

Abstract
Background
Recently, numerous laboratory studies have indicated that many microRNAs (miRNAs) are involved in and associated with human diseases and can serve as potential biomarkers and drug targets. Therefore, developing effective computational models for the prediction of novel associations between diseases and miRNAs could be beneficial for achieving an understanding of disease mechanisms at the miRNA level and the interactions between diseases and miRNAs at the disease level. Thus far, only a few miRNA-disease association pairs are known, and models analyzing miRNA-disease associations based on lncRNA are limited.
Results
In this study, a new computational method based on a distance correlation set is developed to predict miRNA-disease associations (DCSMDA) by integrating known lncRNA-disease associations, known miRNA-lncRNA associations, disease semantic similarity, and various lncRNA and disease similarity measures. The novelty of DCSMDA is due to the construction of a miRNA-lncRNA-disease network, which reveals that DCSMDA can be applied to predict potential lncRNA-disease associations without requiring any known miRNA-disease associations. Although the implementation of DCSMDA does not require known disease-miRNA associations, the area under curve is 0.8155 in the leave-one-out cross validation. Furthermore, DCSMDA was implemented in case studies of prostatic neoplasms, lung neoplasms and leukaemia, and of the top 10 predicted associations, 10, 9 and 9 associations, respectively, were separately verified in other independent studies and biological experimental studies. In addition, 10 of the 10 (100%) associations predicted by DCSMDA were supported by recent bioinformatical studies.
Conclusions
According to the simulation results, DCSMDA can be a great addition to the biomedical research field.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2146-x) contains supplementary material, which is available to authorized users.
Keywords: MiRNA-disease association predictions, Distance correlation set, Disease-lncRNA-miRNA network, Similarity measure
Background
For a long time, RNA was considered a DNA-to-protein gene sequence transporter [1]. The sequencing of the human genome indicates that only approximately 2% of the sequences in human RNA are used to encode proteins [2]. Furthermore, numerous studies performing biological experiments have indicated that noncoding RNA (ncRNA) plays an important role in numerous critical biological processes, such as chromosome dosage compensation, epigenetic regulation and cell growth [3–5]. MicroRNAs (miRNAs) are endogenous single-stranded ncRNA molecules approximately 22 nt in length that regulate the expression of target genes by base pairing with the 3′-untranslated regions (UTRs) of the target genes [6, 7]. Recently, several studies have reported that more than one-third of genes are regulated by miRNAs [8], and more than 1000 miRNAs have been identified using various experimental methods and computational models [9, 10]. In addition, accumulating evidence indicates that many microRNAs (miRNAs) are involved in and associated with human diseases, such as myocardial disease, Alzheimer’s disease, cardiovascular disease and heart disease [11–14]. Therefore, identifying disease-miRNA associations could not only improve our knowledge of the underlying disease mechanism at the miRNA level but also facilitate disease biomarker detection and drug discovery for disease diagnosis, treatment, prognosis and prevention. However, compared with the rapidly increasing number of newly discovered miRNAs, only a few miRNA-disease associations are known [15, 16]. Developing efficient, successful computational approaches that predict potential miRNA-disease associations is challenging and urgently needed.
Recently, several heterogeneous biological datasets, such as HMDD and miR2Disease, have been constructed [17–19], and several computational methods are used to predict potential miRNA-disease associations based these datasets [20–22]. For example, Jiang et al. developed a scoring system to assess the likelihood that a microRNA is involved in a specific disease phenotype based on the assumption that functionally related microRNAs tend to be associated with phenotypically similar diseases [23]. K. Han et al. developed a prediction method called DismiPred that combines functional similarity and common association information to predict potential miRNA-disease associations based on the central hypothesis offered in several previous studies that miRNAs with similar functions are often involved in similar diseases [24]. Furthermore, Xuan et al. proposed a method called HDMP to predict potential disease-miRNA associations based on weighted k most similar neighbours [25] and developed a method for predicting potential disease-associated microRNAs based on random walk (MIDP) [26]. Chen et al. proposed a prediction method called RWRMDA by implementing random walk on the miRNA functional similarity network and further proposed a model called RLSMDA based on semi-supervised learning by integrating a disease-disease semantic similarity network, miRNA-miRNA functional similarity network, and known human miRNA-disease associations for the prediction of potential disease-miRNA associations [27]. In 2016, based on the assumption that functionally similar miRNAs tend to be involved in similar diseases, Chen et al. developed a prediction model called WBSMDA by integrating known miRNA-disease associations, miRNA functional similarity networks, disease semantic similarity networks, and Gaussian interaction profile kernel similarity networks to uncover potential disease-miRNA associations [28].
In the abovementioned computational models, known miRNA-disease associations are required. However, few lncRNA-disease associations have been recorded in several biological datasets, such as MNDR and LncRNADisease [29, 30], and several studies have shown that lncRNA-miRNA associations are involved in and associated with human diseases [31–33]. Thus, in this article, a new model based on the Distance Correlation Set for MiRNA-Disease Association inference (DCSMDA) was developed to predict potential miRNA-disease associations by integrating known lncRNA-disease and lncRNA-miRNA associations, the semantic similarity and functional similarity of the disease pairs, the functional similarity of the miRNA pairs, and the Gaussian interaction profile kernel similarity for the lncRNA, miRNA and disease. Compared with existing state-of-the-art models, the advantage of DCSMDA is its integration of the similarity of the disease pairs, lncRNA pairs, miRNA pairs, and introduction of the distance correlation set; thus, DCSMDA does not require known miRNA-disease associations. Moreover, leave-one-out cross-validation (LOOCV) was implemented to evaluate the performance of DCSMDA based on known miRNA-disease associations downloaded from the HMDD database, and DCSMDA achieved a reliable area under the ROC curve (AUC) of 0.8155. Moreover, case studies of lung neoplasms, prostatic neoplasms and leukaemia were implemented to further evaluate the prediction performance of DCSMDA, and 9, 10 and 9 of the top 10 predicted associations in these three important human complex diseases have been confirmed by recent biological experiments. In addition, a case study identifying the top 10 lncRNA-disease associations showed that 10 of the 10 (100%) associations predicted by DCSMDA were supported by recent bioinformatical studies and the latest HMDD dataset, effectively demonstrating that DCSMDA had a good prediction performance in inferring potential disease-miRNA associations.
Results
To evaluate the prediction performance of DCSMDA, first, our method was compared with other state-of-the-art methods in the framework of the LOOCV, and then, we analyzed the stability of DCSMDA using three lncRNA-disease datasets. Second, we analyzed the effect of the pre-determined threshold parameter b. Finally, several additional experiments were performed to validate the feasibility of our method.
Performance comparison with other methods
Since our method is unsupervised (i.e., known miRNA-disease associations are not used in the training) and the few proposed prediction models for the large-scale forecasting of the associations between miRNAs and diseases are simultaneously based on known miRNA-lncRNA associations and known lncRNA-disease associations, to validate the prediction performance of our novel model, we compared the prediction performance of DCSMDA with that of three state-of-the-art computational prediction models, including WBSMDA [28], RLSMDA [27] and HGLDA [31]; WBSMDA and RLSMDA are semi-supervised methods that do not require any negative samples, and HGLDA is an unsupervised method developed to predict potential lncRNA-disease associations by integrating known miRNA-disease associations and lncRNA-miRNA interactions.
To compare the performance of DCSMDA with that of WBSMDA and RLSMDA, we adopted the DS5 dataset and the framework of the LOOCV. While the LOOCV was implemented for these three methods, each known miRNA-disease association was left out in turn as the test sample, and we further evaluated how well this test association ranked relative to the candidate sample. Here, the candidate samples comprised all potential miRNA-disease associations without any known association evidence. Then, the testing samples with a prediction rank higher than the given threshold were considered successfully predicted. If the testing samples with a prediction rank higher than the given threshold were considered successfully predicted, then DCSMDA, RLSMDA and WBSMDA were checked in the LOOCV.
To compare the performance of DCSMDA with that of HGLDA, we adopted the DS3 dataset and the framework of the LOOCV. While the LOOCV was implemented for HGLDA, each known lncRNA-disease association was removed individually as a testing sample, and we further evaluated how well this test lncRNA-disease association ranked relative to the candidate sample. Here, the candidate samples comprised all potential lncRNA-disease associations without any known association evidence.
Thus, we could further obtain the corresponding true positive rates (TPR, sensitivity) and false positive rates (FPR, 1-specificity) by setting different thresholds. Here, sensitivity refers to the percentage of test samples that were predicted with ranks higher than the given threshold, and the specificity was computed as the percentage of negative samples with ranks lower than the threshold. The receiver-operating characteristic (ROC) curves were generated by plotting the TPR versus the FPR at different thresholds. Then, the AUCs were further calculated to evaluate the prediction performance of DCSMDA.
An AUC value of 1 represented a perfect prediction, while an AUC value of 0.5 indicated a purely random performance. The performance comparison in terms of the LOOCV results is shown in Fig. 1. In the LOOCV, the DCSMDA (when b was set to 6), RLSMDA, WBSMDA and HGLDA achieved AUCs of 0.8155, 0.7826, 0.7582 and 0.7621, respectively. DCSMDA predicted potential miRNA-disease associations without requiring known miRNA-disease associations. To the best of our knowledge, no methods that rely on known miRNA-disease associations exist. More importantly, considering that known disease-lncRNA associations remain very limited, the performance of DCSMDA can be further improved as additional known miRNA-disease associations are obtained in the future.
Fig. 1.
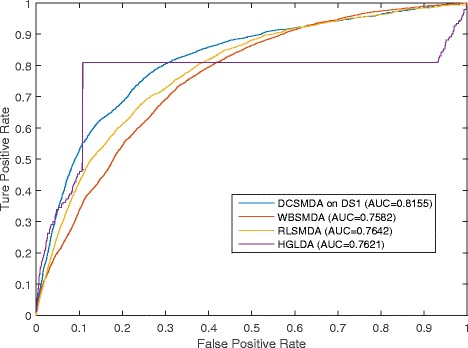
Performance comparisons between DCSMDA, RLSMDA and HGLDA in terms of ROC curve and AUC based on LOOCV
The stability analysis of DCSMDA
Because the current lncRNA-disease databases remain in their infancy and most existing methods are always evaluated using a specific dataset, the stability of the different datasets is ignored. To enhance the credibility of the prediction results, DCSMDA was further implemented using three different known lncRNA-disease association datasets, including DS1, DS2, and DS3, and the known lncRNA-miRNA association dataset DS4.
The comparison results of the ROC are shown in Fig. 2, and the corresponding AUCs are 0.8155, 0.8089 and 0.7642 when DCSMDA (b was set to 6) was evaluated in the framework of the LOOCV using the three different lncRNA-disease association datasets. DCSMDA achieved a reliable and effective prediction performance.
Fig. 2.
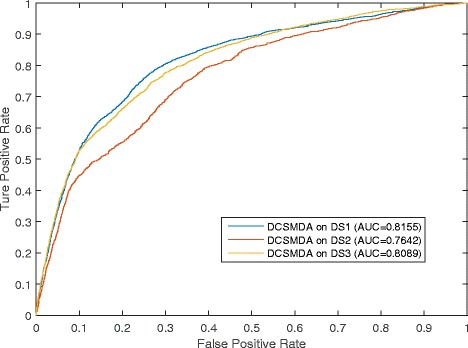
Comparison of different lncRNA-disease datasets to the prediction performance of DCSMDA
Effects of the pre-given threshold parameter b
In DCSMDA, the pre-determined threshold b plays a critical role, and the value of b influences the performance of predicting potential miRNA-disease associations. In this section, we implemented a series of comparison experiments to evaluate the effects of b on the prediction performance of DCSMDA. The LOOCV was implemented, experiments were performed, and b was assigned different values. Considering the time complexity, and that the value of SPM(i, j) always equals 6, when b ≥6, we set b to a value no greater than 6 in our experiments.
As shown in Fig. 3, DCSMDA showed an increasing trend in its prediction performance as the value of the pre-determined threshold parameter b increased and achieved the best prediction performance when b was set to 6. When b was set to 6, DCSMDA achieved an AUC of 0.8089 using DS3 and DS4. In the analysis, we found that the main reason was that the number of known miRNA-lncRNA associations and lncRNA-disease associations was small; thus, when b is set to a larger value, more nodes could be linked to each other in the miRNA-lncRNA-disease interactive network, improving the prediction performance of DCSMDA. Therefore, we finally set b = 6 in our experiments.
Fig. 3.
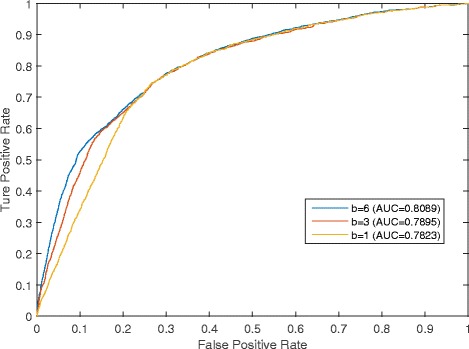
Comparison of effects of the pre-given threshold parameter b to the prediction performance of DCSMDA while b was assigned different values
Case study
Currently, cancer is the leading cause of death in humans worldwide [34–36], and the incidence of cancer is high in both developed and developing countries. Therefore, to estimate the effective predictive performance of DCSMDA, case studies of two important cancers and leukaemia were implemented. The prediction results were verified using recently published experimental studies (see Table 1).
Table 1.
DCSMDA was applied to case studies of three important cancers. In total, 10, 9 and 8 of the top 10 predicted pairs for these diseases were confirmed based on recent experimental studies
| Disease | miRNA | Evidence (PMID and PMCID) |
|---|---|---|
| Prostatic Neoplasms | hsa-mir-15a | PMID: 25418933 |
| Prostatic Neoplasms | hsa-mir-15b | PMID: 24661838 |
| Prostatic Neoplasms | hsa-mir-16 | PMID: 21880514 |
| Prostatic Neoplasms | hsa-mir-195 | PMID: 26080838 |
| Prostatic Neoplasms | hsa-mir-424 | PMID: 27820701 |
| Prostatic Neoplasms | hsa-mir-497 | PMID: 23886135 |
| Prostatic Neoplasms | hsa-mir-125a | PMCID: PMC3979818 |
| Prostatic Neoplasms | hsa-mir-106b | PMID: 26124181 |
| Prostatic Neoplasms | hsa-mir-17 | PMCID: PMC3008681 |
| Prostatic Neoplasms | hsa-mir-93 | PMID: 26124181 |
| Lung Neoplasms | hsa-mir-15a | PMID: 26314859 |
| Lung Neoplasms | hsa-mir-195 | PMID: 25840419 |
| Lung Neoplasms | hsa-mir-424 | PMID: 27666545 |
| Lung Neoplasms | hsa-mir-497 | PMCID: PMC4537005 |
| Lung Neoplasms | hsa-mir-16 | PMID: 21192009 |
| Lung Neoplasms | hsa-mir-15b | Unconfirmed |
| Lung Neoplasms | hsa-mir-125a | PMID: 24044511 |
| Lung Neoplasms | hsa-mir-106a | PMID: 18328430 |
| Lung Neoplasms | hsa-mir-106b | PMID: 18328430 |
| Lung Neoplasms | hsa-mir-93 | PMID: 24037530 |
| Leukaemia | hsa-mir-424 | PMID: 27013583 |
| Leukaemia | hsa-mir-195 | PMCID: PMC4713510 |
| Leukaemia | hsa-mir-16 | PMID:22912766 |
| Leukaemia | hsa-mir-15a | PMID: 24392455 |
| Leukaemia | hsa-mir-15b | PMCID: PMC4577143 |
| Leukaemia | hsa-mir-497 | Unconfirmed |
| Leukaemia | hsa-mir-125a | PMID: 22456625 |
| Leukaemia | hsa-mir-19b | PMID: 28765931 |
| Leukaemia | hsa-mir-19a | PMID: 28765931 |
| Leukaemia | hsa-mir-17 | PMID: 20439436 |
Prostate cancer (prostatic neoplasms), which is the second leading cause of cancer-related death in males, is among the most common malignant cancers and the most commonly diagnosed cancer in men worldwide. In 2012, prostate cancer occurred in 1.1 million men and caused 307,000 deaths. Accumulating evidence shows that microRNAs are strongly associated with prostate cancer. Therefore, DCSMDA was implemented to predict potential prostate cancer-related miRNAs. Consequently, ten of the top ten predicted prostate cancer-related miRNAs were validated by recent biological experimental studies (see Table 1). For example, Junfeng Jiang et al. reconstructed five prostate cancer co-expressed modules using functional gene sets defined by Gene Ontology (GO) annotation (biological process, GO_BP) and found that hsa-mir15a (ranked 1st) regulated these five candidate modules [37]. Medina-Villaamil V et al. analyzed circulating miRNAs in whole blood as non-invasive markers in patients with localized prostate cancer and healthy individuals and found that hsa-mir-15b (ranked 2nd) showed a statistically significant differential expression between the different risk groups and healthy controls [38]. Furthermore, Chao Cai et al. confirmed the tumour suppressive role of hsa-mir-195 (ranked 4th) using prostate cancer cell invasion, migration and apoptosis assays in vitro and tumour xenograft growth, angiogenesis and invasion in vivo by performing both gain-of-function and loss-of-function experiments [39].
Lung cancer (lung neoplasms) has the poorest prognosis among cancers and is the largest threat to people’s health and life. The incidence and mortality of lung cancer are rapidly increasing in China, and approximately 1.4 million deaths are due to lung cancer annually. Recent studies show that miRNAs play critical roles in the progression of lung cancer. Therefore, we used lung cancer as a case study and implemented DCSMDA; nine predicted lung cancer-associated miRNAs of the top ten prediction list were verified based on experimental reports. For example, Bozok Çetintaş V et al. analyzed the effects of selected miRNAs on the development of cisplatin resistance and found that hsa-mir-15a (ranked 1st) was among the most significantly downregulated miRNAs conferring resistance to cisplatin in Calu1 epidermoid lung carcinoma cells [40]. Hsa-mir-195, which ranked 2nd, was further confirmed to suppress tumour growth and was associated with better survival outcomes in several malignancies, including lung cancer [41]. Additionally, according to the biological experiments reported in several studies, hsa-mir-424 (ranked 3rd) plays an important role in lung cancer [42].
Leukaemia refers to a group of diseases that usually begin in the bone marrow and result in high numbers of abnormal white blood cells. The exact cause of leukaemia is unknown, and a combination of genetic factors and environmental factors is believed to play a role. In 2015, leukaemia presented in 2.3 million people and caused 353,500 deaths. Several studies suggest that miRNAs are effective prognostic biomarkers in leukaemia. For example, independent experimental observations showed relatively lower expression levels of mir-424 (ranked 1st) in TRAIL-resistant and semi-resistant acute myeloid leukaemia (AML) cell lines and newly diagnosed patient samples. The overexpression of mir-424 by targeting the 3′ UTR of PLAG1 enhanced TRAIL sensitivity in AML cells [43]. Hsa-mir-16 ranked 3rd, its expression was inversely correlated with Bcl2 expression in leukaemia, and both microRNAs negatively regulate B cell lymphoma 2 (Bcl2) at a posttranscriptional level. Bcl2 repression by these microRNAs induces apoptosis in a leukaemic cell line model [44]. The lncRNA H19 is considered an independent prognostic marker in patients with tumours. The expression of lncRNA H19 is significantly upregulated in bone marrow samples from patients with AML-M2. The results of the current study suggest that lncRNA H19 regulates the expression of inhibitor of DNA binding 2 (ID2) by competitively binding to hsa-mir-19b (ranked 8) and hsa-mir-19a (ranked 9), which may play a role in AML cell proliferation [45].
In addition, DCSMDA predicted all potential associations between the diseases and miRNAs in G3 simultaneously. In addition, notably, potential associations with a high predicted value can be publicly released and benefit from biological experimental validation. To further illustrate the effective performance of DCSMDA, the predicted results were sorted from best to worse, and the top 10 results were selected for analysis (see Table 2). Consequently, 100% of the results were confirmed by recent biological experiments and the HMDD dataset, and thus, DCSMDA can be used as an efficient computational tool in biomedical research studies.
Table 2.
The top 10 predicted miRNA-disease associations by DCSMDA
| Disease | MiRNA | Evidence |
|---|---|---|
| Carcinoma, Hepatocellular | hsa-mir-15a | HMDD |
| Carcinoma, Hepatocellular | hsa-mir-15b | HMDD |
| Carcinoma, Hepatocellular | hsa-mir-16 | HMDD |
| Carcinoma, Hepatocellular | hsa-mir-195 | HMDD |
| Carcinoma, Hepatocellular | hsa-mir-424 | PMID: 26823812 |
| Carcinoma, Hepatocellular | hsa-mir-497 | HMDD |
| Colorectal Neoplasms | hsa-mir-497 | HMDD |
| Colorectal Neoplasms | hsa-mir-15b | PMID: 23267864 |
| Colorectal Neoplasms | hsa-mir-16 | HMDD |
| Colorectal Neoplasms | hsa-mir-195 | HMDD |
Discussion
Accumulating evidence shows that miRNAs play a very important role in several key biological functions and signalling pathways. A large-scale systematic analysis of miRNA-disease data performed by combining relevant biological data is highly important for humans and attractive topics in the field of computational biology. However, only a few prediction models have been proposed for the large-scale forecasting of associations between miRNAs and diseases based on lncRNA information. To utilize the wealth of disease-lncRNA, miRNA-lncRNA and disease-lncRNA association data recorded in four datasets and recently published experimental studies, in this article, we proposed a novel prediction model called DCSMDA to infer the potential associations between diseases and miRNAs. We first constructed a miRNA-lncRNA-disease interactive network and further integrated a distance correlation set, disease semantic similarity, functional similarity and Gaussian interaction profile kernel similarity for DCSMDA. The important difference between DCSMDA and previous computational models is that DCSMDA does not rely on any known miRNA-disease associations and predicts disease-miRNA associations based only on known disease-lncRNA associations and known lncRNA-miRNA associations. To evaluate the prediction performance of DCSMDA, the validation frameworks of the LOOCV were implemented using the HMDD database. Furthermore, case studies were further implemented using three important diseases and the top 10 predicted miRNA-disease associations based on recently published experimental studies and databases. The simulation results showed that DCSMDA achieved a reliable and effective prediction performance. Hence, DCSMDA could be used as an effective and important biological tool that benefits the early diagnosis and treatment of diseases and improves human health in the future.
However, although DCSMDA is a powerful method for predicting novel relationships between diseases and miRNAs, there are several limitations in our method. First, the value of the threshold parameter b plays an important role in DCSMDA, and the selection of a suitable value for b is a critical problem that should be addressed in future studies. Second, although DCSMDA does not rely on any known experimentally verified miRNA-disease relationships, the performance of DCSMDA was not very satisfactory compared with that of several existing methods, such as LRSMDA and WBSMDA [27, 28]. Introducing more reliable measures for the calculations of the disease similarity, miRNA similarity, and lncRNA similarity and developing a more reliable similarity integration method could improve the performance of DCSMDA. Finally, DCSMDA cannot be applied to unknown diseases or miRNAs that are not present in the disease-miRNA or lncRNA-miRNA databases; such genes are poorly investigated and have no known disease-lncRNA and lncRNA-miRNA associations. The performance of DCSMDA will be further improved once more known associations are obtained.
Conclusion
In this article, we mainly achieved the following contributions: (1) we constructed a miRNA-lncRNA-disease interactive network based on common assumptions that similar diseases tend to show similar interaction and non-interaction patterns with lncRNAs, and similar miRNAs tend to show similar interaction and non-interaction patterns with lncRNAs; (2) the concept of a distance correlation set was introduced; (3) the sematic disease similarity, functionally similarity (including disease functionally similarity and miRNA functionally similarity) and Gaussian interaction profile kernel similarity (including disease Gaussian interaction profile kernel similarity, miRNA Gaussian interaction profile kernel similarity and lncRNA Gaussian interaction profile kernel similarity) were integrated; (4) the concept of an optimized matrix was introduced by integrating the Gaussian interaction profile kernel similarity of the miRNA pairs and disease pairs; (5) negative samples are not required in DCSMDA; and (6) DCSMDA can be applied to human diseases without relying on any known miRNA-disease associations.
Methods
Known disease-lncRNA associations
Because the number of lncRNA-disease associations is limited and many heterogeneous biological datasets have been constructed, we collected 8842 known disease-lncRNA associations from the MNDR dataset (http://www.bioinformatics.ac.cn/mndr/index.html) and 2934 known disease-lncRNA associations from the LncRNADisease dataset (http://www.cuilab.cn/lncrnadisease). Since the disease names in the LncRNADisease database differ from those in the MNDR dataset, we mapped the diseases in these two disease-lncRNA association datasets to their MeSH descriptors. After eliminating diseases without any MeSH descriptors, merging the diseases with the same MeSH descriptors and removing the lncRNAs that were not present in the lncRNA-miRNA dataset (DS4) used in this paper, 583 known lncRNA-disease associations (DS1) were obtained from the LncRNADisease dataset (see Additional file 1), and 702 known lncRNA-disease associations (DS2) were obtained from the MNDR dataset (see Additional file 2). Furthermore, after integrating the DS1 and DS2 datasets and removing the duplicate associations, we obtained the DS3 dataset, which included 1073 disease-lncRNA associations (see Additional file 3).
Known lncRNA-miRNA associations
To construct the lncRNA-miRNA network, the lncRNA-miRNA association dataset DS4 was obtained from the starBasev2.0 database (http://starbase.sysu.edu.cn/) in February 2, 2017 and provided the most comprehensive experimentally confirmed lncRNA-miRNA interactions based on large-scale CLIP-Seq data. After the data pre-processing (including the elimination of duplicate values, erroneous data, and disorganized data), removing the lncRNAs that did not exist in the DS3 dataset and merging the miRNA copies that produced the same mature miRNA, we finally obtained 1883 lncRNA-miRNA associations (DS4) (see Additional file 4).
Known disease-miRNA associations
To validate the performance of DCSMDA, the known human miRNA-disease associations were downloaded from the latest version of the HMDD database, which is considered the golden-standard dataset. In this dataset, after eliminating the duplicate associations and miRNA-disease associations involved with other diseases or lncRNAs not contained in the DS3 or DS4, we finally obtained 3252 high-quality lncRNA-disease associations (DS5) (see Additional file 5).
Construction of the disease-lncRNA-miRNA interaction network
To clearly demonstrate the process of constructing the disease-lncRNA-miRNA interaction network, we use the disease-lncRNA dataset DS3 and the lncRNA-miRNA dataset DS4 as examples. We defined L to represent all the different lncRNA terms in DS3 and DS4 and then constructed the disease-lncRNA-miRNA interactive network based on DS3 and DS4 according to the following 3 steps:
Step 1 (Construction of the disease-lncRNA network): Let D and L be the number of different diseases and lncRNAs obtained from DS3, respectively. SD = {d1, d2,..., dD} represents the set of all D different diseases in DS3. SL = {l1, l2,..., lL} represents the set of all L different lncRNAs in DS3, and for any given di ∈ SD and lj∈SL, we can construct the D*L dimensional matrix KAM1 as follows:
| 1 |
Step 2 (Construction of the lncRNA-miRNA network): Let M be the number of different miRNAs obtained from DS4. SM = {m1, m2,..., mM} represents the set of all M different miRNAs in DS4, and for any given mi∈SM and lj∈SL, we can construct the M*L dimensional matrix KAM2 as follows:
| 2 |
Step 3 (Constriction of the disease-lncRNA-miRNA interactive network): Based on the disease-lncRNA network and lncRNA-miRNA network, we can obtain the undirected graph G3 = (V3, E3), where V3 = S D ∪S L ∪S M = {d1, d2,..., dD, lD + 1, lD + 2..., lD + L, mD + L + 1, mD + L + 2..., mD + L + M} is the set of vertices, E3 is the edge set of G3, and di∈SD, lj∈SL, mk∈SM. Here, an edge exists between di and lj in E3KAM1(di, lj) = 1, an edge exists between lj and mk in E3 if KAM2(mk, lj) = 1. Then, for any given a, b∈V3, we can define the Strong Correlation (SC) between a and b as follows:
| 3 |
Notably, although we did not use any known disease-miRNA associations, the diseases and miRNAs can still be indirectly linked by integrating the edges between the disease nodes, the lncRNA nodes and edges between the miRNA nodes and lncRNA nodes in G3.
Disease semantic similarity
We downloaded the MeSH descriptors of the diseases from the National Library of Medicine (http://www.nlm.nih.gov/), which introduced the concept of Categories and Subcategories and provided a strict system for disease classification. The topology of each disease was visualized as a Directed Acyclic Graph (DAG) in which the nodes represented the disease MeSH descriptors, and all MeSH descriptors in the DAG were linked from more general terms (parent nodes) to more specific terms (child nodes) by a direct edge (see Fig. 4). Let DAG(A) = (A, T(A), E(A)), where A represents disease A, T(A) represents the node set, including node A and its ancestor nodes, and E(A) represents the corresponding edge set. Then, we defined the contribution of disease term d in DAG(A) to the semantic value of disease A as follows:
| 4 |
Fig. 4.
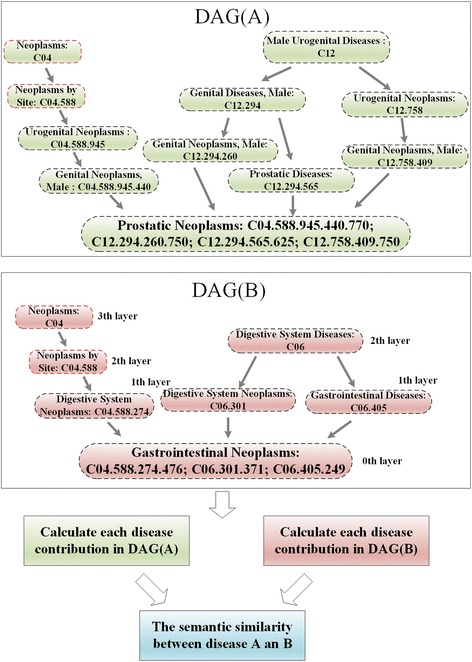
The disease DAGs of Prostatic Neoplasms and Gastrointestinal Neoplasms
For example, the semantic value of the disease ‘Gastrointestinal Neoplasms’ shown in Fig. 4 is calculated by summing the weighted contribution of ‘Neoplasms’ (0.125), ‘Neoplasms by Site’ (0.25), ‘Digestive System Diseases’ (0.25), ‘Digestive System Neoplasms’ (0.5), ‘Digestive System Neoplasms’ (0.5) and ‘Gastrointestinal Diseases’ (0.5) to ‘Gastrointestinal Neoplasms’ and the contribution to ‘Gastrointestinal Neoplasms’ (1) by ‘Gastrointestinal Neoplasms’.
Then, the sematic value of disease A can be obtained by summing the contribution from all disease terms in = DAG(A), and the semantic similarity between the two diseases di and dj can be calculated as follows:
| 5 |
where SSD is the disease semantic similarity matrix.
MiRNA Gaussian interaction profile kernel similarity
Based on the assumption that similar miRNAs tend to show similar interaction and non-interaction patterns with lncRNAs, in this section, we introduce the Gaussian interaction profile kernel used to calculate the network topologic similarity between miRNAs and used the vector MLP(mi) to denote the ith row of the adjacency matrix KAM2. Then, the Gaussian interaction profile kernel similarity for all investigated miRNAs can be calculated as follows:
| 6 |
where parameter M is the number of miRNAs in DS4.
Disease Gaussian interaction profile kernel similarity
Based on the assumption that similar diseases tend to show similar interaction and non-interaction patterns with lncRNAs, the Gaussian interaction profile kernel similarity for all investigated diseases can be calculated as follows:
| 7 |
where parameter D is the number of diseases in DS3, and DLP(di) represent the ith row of the matrix KAM1. Then, based on previous work [46], we can improve the predictive accuracy problems by logistic function transformation as follows:
| 8 |
lncRNA Gaussian interaction profile kernel similarity
Based on the assumption that similar lncRNAs tend to show similar interaction and non-interaction patterns with miRNAs and similar lncRNAs tend to show similar interaction and non-interaction patterns with diseases, the Gaussian interaction profile kernel similarity matrix for all investigated lncRNAs in DS3 can be computed in a similar way as that for disease, as follows:
| 9 |
where parameter L is the number of lncRNAs in DS3, and LDP(li) represents the ith column of the matrix KAM1.
Obviously, the Gaussian interaction profile kernel similarity for all investigated lncRNAs in DS4 can be computed as follows:
| 10 |
where LMP(li) represents the ith column of the matrix KAM2.
Disease functional similarity based on the lncRNAs
To calculate the functional similarity of the diseases, we first constructed the undirected graph G1 = (V1, E1) based on KAM1, where V1 = SD∪SM = {d1, d2, …, dD, lD + 1, lD + 2,…, lD + M} is the set of vertices, E1 is the set of edges, and for any two nodes a, b∈V1, an edge exists between a and b in E1 if KAM1(a, b) = 1. Therefore, we can calculate the similarities between two disease nodes by comparing and integrating the similarities of the lncRNA nodes associated with these two disease nodes based on the assumption that similar diseases tend to show similar interaction and non-interaction patterns with lncRNAs. The procedure used to calculate the disease functional similarity is shown in Fig. 5.
Fig. 5.
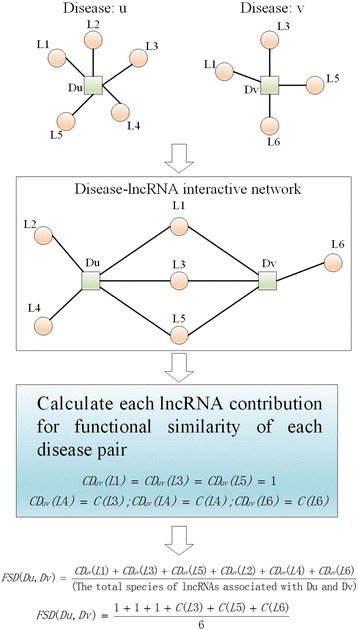
The Flow chart of the disease functional similarity calculation model
Because different lncRNA terms in DS3 may relate to several diseases, assigning the same contribution value to all miRNAs is not suitable, and therefore, we defined the contribution value of each lncRNA as follows:
| 11 |
Based on the definition of C(li), we can define the contribution value of each lncRNA to the functional similarity of each disease pair as follows:
| 12 |
Finally, we can define the functional similarity between diseases di and dj by integrating lncRNAs related to di, dj or both as follows:
| 13 |
where D(di) and D(dj) represent all lncRNAs related to di and dj in E1, respectively.
MiRNA functional similarity based on lncRNAs
Based on the assumption that similar miRNAs tend to show similar interaction and non-interaction patterns with lncRNAs, we can also calculate the miRNA functional similarity in the lncRNA-miRNA interactive network. Similar to the procedure used to calculate the disease functional similarity, first, we constructed the undirected graph G2 = (V2, E2), where V2 = SM∪SL = {m1, m2,…, lM + 1, lM + 2,…, lM + L} is the set of vertices, E2 is the set of edges, and for any two nodes a, b ∈ V2, an edge exists between a and b in E2 if KAM2(a, b) = 1. Then, we defined the contribution of each lncRNA to the functional similarity of each miRNA pair as follows:
| 14 |
Additionally, we can define the functional similarity between mi and mj as follows:
| 15 |
where D(mi) represents all lncRNAs related to mi, and D(mj) represents lncRNAs relate to mj in E2.
Integrated similarity
The processes used to calculate the integrated similarities of the diseases, lncRNAs and miRNAs are illustrated in Fig. 6. Combining the disease semantic similarity, the disease Gaussian interaction profile kernel similarity and the disease functional similarity mentioned above, we can construct the disease integrated similarity matrix FDD as follows:
| 16 |
Fig. 6.
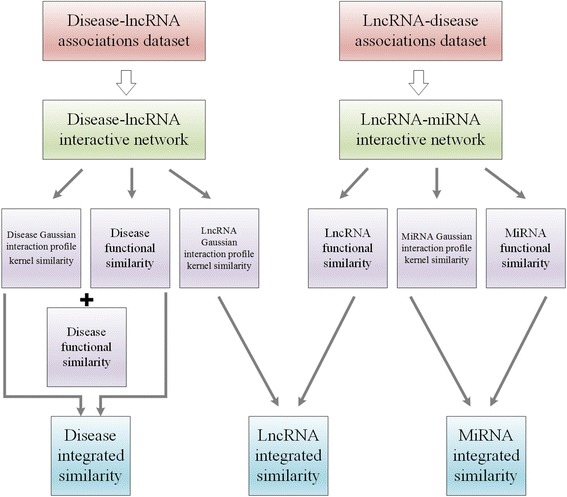
Flow chart of calculation of diseases integrated similarity, lncRNA integrated similarity and miRNA integrated similarity
Additionally, based on the miRNA Gaussian interaction profile kernel similarity and the miRNA functional similarity, we can construct the miRNA integrated similarity matrix FMM as follows:
| 17 |
Furthermore, based on the Gaussian interaction profile kernel similarity matrices LGS1 and LGS2, we can construct the lncRNA integrated similarity matrix FLL as follows:
| 18 |
Prediction of disease-miRNA associations based on a distance correlation set
In this section, we developed a novel computational method, i.e., DCSMDA, to predict potential disease-miRNA associations by introducing a distance correlation set based on the following assumptions: similar diseases tend to show similar interaction and non-interaction patterns with lncRNAs, and similar lncRNAs tend to show similar interaction and non-interaction patterns with miRNAs. As illustrated in Fig. 7, the DCSMDA procedure consists of the following 5 major steps:
Fig. 7.
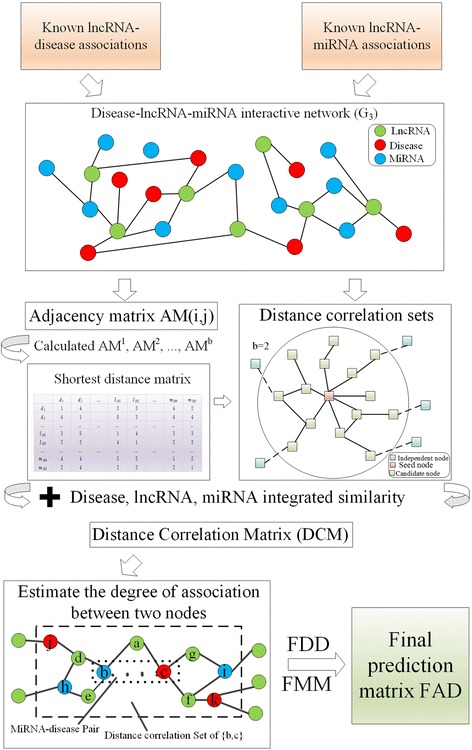
The procedures of DCSMDA
Step 1 (Construction of the adjacency matrix based on G3): First, we construct a (D + L + M) * (D + L + M) Adjacency Matrix (AM) based on the undirected graph G3 and SC, and then for any two nodes vi, vj∈V3, we can define the AM(i, j) as follows:
| 19 |
where i∈[1, D + L + M] and j∈[1, D + L + M], and to calculate the shortest distance matrix in step 2, we define AM (i, j) = 1 if i = j.
Step 2 (Construction of the shortest distance matrix based on adjacency matrix AM): First, we set parameter b to control the bandwidth of the distance correlation set and let b be a pre-determined positive integer, and then, we can obtain b matrices, such as AM1, AM2,..., AMb, based on the above formula (19), and the Shortest Path Matrix is calculated as follows:
| 20 |
where i∈[1, D + M + L], j∈[1, D + M + L], k∈[2, b], and k satisfies the following: AM k(i, j)≠0, while AM 1(i, j) = AM 2(i, j) = … = AM k-1(i, j) = 0.
Step 3 (Calculation of distance correlation sets and distance coefficient of each node pair in G3):
For each node vi ∈ V3, we can obtain distance correlation set DCS(i) according to the shortest distance matrix as follows:
| 21 |
where DCS(i) of each node contains itself and all nodes with the shortest distance less than b.
For instance, in the disease-miRNA-lncRNA interaction network illustrated in Fig. 7, DCS (seed node) is all candidate nodes when b is set to 2.
Then, we can calculate the distance coefficient (DC) of the node pair (vi, vj) as follows:
| 22 |
Furthermore, we can construct a Distance Correlation Matrix (DCM) based on the disease integrated similarity, the lncRNA integrated similarity, and the miRNA integrated similarity as follows:
| 23 |
where i∈[1, D + L + M] and j∈[1, D + L + M].
Step 4 (Estimation of the association degree between a pair of nodes): Based on formula (23), we can estimate the association degree between vi and vj as follows:
| 24 |
Thus, we can obtain prediction matrix PM, where the entity PM (i, j) in row i column j represents the predicted association between node vi and vj.
Step 5 (Calculation of the final prediction result matrix between the miRNAs and diseases): Let , where C11 is a D×D matrix, C12 is a D×L matrix, C13 is a D×M matrix, C21 is an L×D matrix, C22 is an L ×L matrix, C23 is an L×M matrix, C31 is an M×D matrix, C32 is an M×L matrix and C33 is an M ×M matrix. Obviously, C13 is our predicted result, which provides the association probability between each disease and miRNA. A previous study [27] demonstrated that the Gaussian interaction profile kernel similarity is a high-efficiency tool for optimizing the result of prediction, and therefore, we used the miRNA Gaussian interaction profile kernel similarity and the disease Gaussian interaction profile kernel similarity to optimize the result of the DCSMDA as follows:
| 25 |
where the matrix FAD denotes the relationship between the miRNA-disease pairs.
Additional files
The known lncRNA-disease associations for constructing the DS1. We list 583 known lncRNA-disease associations which were collected from LncRNAdisease dataset to construct the DS1. (XLS 58 kb)
The known lncRNA-disease associations for constructing the DS2. We list 702 known lncRNA-disease associations which were collected from MNDR dataset to construct the DS2. (XLS 63 kb)
The integrated lncRNA-disease associations for constructing the DS3. We list 1073 lncRNA-disease associations which were collected by integrating the datasets of DS1 and DS2. (XLS 83 kb)
The known lncRNA-miRNA associations for constructing the DS4. We list 1883 known lncRNA-miRNA associations which were collected from starBasev2.0 database to construct the DS4. (XLS 123 kb)
The known miRNA-disease associations for constructing the DS5. We list 3252 high-quality miRNA-disease associations which were collected from HMDD database to validate the performance of our method. (XLS 191 kb)
Acknowledgements
The authors thank the anonymous referees for suggestions that helped improve the paper substantially.
Funding
The project is partly sponsored by the Construct Program of the Key Discipline in Hunan province, the National Natural Science Foundation of China (No.61640210, No.61672447), the CERNET Next Generation Internet Technology Innovation Project (No. NGII20160305), the Science & Education Joint Project of Hunan Natural Science Foundation (No.2017JJ5036), and the Upgrading Project of Industry-University- Research of Xiangtan University (No.11KZ|KZ03051).
Availability of data and materials
All data generated or analyzed during this study are included in this published article [Additional file 1, Additional file 2, Additional file 3, Additional file 4 and Additional file 5].
Author’s contributions
HCZ conceived the study. HCZ, LAK and LW developed the method. PYP and ZWX implemented the algorithms. HCZ and TRP analyzed the data. LW supervised the study. HCZ and LW wrote the manuscript. ZLW, PYP and LW reviewed and improved the manuscript, ZLW provided supplementary data. All authors read and approved the final manuscript.
Abbreviations
- AUC
Areas under ROC curve
- DCSMDA
Distance Correlation Set is developed to predict MiRNA-Disease Associations
- FPR
False positive rates
- GO
Gene Ontology
- miRNA
MicroRNA
- ncRNA
Noncoding RNA
- ROC
Receiver-operating characteristics
- TPR
True positive rates
- LOOCV
Leave-One Out Cross Validation
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2146-x) contains supplementary material, which is available to authorized users.
Contributor Information
Haochen Zhao, Email: zhaohc940702@163.com.
Linai Kuang, Email: 41118759@qq.com.
Lei Wang, Email: wanglei@xtu.edu.cn.
Pengyao Ping, Email: 956272448@qq.com.
Zhanwei Xuan, Email: 1309240100@qq.com.
Tingrui Pei, Email: peitingrui@xtu.edu.cn.
Zhelun Wu, Email: zhelunw@princeton.edu.
References
- 1.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crick FHC, Barnett L, Brenner S, Watts-Tobin RJ. General nature of the genetic code for proteins. Nature. 1961;192(4809):1227–1232. doi: 10.1038/1921227a0. [DOI] [PubMed] [Google Scholar]
- 3.Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(suppl_1):R17. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 4.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 5.Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol. 2015;22(1):5. doi: 10.1038/nsmb.2942. [DOI] [PubMed] [Google Scholar]
- 6.Ambros V. The functions of animal micrornas. Nature. 2004;431(7006):350. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 7.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human mirnas and indications for an involvement of mirna in cell growth and apoptosis. Nucleic Acids Res. 2005;33(4):1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taguchi Y. Inference of target gene regulation via mirnas during cell senescence by using the mirage server. Aging Dis. 2012;3(4):301. [PMC free article] [PubMed] [Google Scholar]
- 9.Peng H, Lan C, Zheng Y, Hutvagner G, Tao D, Li J. Cross disease analysis of co-functional microrna pairs on a reconstructed network of disease-gene-microrna tripartite. Bmc Bioinformatics. 2017;18(1):193. doi: 10.1186/s12859-017-1605-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber MJ. New human and mouse microrna genes found by homology search. FEBS J. 2005;272(1):59. doi: 10.1111/j.1432-1033.2004.04389.x. [DOI] [PubMed] [Google Scholar]
- 11.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. Microrna-21 contributes to myocardial disease by stimulating map kinase signalling in fibroblasts. Nature. 2008;456(7224):980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 12.Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of mirna changes in alzheimer's disease brain and csf yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14(1):27–41. doi: 10.3233/JAD-2008-14103. [DOI] [PubMed] [Google Scholar]
- 13.Corsten MF, Dennert R, Jochems S, Kuznetsova T, Devaux Y, Hofstra L, et al. Circulating microrna-208b and microrna-499 reflect myocardial damage in cardiovascular disease. Circ Cardiovasc Genet. 2010;3(6):499. doi: 10.1161/CIRCGENETICS.110.957415. [DOI] [PubMed] [Google Scholar]
- 14.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, et al. Altered microrna expression in human heart disease. Physiol Genomics. 2007;31(3):367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 15.Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, et al. An analysis of human microrna and disease associations. PLoS One. 2008;3(10):e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Liu MX, Yan GY. Rwrmda: predicting novel human microrna-disease associations. Mol BioSyst. 2012;8(10):2792. doi: 10.1039/c2mb25180a. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, et al. HMDD v2.0: a database for experimentally supported human microrna and disease associations. Nucleic Acids Res. 2014;42(Database issue):1070–1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microrna functional similarity and functional network based on microrna-associated diseases. Bioinformatics. 2010;26(13):1644–1650. doi: 10.1093/bioinformatics/btq241. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, et al. Mir2disease: a manually curated database for microrna deregulation in human disease. Nucleic Acids Res. 2009;37(1):D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Q, Li J, Hong Q, Lin Z, Wu Y, Shi H, et al. Prediction of microrna-disease associations based on social network analysis methods. Biomed Res Int. 2015;2015(10):810514. doi: 10.1155/2015/810514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You ZH, Wang LP, Chen X, et al. PRMDA: personalized recommendation-based MiRNA-disease association prediction[J]. Oncotarget. 2017;8(49):85568-83. [DOI] [PMC free article] [PubMed]
- 22.Shi H, Xu J, Zhang G, Xu L, Li C, Wang L, et al. Walking the interactome to identify human mirna-disease associations through the functional link between mirna targets and disease genes. BMC Syst Biol. 2013;7(1):1–12. doi: 10.1186/1752-0509-7-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Q, Hao Y, Wang G, Juan L, Zhang T, Teng M, et al. Prioritization of disease micrornas through a human phenome-micrornaome network. BMC Syst Biol. 2010;4(S1):S2. doi: 10.1186/1752-0509-4-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han K, Xuan P, Ding J, Zhao ZJ, Hui L, Zhong YL. Prediction of disease-related micrornas by incorporating functional similarity and common association information. Gen Mol Res Gmr. 2014;13(1):2009–2019. doi: 10.4238/2014.March.24.5. [DOI] [PubMed] [Google Scholar]
- 25.Xuan P, Han K, Guo M, Guo Y, Li J, Ding J, et al. Prediction of micrornas associated with human diseases based on weighted k most similar neighbors. PLoS One. 2013;8(9):e70204. doi: 10.1371/journal.pone.0070204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xuan P, Han K, Guo Y, Li J, Li X, Zhong Y, et al. Prediction of potential disease-associated micrornas based on random walk. Bioinformatics. 2015;31(11):1805–1815. doi: 10.1093/bioinformatics/btv039. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Yan GY. Semi-supervised learning for potential human microrna-disease associations inference. Sci Rep. 2014;4:5501. doi: 10.1038/srep05501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Yan CC, Zhang X, You ZH, Deng L, Liu Y, et al. Wbsmda: within and between score for mirna-disease association prediction. Sci Rep. 2016;6:21106. doi: 10.1038/srep21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Chen L, Chen B, Li X, Kang J, Fan K, et al. Mammalian ncrna-disease repository: a global view of ncrna-mediated disease network. Cell Death Dis. 2013;4(8):e765. doi: 10.1038/cddis.2013.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, et al. lncrna-disease: a database for long-non-coding rna-associated diseases. Nucleic Acids Res. 2013;41(Database issue):983–986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X. Predicting lncrna-disease associations and constructing lncrna functional similarity network based on the information of mirna. Sci Rep. 2015;5:13186. doi: 10.1038/srep13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang WT, Guo XQ, Dai JP, Chen RS. Microrna and lncrna in neurodegenerative diseases*: microrna and lncrna in neurodegenerative diseases. Prog Biochem Biophys. 2010;37(8):826–833. doi: 10.3724/SP.J.1206.2010.00104. [DOI] [Google Scholar]
- 33.Guo L, Peng Y, Meng Y, et al. Expression profiles analysis reveals an integrated miRNA-lncRNA signature to predict survival in ovarian cancer patients with wild-type BRCA1/2. Oncotarget. 2017;8(40):68483. [DOI] [PMC free article] [PubMed]
- 34.Spiess PE, Dhillon J, Baumgarten AS, Johnstone PA, Giuliano AR. Pathophysiological basis of human papillomavirus in penile cancer: key to prevention and delivery of more effective therapies. CA-A Cancer J Clinicians. 2016;6:481–495. doi: 10.3322/caac.21354. [DOI] [PubMed] [Google Scholar]
- 35.Ruprecht B, Zaal EA, Zecha J, Wu W, Berkers CR, Kuster B, Lemeer S. Lapatinib resistance in breast Cancer cells is accompanied by phosphorylation-mediated reprogramming of glycolysis. Cancer Res. 2017;77(8):1842–1853. doi: 10.1158/0008-5472.CAN-16-2976. [DOI] [PubMed] [Google Scholar]
- 36.Barton MK. Local consolidative therapy may be beneficial in patients with oligometastatic non-small cell lung cancer. CA-A Cancer J Clinicians. 2017;2:89–90. doi: 10.3322/caac.21363. [DOI] [PubMed] [Google Scholar]
- 37.Jiang J, Jia P, Zhao Z, Shen B. Key regulators in prostate cancer identified by co-expression module analysis. BMC Genomics. 2014;15(1):1015. doi: 10.1186/1471-2164-15-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medina-Villaamil V, Martínez-Breijo S, Portela-Pereira P, Quindós-Varela M, Santamarina-Caínzos I, Antón-Aparicio LM, et al. Circulating micrornas in blood of patients with prostate cancer. Actas Urol Esp. 2014;38(10):633–639. doi: 10.1016/j.acuro.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 39.Cai C, Chen QB, Han ZD, Zhang YQ, He HC, Chen JH, et al. Mir-195 inhibits tumor progression by targeting rps6kb1 in human prostate cancer. Clin Cancer Res. 2015;21(21):4922. doi: 10.1158/1078-0432.CCR-15-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bozok ÇV, Tetik VA, Düzgün Z, Tezcanlı KB, Açıkgöz E, Aktuğ H, et al. Mir-15a enhances the anticancer effects of cisplatin in the resistant non-small cell lung cancer cells. Tumor Biol. 2016;37(2):1739–1751. doi: 10.1007/s13277-015-3950-9. [DOI] [PubMed] [Google Scholar]
- 41.Liu B, Qu J, Xu F, Guo Y, Wang Y, Yu H, et al. Mir-195 suppresses non-small cell lung cancer by targeting chek1. Oncotarget. 2015;6(11):9445–9456. doi: 10.18632/oncotarget.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Lan H, Zhang M, An N, Yu R, He Y, et al. Effects of mir-424 on proliferation and migration abilities in non-small cell lung cancer a549 cells and its molecular mechanism. Zhongguo Fei Ai Za Zhi. 2016;19:571–576. doi: 10.3779/j.issn.1009-3419.2016.09.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun YP, Lu F, Han XY, et al. MiR-424 and miR-27a increase TRAIL sensitivity of acute myeloid leukemia by targeting PLAG1. Oncotarget. 2016;7(18):25276-90. [DOI] [PMC free article] [PubMed]
- 44.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. Mir-15 and mir-16 induce apoptosis by targeting bcl2. Proc Natl Acad Sci U S A. 2005;102(39):13944. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao TF, Jia HZ, Zhang ZZ, Zhao XS, Zou YF, Zhang W, et al. Lncrna h19 regulates id2 expression through competitive binding to hsa-mir-19a/b in acute myelocytic leukemia. Mol Med Rep. 2017;16(3):3687. doi: 10.3892/mmr.2017.7029. [DOI] [PubMed] [Google Scholar]
- 46.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010;6(1):e1000641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The known lncRNA-disease associations for constructing the DS1. We list 583 known lncRNA-disease associations which were collected from LncRNAdisease dataset to construct the DS1. (XLS 58 kb)
The known lncRNA-disease associations for constructing the DS2. We list 702 known lncRNA-disease associations which were collected from MNDR dataset to construct the DS2. (XLS 63 kb)
The integrated lncRNA-disease associations for constructing the DS3. We list 1073 lncRNA-disease associations which were collected by integrating the datasets of DS1 and DS2. (XLS 83 kb)
The known lncRNA-miRNA associations for constructing the DS4. We list 1883 known lncRNA-miRNA associations which were collected from starBasev2.0 database to construct the DS4. (XLS 123 kb)
The known miRNA-disease associations for constructing the DS5. We list 3252 high-quality miRNA-disease associations which were collected from HMDD database to validate the performance of our method. (XLS 191 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article [Additional file 1, Additional file 2, Additional file 3, Additional file 4 and Additional file 5].