.
LC–MS/MS analysis
Five microgram of protein digest was loaded onto a Dionex Ultimate 3000 RSLS nano flow system (Dionex, Camberly UK). After loading onto a Dionex 0.1 × 20 mm 5 μm C18 nano trap column at a flow rate of 5 μl/min in 0.1% formic acid and 2% acetonitrile, samples were applied onto an Acclaim PepMap C18 nano column 75 μm × 50 cm (Dionex, Sunnyvale, CA, USA), 2 μm 100 Å at a flow rate of 0.3 μl/min. The trap and nano flow column were maintained at 35 °C. The samples were eluted with a gradient of solvent A: 0.1% formic acid and 2% acetonitrile versus solvent B: 0.1% formic acid and 80% acetonitrile starting at 1% B for 5 min rising to 5% B at 10 min then to 25% B at 360 min and 65% B at 480 min. The column was then washed and re-equilibrated prior to injection of the next sample. The eluent was ionized using a Proxeon nano spray ESI source operating in positive ion mode into an Orbitrap Velos FTMS (Thermo Finnigan, Bremen, Germany). Ionization voltage was 2.6 kV and the capillary temperature was 275 °C. The mass-spectrometer was operated in MS/MS mode scanning from 380 to 1600 amu. The resolution of ions in MS1 was 60,000 and 7500 for higher-energy collisional dissociation (HCD) MS2. The top 20 multiply charged ions were selected from each scan for MS/MS analysis using HCD at 40% collision energy. AGC settings were 1,000,000 for full scan in the FTMS and 200,000 for MSn. Dynamic exclusion was enabled with a repeat count of 1, exclusion duration of 30 s.
MS data processing
Protein identification was performed with Proteome Discoverer 1.4 (Thermo Scientific) using the SEQUEST search engine [28]. Protein search was performed against the SwissProt human protein database [29] (30.05.2016) containing 20,197 reviewed entries. The following search parameters were applied: (i) precursor mass tolerance: 10 ppm and fragment mass tolerance: 0.05 Da; (ii) full tryptic digestion; (iii) max missed cleavage sites: 2; (iv) static modifications: carbamidomethylation of cysteine; (v) dynamic modifications: oxidation of methionine; (vi) event detector mass precision: 2 ppm; (vii) precursor mass: 600–5000 Da; (viii) collision energy: 0–1000 eV; (ix) target FDR (strict): 0.01; (x) target FDR (relaxed): 0.05; (xi) FDR validation based on: q-value. Obtained results were further processed by applying the following filters: (i) high, medium and low confidence peptides (FDR < 10%); (ii) peptide rank up to 5; (iii) peptide grouping was enabled and protein grouping was disabled. The list of peptides was exported and processed using a clustering approach that allows combining the data sets acquired in the course of multiple MS runs. This approach groups features (as defined by calibrated retention time and mass) into clusters, followed by the sequence assignment. In this way, data are harmonized, and consistency in sequence assignment when multiple samples are analyzed in individual experiments is achieved. In brief, data were processed in the following steps: (1) calibration: to adjust for sample to sample LC retention time (rt) shifts, rt was calibrated using LOWESS (locally weighted scatterplot smoothing) non-parametric regression method [30]. To identify and calibrate respective peptides across the whole study, a peptide list from one sample (i.e. sample 27—control) covering the full mass and rt range was selected as reference, based on which alignment/calibration of rt of all samples was performed. (2) Clustering: peptides, as defined by mass and calibrated rt, from all exports from Proteome Discoverer were compiled into a single file. This compiled list was used as an input file for clustering. Clustering was then performed according to the following algorithm: all data points (pairs of mass and rt) were placed in a 2-dimensional plane. Clusters (mass window of ± 5 ppm and retention time window of ± 5% of the feature’s rt) were defined and moved until capturing most data points. The center of these clusters (pairs of mass and rt) were compiled in a “cluster list”, with each of the clusters assigned with a unique identifier. Only clusters with at least 2 members were considered for further analysis. (3) Matching and sequence assignment: as described above, at this stage each cluster includes a set of features (peptides) within a certain “window” of mass and rt. For each cluster, the sequence with the highest frequency across the study was selected as representative. In case of a tie, the sequence with the highest Xcorr was selected. (4) Protein annotation: for each assigned sequence, Uniprot ID was annotated with protein name and gene symbol, based on the respective Uniprot database. In the case of one peptide being assigned to multiple proteins, the protein represented with highest frequency across the cluster list was selected. (5) Retrieve peptide information for further quantification: peptide area, confidence, Xcorr and mass/retention time information were retrieved from the peptide lists of each sample according to their cluster IDs. Only peptides < 5 ppm difference between experimental and theoretical mass were considered. (6) Quantification: Samples were randomly divided into discovery and validation set based on the rule 2/3 (n = 22, 13 cases and 8 controls) and 1/3 (n = 11, 6 cases and 5 controls), respectively. Each set of samples was analyzed separately as follows: for a limited number of sequences for which no peptide area could be retrieved by proteome discoverer (this is a well-known, but not yet corrected problem of this software), the missing values were replaced by the mean area values of that group. When the peptide was not identified in the particular sample, the missing values were replaced with zero. For the discovery set, only peptides reported in more than 50% of the samples of at least one group were included in the differential expression analysis, while no filters were applied in the test set analysis. Subsequently, part per million (ppm)-normalization of the peptide peak areas was conducted according to the following formula: normalized peak area = (peptide peak area/total peak area) × 106. Protein abundance in each sample was calculated as the sum of all normalized peptide areas for a given protein, as described previously [31]. Statistical analysis was based on the Mann–Whitney test performed using R package. Proteins with p-value ≤ 0.05 were considered as statistically significant.
Functional analysis
Functional analysis was performed with the ClueGO plug-in [32] in Cytoscape 3.4.0 [33] as well as the Ingenuity Pathway Analysis software (QIAGEN Inc.) [34]. In regard to the ClueGO analysis, ontologies were retrieved from REACTOME pathways database (updated on September 4, 2017) and only statistical significant pathways (Bonferroni corrected p-value ≤ 0.05, two-sided hypergeometric test) were taken into account. For the remaining parameters, default settings were used. Results were simplified based on biological relevance and only the leading term from each group is presented.
Correlation analysis
Spearman rank correlation analysis was performed using IBM SPSS Statistics for Windows, Version 22.0 after logarithmic transformation of the values.
Results
Discovery set analysis: establishing validity of the proteomics dataset
In total, 32 plasma samples, 13 controls and 19 CVD cases, were analyzed by high-resolution LC–MS/MS. To establish confidence in the validity of the results from the differential expression analysis, the samples were randomly divided into discovery (2/3 of the samples; corresponding to 21 samples—13 cases and 8 controls) and test (1/3 of the samples; corresponding to 11 samples—6 cases and 5 controls) sets. In the discovery set, only peptides detected in at least 50% of samples per group (cases or controls) were considered, resulting in a total of 3796 identified proteins. Of these, 838 protein identifications were based on at least 2 peptides. The full list of identified peptides and proteins is provided in Additional file 1. To confirm the biological relevance of this dataset, pathway annotation was performed using the ClueGO plug-in of Cytoscape platform (Additional file 2). As shown, the detected proteins segregated into (plasma) biologically relevant pathways such as complement cascade, scavenging heme from plasma, fibrin clot formation and platelet degranulation (these being among the statistically significant formed pathways—Additional file 2). When only identifications based on at least 2 peptides were included in the analysis, HDL assembly and TGF-b signaling also emerged among the statistically significant pathways (Additional file 2). Collectively, this phenotyping of the findings via pathway annotation verified that the proteins identified via the applied proteomics approach are biologically relevant and the ones expected to be found in plasma based on the current knowledge.
To assess the reliability of the applied quantification approach, the relative abundance of proteins from the discovery set was compared to the respective absolute concentrations reported in the plasma proteome database (PPD) (http://plasmaproteomedatabase.org/index.html) [35]. For this comparison, PPD entries originating from spectral counting experiments on plasma were considered. This corresponded to 299 proteins (286 proteins in the controls, 288 proteins in the cases) reported in both PPD and our study (listed in Additional file 3). Spearman rank correlation analysis revealed significant correlation between the reported PPD concentrations and the observed protein abundances in our datasets (rs = 0.521, p-value < 0.0001 for controls; and rs = 0.536, p-value < 0.0001 for cases). Scatter plots of these comparisons are presented in Fig. 2. This result supports that the applied quantification strategy is appropriate incorporating state of the art knowledge.
Fig. 2.
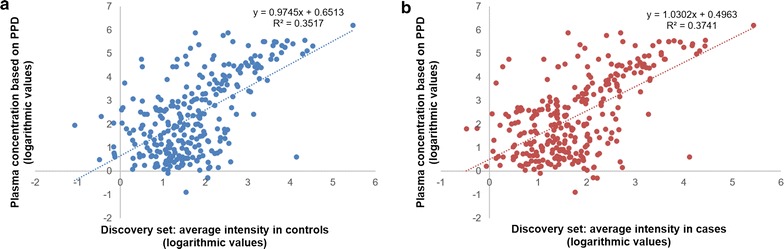
Scatter plots showing the relative abundances of identified proteins and their plasma concentration in PPD. a Proteins identified in controls and b proteins identified in cases. Scatter plots were created after logarithmic transformation of the values
Test set analysis: differential expression analysis reveals expected and novel protein changes
To increase reliability in the differential expression analysis in the discovery set, given also the relatively small sample size, only proteins identified based on at least 2 peptides were considered, and differential expression was defined based on statistical significance (Mann–Whitney p-value ≤ 0.05) and change by at least twofold in the cases versus controls. A total of 100 proteins were found to meet these criteria (listed in Additional file 1). These findings were subsequently examined for validity in an independent test set of 11 samples, including 5 controls and 6 cases (the full list of peptides and proteins identified in the test set is provided in Additional file 3). Spearman rank correlation analysis revealed significant correlation between the protein abundances of discovery and test sets (rs = 0.850, p-value < 0.0001 for controls; and rs = 0.893, p-value < 0.0001 for cases). Out of the 100 differentially expressed proteins, 39 proteins were detected at the same expression trend (up- or down-regulated) and with a fold change of at least 2, when comparing cases and controls in the test set (Table 2, Additional file 3). Of these 39 proteins, 9 showed statistically significant changes (Mann–Whitney p-value ≤ 0.05) also in the test set (Table 2, Additional file 3) and thirteen were described in the past as associated with cardiovascular disease, further supporting the validity of our approach. As shown in Table 2, the latter proteins included alpha-2-macroglobulin; apolipoprotein B; heparin cofactor 2; lipopolysaccharide-binding protein; latent-transforming growth factor beta-binding protein 2; and others.
Table 2.
Shortlisted differentially expressed proteins following the same expression trend in both discovery and test sets
| Uniprot ID | Name | Discovery set | Test set | CVD relevant publications | ||||
|---|---|---|---|---|---|---|---|---|
| Number of peptides | Cases/controls | Mann–Whitney p-value | Number of peptides | Cases/controls | Mann–Whitney p-value | |||
| Q96M60 | Protein FAM227B | 2 | 0.44 | 7.58E−04 | 2 | 0.38 | 8.11E−03 | |
| Q03164 | Histone-lysine N-methyltransferase 2A | 2 | 0.38 | 9.84E−04 | 2 | 0.12 | 8.11E−03 | |
| Q9NVW2 | E3 ubiquitin-protein ligase RLIM | 3 | 2.66 | 4.23E−03 | 4 | 2.46 | 8.11E−03 | |
| O60494 | Cubilin | 2 | 15.19 | 1.18E−03 | 2 | 27.19 | 1.29E−02 | |
| P01023 | Alpha-2-macroglobulin | 64 | 2.37 | 1.27E−03 | 82 | 2.50 | 1.37E−02 | [39, 40] |
| Q02880 | DNA topoisomerase 2-beta | 3 | 3.86 | 2.98E−03 | 9 | 3.25 | 1.37E−02 | |
| Q8WWI1 | LIM domain only protein 7 | 2 | 3.26 | 4.23E−03 | 2 | 3.33 | 2.25E−02 | [76] |
| O75665 | Oral-facial-digital syndrome 1 protein | 2 | 3.01 | 1.86E−02 | 2 | 3.36 | 2.25E−02 | [77] |
| P36957 | Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial | 2 | 2.67 | 4.61E−02 | 2 | 5.50 | 3.53E−02 | [78] |
| O75445 | Usherin | 2 | 2.50 | 3.35E−03 | 5 | 9.25 | 5.52E−02 | |
| P05546 | Heparin cofactor 2 | 8 | 2.99 | 3.36E−03 | 17 | 3.99 | 5.52E−02 | [37, 79] |
| Q8IZU0 | Protein FAM9B | 2 | 2.36 | 3.36E−03 | 2 | 4.14 | 5.52E−02 | |
| A0AUZ9 | KAT8 regulatory NSL complex subunit 1-like protein | 2 | 3.30 | 1.53E−02 | 2 | 2.11 | 5.52E−02 | |
| Q92766 | Ras-responsive element-binding protein 1 | 2 | 2.31 | 3.88E−02 | 2 | 3.35 | 6.73E−02 | |
| Q5TEZ5 | Uncharacterized protein C6orf163 | 4 | 2.13 | 1.25E−02 | 4 | 2.28 | 8.28E−02 | |
| P54317 | Pancreatic lipase-related protein 2 | 2 | 0.01 | 4.51E−03 | 3 | Only in controls | 9.96E−02 | |
| Q9H6S0 | Probable ATP-dependent RNA helicase YTHDC2 | 3 | 5.23 | 4.60E−03 | 6 | 2.31 | 1.21E−01 | |
| P01266 | Thyroglobulin | 3 | 0.33 | 9.90E−03 | 4 | 0.46 | 1.21E−01 | |
| P08185 | Corticosteroid-binding globulin | 2 | 2.65 | 3.26E−02 | 4 | 3.35 | 1.21E−01 | [80] |
| Q5T200 | Zinc finger CCCH domain-containing protein 13 | 3 | 8.07 | 4.18E−03 | 3 | 5.71 | 1.35E−01 | |
| P18428 | Lipopolysaccharide-binding protein | 5 | 0.29 | 7.55E−04 | 8 | 0.43 | 1.71E−01 | [81] |
| P98164 | Low-density lipoprotein receptor-related protein 2 | 5 | 4.00 | 1.25E−02 | 10 | 3.92 | 1.71E−01 | [58] |
| Q5T011 | Protein SZT2 | 2 | 0.33 | 1.86E−02 | 3 | 0.37 | 1.71E−01 | |
| P04114 | Apolipoprotein B-100 | 68 | 2.47 | 3.26E−02 | 135 | 2.16 | 1.71E−01 | [41, 42, 46] |
| Q9NS86 | LanC-like protein 2 | 3 | 2.54 | 4.64E−02 | 3 | 2.76 | 1.71E−01 | |
| Q9H7L9 | Sin3 histone deacetylase corepressor complex component SDS3 | 2 | 4.70 | 1.84E−02 | 4 | 2.50 | 2.00E−01 | |
| O60884 | DnaJ homolog subfamily A member 2 | 2 | 6.36 | 2.67E−03 | 2 | 2.21 | 2.31E−01 | |
| Q5T481 | RNA-binding protein 20 | 4 | 4.84 | 9.84E−04 | 4 | 4.61 | 2.35E−01 | [47, 82] |
| Q5VZL5 | Zinc finger MYM-type protein 4 | 2 | 0.36 | 5.30E−03 | 2 | 0.38 | 2.62E−01 | |
| O15553 | Pyrin | 2 | 11.75 | 1.72E−03 | 2 | 2.65 | 2.74E−01 | [49–51, 83] |
| Q96PV0 | Ras/Rap GTPase-activating protein SynGAP | 2 | 0.15 | 7.33E−03 | 3 | 0.49 | 3.11E−01 | |
| Q8N436 | Inactive carboxypeptidase-like protein X2 | 2 | 0.46 | 1.84E−02 | 3 | 0.37 | 3.11E−01 | |
| Q14767 | Latent-transforming growth factor beta-binding protein 2 | 4 | 0.28 | 7.58E−04 | 6 | 0.43 | 3.15E−01 | [84] |
| Q8IWB6 | Inactive serine/threonine–protein kinase TEX14 | 2 | 4.64 | 3.84E−02 | 4 | 2.43 | 3.60E−01 | |
| Q5T9S5 | Coiled-coil domain-containing protein 18 | 2 | 0.37 | 2.01E−02 | 3 | 0.25 | 3.89E−01 | |
| Q8N163 | Cell cycle and apoptosis regulator protein 2 | 2 | 0.21 | 3.51E−02 | 3 | 0.17 | 3.89E−01 | |
| Q9GZU1 | Mucolipin-1 | 2 | 0.35 | 1.46E−02 | 3 | 0.48 | 4.10E−01 | |
| O43301 | Heat shock 70 kDa protein 12A | 2 | 3.16 | 1.68E−02 | 2 | 3.91 | 4.11E−01 | [85] |
| A6NES4 | Maestro heat-like repeat-containing protein family member 2A | 2 | 0.28 | 7.55E−04 | 3 | 0.24 | 6.48E−01 | |
Mann–Whitney p-values in italics font indicate values ≤ 0.05
Interestingly, molecular pathways that these 39 proteins participate in, based on the Ingenuity Pathway Analysis (IPA), included acute phase signaling; LXR/RXR activation; interleukin-6 signaling; coagulation system; and iNOS signaling (Additional file 4; canonical pathways), suggesting potential impact on these processes based on the observed protein abundance changes in CVD. In fact, and despite the small input dataset (39 proteins), most of the aforementioned pathways (with the exception of iNOS signaling) were predicted to be changing at statistically significant levels in the CVD cases versus controls (Additional file 4).
Along the same lines, IPA predicted significant enrichment of various cardiovascular system- and lipid metabolism-related processes based on the 39 verified proteins; these included uptake of lipid; activation of vascular endothelial cells; atherogenesis; hypercholesterolemia; vasculogenesis; dilated cardiomyopathy; all emerging as statistically significant functions reflected in the verified proteins (Additional file 4; biological functions).
Discussion
The proteomic findings presented in this study provide a good source of information for further analysis and systems biology approaches. Proteomic data were generated from well characterized clinical specimens following a high resolution proteomic approach. The latter has been extensively optimized to provide a comprehensive view of the proteome, without performing abundant plasma protein depletion, frequently associated with technical variability and loss of information (from non-specific protein removal due to antibody cross-reactivity or protein binding) [36]. The identified proteins were investigated regarding their biological relevance, by pathway annotation, and, regarding the reliability of the quantification approach, by cross-correlation with the respective PPD absolute concentrations. As expected, biological pathways, such as acute phase response, interleukin-6, iNOS, and atherosclerosis signaling, and functions, such as activation of vascular endothelial cells, atherogenesis, hypercholesterolemia and transport of lipid, were predicted to be deregulated in CVD based on the observed protein changes.
To our knowledge, this is the first untargeted proteomic study on patients with various CVD-related conditions and severities. Thus, this study is expected to represent a valid foundation and initiation point for further investigation of common molecular mechanisms that underline the variety of CVD traits and conditions.
Interestingly, 13 out of the 39 verified proteins found in our analysis have been already associated with CVD in the literature (Table 2). As examples, Li et al. recently reported that plasma levels of alpha-2-macroglobulin positively correlated with higher vulnerability of carotid plaques, and increased risk of stroke [37]. In a proteomic study of plasma micro-particles, alpha-2-macroglobulin was also found upregulated in patients after deep venous thrombosis in comparison to healthy subjects [38]. In addition, higher alpha-2-macroglobulin serum levels were indicators of cardiac complications in HIV patients [39] and myocardial infarction in diabetic patients [40].
Along the same lines, apolipoprotein B, which we found upregulated in CVD, is widely considered as a major causal agent of atherosclerosis, is a structural component of plasma lipoproteins [i.e. chylomicron remnants, very low-density lipoprotein (VLDL), intermediate-density lipoprotein, LDL, and lipoprotein(a)] and mediates cholesterol transport and removal in vascular wall [41]. Apoliprotein B-100, the form identified in our study, plays a key role in the binding of LDL particles (prominent driver of atherogenesis) to the LDL receptor, allowing cells to internalize LDL and thus to absorb cholesterol [42]. Plasma levels of apoliprotein B and ratio of apoliprotein B/apoliporotein A–I (controls: 0.21, cases: 0.29) are increased in CVD compared to controls in our analysis. This finding is in agreement with studies correlating increased plasma apolipoprotein B and apoliprotein B/apoliporotein A–I ratio with myocardial infarction [43–45] and ischemic stroke [46].
In addition, mutations and polymorphisms in genes that code for some of the validated differentially expressed proteins detected in the present analysis have been connected with CVD. This suggests that these proteins may play important roles in the pathogenesis of CVD, and/or might also serve as early disease markers, hence are worth further investigation. Examples include: Rbm20 (encoding for RNA-binding protein 20) whose mutations in exon 9 of the gene have been linked to familial dilated cardiomyopathy and associated with young age at diagnosis, end-stage heart failure, and high mortality [47, 48]; and MEFV (pyrin encoding gene) whose mutations are a risk factor for early coronary artery disease [49] and are associated with childhood polyarteritis nodosa [50] and vascular complications in Behçet’s disease [51].
Within the verified proteins of our study and with no previous report in CVD to the best of our knowledge, low-density lipoprotein receptor-related protein 2 (LRP2) was also detected at increased levels in the plasma of CVD subjects in comparison to controls. With LRP2 interacting with both apolipoprotein B-100, mediating endocytosis of low density lipoproteins [52], and cubilin, (also upregulated in CVD plasma compared to controls), facilitating endocytosis of high density lipoproteins [53], we may hypothesize a central role of this protein in the accumulation of lipoproteins and the abnormal cholesterol metabolism occurring during CVD [54].
The central role of LRP2 in CVD is further supported via its function as an auxiliary receptor that controls sonic hedgehog (Shh) signaling [55]. Both knock-out mice for Shh [56] and mice carrying mutations in Lrp2 [57] exhibit cardiac outflow tract septation defects. Moreover, Baardman et al. showed that Lrp2 knock-out mice carry a variety of severe cardiovascular abnormalities, such as aortic arch anomalies, ventricular septal defects, overriding of the tricuspid valve and marked thinning of the ventricular myocardium [58]. Collectively, and considering the observed cardioprotective and angiogenic effects of Shh signaling in adulthood [59–61], further investigation of LRP2 as a potential central and pleiotropic node in CVD is suggested.
One additional finding with no previous association to CVD, is Ras-responsive element-binding protein 1 (RREB1), found at increased levels in the plasma of CVD patients compared to controls. Interestingly, RREB1 is an upstream regulator of the renin-angiotensin system acting as a transcriptional suppressor of the angiotensinogen gene [62, 63]. Since angiotensinogen is the precursor molecule of angiotensins I and II that induce vasoconstriction and blood pressure increase [64, 65], we may hypothesize that RREB1 may reflect the upregulation of a compensatory mechanism targeting to reduce hypertension.
Protein SZT2, that was found downregulated in CVD patients’ plasma, has been mainly associated with epileptogenesis and human brain development [66]. Recent reports support that SZT2 deficiency leads to increased mechanistic target of rapamycin complex 1 (mTORC1) signaling [67, 68]. Additionally, hypertrophic signals (such as pressure overload, β-adrenergic stimulation and angiotensin II) can also induce mTORC1 signaling in the heart, resulting in significant phenotypic impact: mTORC1 can induce cardiac hypertrophy as a compensatory mechanism to maintain function during pressure overload but also, and depending on the molecular background, can induce pathological cardiac hypertrophy [69]. Therefore, collectively, we may propose that the observed SZT2 decrease in CVD may be linked to increased mTORC1 signaling, a hypothesis meriting further investigation.
Another protein with no previous association to CVD included in the verified list, is lanC-like protein 2 (LANCL2) found to be at increased plasma levels in CVD patients. LANCL2 is the molecular target of abscisic acid, a natural phytohormone and an endogenous hormone with immune modulatory function [70]. Magnone et al. reported that abscisic acid participates in the development of atherosclerosis through the activation of monocytes and vascular smooth muscle cell responses, and its levels in atherosclerotic plaques are significantly higher than in normal vessels [71]. On the other hand, Guri et al. showed that feeding apolipoprotein E-deficient (ApoE−/−) mice with abscisic acid can ameliorate atherosclerosis by suppressing immune cell recruitment into the aortic root wall and upregulating aortic endothelial nitric oxide synthase expression [72]. They also suggested that, considering the findings of the former study by Magnone et al. [71], abscisic acid may be a part of a lesion-reducing mechanism [72]. Whatever the role of abscisic acid in the atherosclerotic plaque is, its function in inflammation (and presumably atherosclerosis) is mediated by LANCL2 [73]. The increased plasma LANCL2 levels of CVD patients found in our study, in combination with the observed increase of abscisic acid in plaques [71], further supports the proposed scheme.
Further proteins identified in this study and of special interest for further evaluation may be those that take part in cardiovascular system-related processes based mainly on animal model studies, but have not been studied in the human disease yet. DNA topoisomerase 2-beta (TOP2B) is such an example. In the study by Zhang et al. cardiomyocyte-specific deletion of Top2b protected mice from the development of progressive heart failure induced by doxorubicin [74]. Doxorubicin inhibits TOP2B, generating DNA double-strand breaks that are cytotoxic, induce pro-apoptotic DNA damage response and can eventually lead to cardiomyocyte cell death [74, 75]. Nevertheless, a functional connection of the observed increase in plasma TOP2B of CVD patients and the disease remains elusive.
Even though informative, our study also has limitations: the sample size is small especially considering the disease complexity, corresponding to a low statistical power. This largely stems from the fact that these samples were collected in the context of a prospective study where collection of tissue was also targeted to confirm disease pathology. To decrease, as possible, potential artifacts as a result of impact from this limitation, we applied stringent criteria for differential expression and targeted to verify findings based on dataset cross-checking (discovery versus test set; test set versus literature). In addition, one further shortcoming is that there is a significant mean age difference between cases and controls (65.8 versus 43.7 respectively). Nevertheless, following a closer data investigation (study expression levels of the specific proteins after omitting outliers per group and significance in the age difference; data not shown), the highlighted changes were not found to be age-associated.
Conclusions
The proteomic analysis performed in this study, comparing plasma samples from CVD patients to control subjects, provides a comprehensive dataset to be used for further integrative studies in the future. The observed protein changes reflect known CVD-related processes such as changes in lipid uptake and inflammatory processes. Several novel findings are also highlighted forming the basis for hypotheses meriting further investigation including a potential pleiotropic role of LPR2 in CVD development but also links of SZT2 to hypertension regulation.
Additional files
Additional file 1. Discovery set analysis results. List of peptides and proteins along with the detailed protein identification and relative quantification data per sample in the discovery set. The list of differentially expressed proteins is also provided.
Additional file 2. Pathway annotation results. Pathway (Cytoscape) analysis of the proteins identified in the discovery set. Pathways predicted based on proteins identified by at least two peptides are also shown.
Additional file 3. Test set analysis results. List of peptides and proteins along with the detailed protein identification and relative quantification data per sample in the test set. The detailed list of the differentially expressed proteins following the same expression trend (≥ twofold change) in both discovery and test sets is also provided.
Additional file 4. Ingenuity Pathway Analysis of the verified proteins. Lists of the canonical pathways and biological functions that resulted from the Ingenuity Pathway Analysis of the 39 verified proteins.
Authors’ contributions
CD, JPS, JZ, BP, HM and AV planned and supervised the study, BP organized the collection of the samples and coordinates the sysvasc project funding the study, CD and BP contributed with the clinical characterization of the participants, MM performed the sample preparation, WM performed the mass spectrometry analysis, VL, AL and MM analyzed the data, VL performed the literature mining and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
Mosaiques Diagnostics GmbH provided support in the form of salaries for authors AL and HM, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by Medical University of Graz Ethics Committee (Approval Number: 26-355 ex 13/14), Graz, Austria. Informed consent to participate in the study has been obtained for all participants.
Funding
The research presented in this manuscript was supported by European Union’s Seventh Framework Programme (FP7/2007–2013) and the project “Systems Biology to Identify Molecular Targets for Vascular Disease Treatment—SysVasc” under Grant Agreement Number 603288. The funding body had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- CVD
cardiovascular disease
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- PPD
plasma proteome database
- GWAS
genome-wide association study
- CE-MS
capillary electrophoresis-mass spectrometry
- LDL
low-density lipoprotein
- HDL
high-density lipoprotein
- DTE
dithioerythritol
- RT
room temperature
- HCD
higher-energy collisional dissociation
- FDR
false discovery rate
- LXR
liver X receptor
- RXR
retinoid X receptor
- iNOS
inducible nitric oxide synthase
- VLDL
very low-density lipoprotein
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12967-018-1476-9) contains supplementary material, which is available to authorized users.
Contributor Information
Vasiliki Lygirou, Email: vlygirou@hotmail.com.
Agnieszka Latosinska, Email: latosinska@mosaiques-diagnostics.com.
Manousos Makridakis, Email: mmakrid@bioacademy.gr.
William Mullen, Email: william.mullen@glasgow.ac.uk.
Christian Delles, Email: Christian.Delles@glasgow.ac.uk.
Joost P. Schanstra, Email: joost-peter.schanstra@inserm.fr
Jerome Zoidakis, Email: izoidakis@bioacademy.gr.
Burkert Pieske, Email: burkert.pieske@charite.de.
Harald Mischak, Email: mischak@mosaiques-diagnostics.com.
Antonia Vlahou, Email: vlahoua@bioacademy.gr.
References
- 1.Cardiovascular diseases (CVDs)—fact sheet. http://www.who.int/mediacentre/factsheets/fs317/en/. Accessed 23 Aug 2017.
- 2.He F, Zuo L. Redox roles of reactive oxygen species in cardiovascular diseases. Int J Mol Sci. 2015;16:27770–27780. doi: 10.3390/ijms161126059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chistiakov DA, Sobenin IA, Orekhov AN. Vascular extracellular matrix in atherosclerosis. Cardiol Rev. 2013;21:270–288. doi: 10.1097/CRD.0b013e31828c5ced. [DOI] [PubMed] [Google Scholar]
- 4.Brown RA, Shantsila E, Varma C, Lip GY. Current understanding of atherogenesis. Am J Med. 2017;130:268–282. doi: 10.1016/j.amjmed.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 5.Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE, Laurent S, et al. 2007 guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) J Hypertens. 2007;25:1105–1187. doi: 10.1097/HJH.0b013e3281fc975a. [DOI] [PubMed] [Google Scholar]
- 6.Dominiczak AF, Herget-Rosenthal S, Delles C, Fliser D, Fournier I, Graber A, Girolami M, Holmes E, Lang F, Molina F, et al. Systems biology to battle vascular disease. Nephrol Dial Transplant. 2010;25:1019–1022. doi: 10.1093/ndt/gfq025. [DOI] [PubMed] [Google Scholar]
- 7.Smith JG, Newton-Cheh C. Genome-wide association studies of late-onset cardiovascular disease. J Mol Cell Cardiol. 2015;83:131–141. doi: 10.1016/j.yjmcc.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ndiaye NC, Azimi Nehzad M, El Shamieh S, Stathopoulou MG, Visvikis-Siest S. Cardiovascular diseases and genome-wide association studies. Clin Chim Acta. 2011;412:1697–1701. doi: 10.1016/j.cca.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 9.Chen HH, Stewart AF. Transcriptomic signature of atherosclerosis in the peripheral blood: fact or fiction? Curr Atheroscler Rep. 2016;18:77. doi: 10.1007/s11883-016-0634-x. [DOI] [PubMed] [Google Scholar]
- 10.Mokou M, Lygirou V, Vlahou A, Mischak H. Proteomics in cardiovascular disease: recent progress and clinical implication and implementation. Expert Rev Proteomics. 2017;14:117–136. doi: 10.1080/14789450.2017.1274653. [DOI] [PubMed] [Google Scholar]
- 11.Hou LN, Li F, Zeng QC, Su L, Chen PA, Xu ZH, Zhu DJ, Liu CH, Xu DL. Excretion of urinary orosomucoid 1 protein is elevated in patients with chronic heart failure. PLoS ONE. 2014;9:e107550. doi: 10.1371/journal.pone.0107550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee MY, Huang CH, Kuo CJ, Lin CL, Lai WT, Chiou SH. Clinical proteomics identifies urinary CD14 as a potential biomarker for diagnosis of stable coronary artery disease. PLoS ONE. 2015;10:e0117169. doi: 10.1371/journal.pone.0117169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matafora V, Zagato L, Ferrandi M, Molinari I, Zerbini G, Casamassima N, Lanzani C, Delli Carpini S, Trepiccione F, Manunta P, et al. Quantitative proteomics reveals novel therapeutic and diagnostic markers in hypertension. BBA Clin. 2014;2:79–87. doi: 10.1016/j.bbacli.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delles C, Schiffer E, von Zur Muhlen C, Peter K, Rossing P, Parving HH, Dymott JA, Neisius U, Zimmerli LU, Snell-Bergeon JK, et al. Urinary proteomic diagnosis of coronary artery disease: identification and clinical validation in 623 individuals. J Hypertens. 2010;28:2316–2322. doi: 10.1097/HJH.0b013e32833d81b7. [DOI] [PubMed] [Google Scholar]
- 15.Rossing K, Bosselmann HS, Gustafsson F, Zhang ZY, Gu YM, Kuznetsova T, Nkuipou-Kenfack E, Mischak H, Staessen JA, Koeck T, Schou M. Urinary proteomics pilot study for biomarker discovery and diagnosis in heart failure with reduced ejection fraction. PLoS ONE. 2016;11:e0157167. doi: 10.1371/journal.pone.0157167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawson J, Walters M, Delles C, Mischak H, Mullen W. Urinary proteomics to support diagnosis of stroke. PLoS ONE. 2012;7:e35879. doi: 10.1371/journal.pone.0035879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuznetsova T, Mischak H, Mullen W, Staessen JA. Urinary proteome analysis in hypertensive patients with left ventricular diastolic dysfunction. Eur Heart J. 2012;33:2342–2350. doi: 10.1093/eurheartj/ehs185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Staessen JA, Thijs L, Gu Y, Liu Y, Jacobs L, Koeck T, Zurbig P, Mischak H, Kuznetsova T. Left ventricular diastolic function in relation to the urinary proteome: a proof-of-concept study in a general population. Int J Cardiol. 2014;176:158–165. doi: 10.1016/j.ijcard.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Htun NM, Magliano DJ, Zhang ZY, Lyons J, Petit T, Nkuipou-Kenfack E, Ramirez-Torres A, von Zur Muhlen C, Maahs D, Schanstra JP, et al. Prediction of acute coronary syndromes by urinary proteome analysis. PLoS ONE. 2017;12:e0172036. doi: 10.1371/journal.pone.0172036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen X, Young R, Canty JM, Qu J. Quantitative proteomics in cardiovascular research: global and targeted strategies. Proteomics Clin Appl. 2014;8:488–505. doi: 10.1002/prca.201400014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burillo E, Lindholt JS, Molina-Sanchez P, Jorge I, Martinez-Pinna R, Blanco-Colio LM, Tarin C, Torres-Fonseca MM, Esteban M, Laustsen J, et al. ApoA-I/HDL-C levels are inversely associated with abdominal aortic aneurysm progression. Thromb Haemost. 2015;113:1335–1346. doi: 10.1160/TH14-10-0874. [DOI] [PubMed] [Google Scholar]
- 22.Suski M, Siudut J, Zabczyk M, Korbut R, Olszanecki R, Undas A. Shotgun analysis of plasma fibrin clot-bound proteins in patients with acute myocardial infarction. Thromb Res. 2015;135:754–759. doi: 10.1016/j.thromres.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Basak T, Tanwar VS, Bhardwaj G, Bhardwaj N, Ahmad S, Garg G, Sreenivas V, Karthikeyan G, Seth S, Sengupta S. Plasma proteomic analysis of stable coronary artery disease indicates impairment of reverse cholesterol pathway. Sci Rep. 2016;6:28042. doi: 10.1038/srep28042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang CS, Wei YS, Tsai HL, Cheong IS, Chang SJ, Chou HC, Lee YR, Chan HL. Proteomic analysis of prognostic plasma biomarkers in peripheral arterial occlusive disease. Mol BioSyst. 2017;13:1297–1303. doi: 10.1039/C7MB00229G. [DOI] [PubMed] [Google Scholar]
- 25.Bruzelius M, Iglesias MJ, Hong MG, Sanchez-Rivera L, Gyorgy B, Souto JC, Franberg M, Fredolini C, Strawbridge RJ, Holmstrom M, et al. PDGFB, a new candidate plasma biomarker for venous thromboembolism: results from the VEREMA affinity proteomics study. Blood. 2016;128:e59–e66. doi: 10.1182/blood-2016-05-711846. [DOI] [PubMed] [Google Scholar]
- 26.Fucikova A, Lenco J, Tambor V, Rehulkova H, Pudil R, Stulik J. Plasma concentration of fibronectin is decreased in patients with hypertrophic cardiomyopathy. Clin Chim Acta. 2016;463:62–66. doi: 10.1016/j.cca.2016.09.024. [DOI] [PubMed] [Google Scholar]
- 27.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 28.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 29.Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cleveland WS. Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74:829–836. doi: 10.1080/01621459.1979.10481038. [DOI] [Google Scholar]
- 31.Glorieux G, Mullen W, Duranton F, Filip S, Gayrard N, Husi H, Schepers E, Neirynck N, Schanstra JP, Jankowski J, et al. New insights in molecular mechanisms involved in chronic kidney disease using high-resolution plasma proteome analysis. Nephrol Dial Transplant. 2015;30:1842–1852. doi: 10.1093/ndt/gfv254. [DOI] [PubMed] [Google Scholar]
- 32.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-into decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanjappa V, Thomas JK, Marimuthu A, Muthusamy B, Radhakrishnan A, Sharma R, Ahmad Khan A, Balakrishnan L, Sahasrabuddhe NA, Kumar S, et al. Plasma proteome database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014;42:D959–D965. doi: 10.1093/nar/gkt1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, Mann M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2016;2:185–195. doi: 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Li J, Liu X, Xiang Y, Ding X, Wang T, Liu Y, Yin M, Tan C, Deng F, Chen L. Alpha-2-macroglobulin and heparin cofactor II and the vulnerability of carotid atherosclerotic plaques: an iTRAQ-based analysis. Biochem Biophys Res Commun. 2017;483:964–971. doi: 10.1016/j.bbrc.2017.01.042. [DOI] [PubMed] [Google Scholar]
- 38.Ramacciotti E, Hawley AE, Wrobleski SK, Myers DD, Jr, Strahler JR, Andrews PC, Guire KE, Henke PK, Wakefield TW. Proteomics of microparticles after deep venous thrombosis. Thromb Res. 2010;125:e269–e274. doi: 10.1016/j.thromres.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramasamy S, Omnath R, Rathinavel A, Kannan P, Dhandapany PS, Annapoorani P, Balakumar P, Singh M, Ganesh R, Selvam GS. Cardiac isoform of alpha 2 macroglobulin, an early diagnostic marker for cardiac manifestations in AIDS patients. AIDS. 2006;20:1979–1981. doi: 10.1097/01.aids.0000247122.97079.71. [DOI] [PubMed] [Google Scholar]
- 40.Annapoorani P, Dhandapany PS, Sadayappan S, Ramasamy S, Rathinavel A, Selvam GS. Cardiac isoform of alpha-2 macroglobulin—a new biomarker for myocardial infarcted diabetic patients. Atherosclerosis. 2006;186:173–176. doi: 10.1016/j.atherosclerosis.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Shapiro MD, Fazio S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. Res. 2017;6:134. doi: 10.12688/f1000research.9845.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Q, Zhang Y, Xu CB. Apolipoprotein B, the villain in the drama? Eur J Pharmacol. 2015;748:166–169. doi: 10.1016/j.ejphar.2014.08.037. [DOI] [PubMed] [Google Scholar]
- 43.Walldius G, Jungner I. The apoB/apoA-I ratio: a strong, new risk factor for cardiovascular disease and a target for lipid-lowering therapy—a review of the evidence. J Intern Med. 2006;259:493–519. doi: 10.1111/j.1365-2796.2006.01643.x. [DOI] [PubMed] [Google Scholar]
- 44.Walldius G, Jungner I, Holme I, Aastveit AH, Kolar W, Steiner E. High apolipoprotein B, low apolipoprotein A–I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): a prospective study. Lancet. 2001;358:2026–2033. doi: 10.1016/S0140-6736(01)07098-2. [DOI] [PubMed] [Google Scholar]
- 45.Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet. 2004;364:937–952. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 46.Dong H, Chen W, Wang X, Pi F, Wu Y, Pang S, Xie Y, Xia F, Zhang Q. Apolipoprotein A1, B levels, and their ratio and the risk of a first stroke: a meta-analysis and case–control study. Metab Brain Dis. 2015;30:1319–1330. doi: 10.1007/s11011-015-9732-7. [DOI] [PubMed] [Google Scholar]
- 47.Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–773. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basar N, Kisacik B, Ercan S, Pehlivan Y, Yilmaz S, Simsek I, Erdem H, Ozer O, Pay S, Onat AM, Dinc A. Familial Mediterranean fever gene mutations as a risk factor for early coronary artery disease. Int J Rheum Dis. 2014;20(12):2113–2117. doi: 10.1111/1756-185X.12356. [DOI] [PubMed] [Google Scholar]
- 50.Yalcinkaya F, Ozcakar ZB, Kasapcopur O, Ozturk A, Akar N, Bakkaloglu A, Arisoy N, Ekim M, Ozen S. Prevalence of the MEFV gene mutations in childhood polyarteritis nodosa. J Pediatr. 2007;151:675–678. doi: 10.1016/j.jpeds.2007.04.062. [DOI] [PubMed] [Google Scholar]
- 51.Atagunduz P, Ergun T, Direskeneli H. MEFV mutations are increased in Behcet’s disease (BD) and are associated with vascular involvement. Clin Exp Rheumatol. 2003;21:S35–S37. [PubMed] [Google Scholar]
- 52.Stefansson S, Chappell DA, Argraves KM, Strickland DK, Argraves WS. Glycoprotein 330/low density lipoprotein receptor-related protein-2 mediates endocytosis of low density lipoproteins via interaction with apolipoprotein B100. J Biol Chem. 1995;270:19417–19421. doi: 10.1074/jbc.270.33.19417. [DOI] [PubMed] [Google Scholar]
- 53.Hammad SM, Barth JL, Knaak C, Argraves WS. Megalin acts in concert with cubilin to mediate endocytosis of high density lipoproteins. J Biol Chem. 2000;275:12003–12008. doi: 10.1074/jbc.275.16.12003. [DOI] [PubMed] [Google Scholar]
- 54.Ouweneel AB, Van Eck M. Lipoproteins as modulators of atherothrombosis: from endothelial function to primary and secondary coagulation. Vascul Pharmacol. 2016;82:1–10. doi: 10.1016/j.vph.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 55.Christ A, Herzog K, Willnow TE. LRP2, an auxiliary receptor that controls sonic hedgehog signaling in development and disease. Dev Dyn. 2016;245:569–579. doi: 10.1002/dvdy.24394. [DOI] [PubMed] [Google Scholar]
- 56.Washington Smoak I, Byrd NA, Abu-Issa R, Goddeeris MM, Anderson R, Morris J, Yamamura K, Klingensmith J, Meyers EN. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev Biol. 2005;283:357–372. doi: 10.1016/j.ydbio.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, et al. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520–524. doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baardman ME, Zwier MV, Wisse LJ, Gittenberger-de Groot AC, Kerstjens-Frederikse WS, Hofstra RM, Jurdzinski A, Hierck BP, Jongbloed MR, Berger RM, et al. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis Model Mech. 2016;9:413–425. doi: 10.1242/dmm.022053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paulis L, Fauconnier J, Cazorla O, Thireau J, Soleti R, Vidal B, Ouille A, Bartholome M, Bideaux P, Roubille F, et al. Activation of Sonic hedgehog signaling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci Rep. 2015;5:7983. doi: 10.1038/srep07983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmed RP, Haider KH, Shujia J, Afzal MR, Ashraf M. Sonic Hedgehog gene delivery to the rodent heart promotes angiogenesis via iNOS/netrin-1/PKC pathway. PLoS ONE. 2010;5:e8576. doi: 10.1371/journal.pone.0008576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lavine KJ, Kovacs A, Ornitz DM. Hedgehog signaling is critical for maintenance of the adult coronary vasculature in mice. J Clin Invest. 2008;118:2404–2414. doi: 10.1172/JCI34561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Date S, Nibu Y, Yanai K, Hirata J, Yagami K, Fukamizu A. Finb, a multiple zinc finger protein, represses transcription of the human angiotensinogen gene. Int J Mol Med. 2004;13:637–642. [PubMed] [Google Scholar]
- 63.Bonomo JA, Guan M, Ng MC, Palmer ND, Hicks PJ, Keaton JM, Lea JP, Langefeld CD, Freedman BI, Bowden DW. The ras responsive transcription factor RREB1 is a novel candidate gene for type 2 diabetes associated end-stage kidney disease. Hum Mol Genet. 2014;23:6441–6447. doi: 10.1093/hmg/ddu362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carey RM. The intrarenal renin–angiotensin system in hypertension. Adv Chronic Kidney Dis. 2015;22:204–210. doi: 10.1053/j.ackd.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 65.Kobori H, Urushihara M. Augmented intrarenal and urinary angiotensinogen in hypertension and chronic kidney disease. Pflugers Arch. 2013;465:3–12. doi: 10.1007/s00424-012-1143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakamura Y, Togawa Y, Okuno Y, Muramatsu H, Nakabayashi K, Kuroki Y, Ieda D, Hori I, Negishi Y, Togawa T, et al. Biallelic mutations in SZT2 cause a discernible clinical entity with epilepsy, developmental delay, macrocephaly and a dysmorphic corpus callosum. Brain Dev. 2018;40:134–139. doi: 10.1016/j.braindev.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543:438–442. doi: 10.1038/nature21423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature. 2017;543:433–437. doi: 10.1038/nature21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018;122:489–505. doi: 10.1161/CIRCRESAHA.117.311147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu P, Hontecillas R, Philipson CW, Bassaganya-Riera J. Lanthionine synthetase component C-like protein 2: a new drug target for inflammatory diseases and diabetes. Curr Drug Targets. 2014;15:565–572. doi: 10.2174/1389450115666140313123714. [DOI] [PubMed] [Google Scholar]
- 71.Magnone M, Bruzzone S, Guida L, Damonte G, Millo E, Scarfi S, Usai C, Sturla L, Palombo D, De Flora A, Zocchi E. Abscisic acid released by human monocytes activates monocytes and vascular smooth muscle cell responses involved in atherogenesis. J Biol Chem. 2009;284:17808–17818. doi: 10.1074/jbc.M809546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guri AJ, Misyak SA, Hontecillas R, Hasty A, Liu D, Si H, Bassaganya-Riera J. Abscisic acid ameliorates atherosclerosis by suppressing macrophage and CD4+ T cell recruitment into the aortic wall. J Nutr Biochem. 2010;21:1178–1185. doi: 10.1016/j.jnutbio.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bassaganya-Riera J, Guri AJ, Lu P, Climent M, Carbo A, Sobral BW, Horne WT, Lewis SN, Bevan DR, Hontecillas R. Abscisic acid regulates inflammation via ligand-binding domain-independent activation of peroxisome proliferator-activated receptor gamma. J Biol Chem. 2011;286:2504–2516. doi: 10.1074/jbc.M110.160077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 75.Henninger C, Fritz G. Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell Death Dis. 2017;8:e2564. doi: 10.1038/cddis.2016.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li C, Peng Y, Zhou B, Bai W, Rao L. Association of LIM domain 7 gene polymorphisms and plasma levels of LIM domain 7 with dilated cardiomyopathy in a Chinese population. Appl Biochem Biotechnol. 2017;182:885–897. doi: 10.1007/s12010-016-2368-1. [DOI] [PubMed] [Google Scholar]
- 77.Bouman A, Alders M, Oostra RJ, van Leeuwen E, Thuijs N, van der Kevie-Kersemaekers AM, van Maarle M. Oral–facial–digital syndrome type 1 in males: congenital heart defects are included in its phenotypic spectrum. Am J Med Genet A. 2017;173:1383–1389. doi: 10.1002/ajmg.a.38179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kessler M, Berger IM, Just S, Rottbauer W. Loss of dihydrolipoyl succinyltransferase (DLST) leads to reduced resting heart rate in the zebrafish. Basic Res Cardiol. 2015;110:14. doi: 10.1007/s00395-015-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aihara K, Azuma H, Akaike M, Kurobe H, Takamori N, Ikeda Y, Sumitomo Y, Yoshida S, Yagi S, Iwase T, et al. Heparin cofactor II is an independent protective factor against peripheral arterial disease in elderly subjects with cardiovascular risk factors. J Atheroscler Thromb. 2009;16:127–134. doi: 10.5551/jat.E695. [DOI] [PubMed] [Google Scholar]
- 80.Potter JM, Mueller UW, Hickman PE, Michael CA. Corticosteroid binding globulin in normotensive and hypertensive human pregnancy. Clin Sci (Lond) 1987;72:725–735. doi: 10.1042/cs0720725. [DOI] [PubMed] [Google Scholar]
- 81.Lepper PM, Kleber ME, Grammer TB, Hoffmann K, Dietz S, Winkelmann BR, Boehm BO, Marz W. Lipopolysaccharide-binding protein (LBP) is associated with total and cardiovascular mortality in individuals with or without stable coronary artery disease—results from the Ludwigshafen Risk and Cardiovascular Health Study (LURIC) Atherosclerosis. 2011;219:291–297. doi: 10.1016/j.atherosclerosis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Hinze F, Dieterich C, Radke MH, Granzier H, Gotthardt M. Reducing RBM20 activity improves diastolic dysfunction and cardiac atrophy. J Mol Med (Berl) 2016;94:1349–1358. doi: 10.1007/s00109-016-1483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salah S, Rizk S, Lotfy HM, El Houchi S, Marzouk H, Farag Y. MEFV gene mutations in Egyptian children with Henoch–Schonlein purpura. Pediatr Rheumatol Online J. 2014;12:41. doi: 10.1186/1546-0096-12-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bai Y, Zhang P, Zhang X, Huang J, Hu S, Wei Y. LTBP-2 acts as a novel marker in human heart failure—a preliminary study. Biomarkers. 2012;17:407–415. doi: 10.3109/1354750X.2012.677860. [DOI] [PubMed] [Google Scholar]
- 85.Han Z, Truong QA, Park S, Breslow JL. Two Hsp70 family members expressed in atherosclerotic lesions. Proc Natl Acad Sci USA. 2003;100:1256–1261. doi: 10.1073/pnas.252764399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Discovery set analysis results. List of peptides and proteins along with the detailed protein identification and relative quantification data per sample in the discovery set. The list of differentially expressed proteins is also provided.
Additional file 2. Pathway annotation results. Pathway (Cytoscape) analysis of the proteins identified in the discovery set. Pathways predicted based on proteins identified by at least two peptides are also shown.
Additional file 3. Test set analysis results. List of peptides and proteins along with the detailed protein identification and relative quantification data per sample in the test set. The detailed list of the differentially expressed proteins following the same expression trend (≥ twofold change) in both discovery and test sets is also provided.
Additional file 4. Ingenuity Pathway Analysis of the verified proteins. Lists of the canonical pathways and biological functions that resulted from the Ingenuity Pathway Analysis of the 39 verified proteins.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional files.