

Abstract
Mesenchymal stem cells (MSC) have been experimentally used for kidney repair, but modest retention limits their efficacy. Cell‐surface coating allows modulating MSC homing and interaction with target cells. We coated mouse adipose tissue‐derived MSC with antibodies directed against kidney injury molecule‐1 (ab‐KIM1), which is upregulated in injured kidneys, and tested the hypothesis that this would enhance their therapeutic effects in ischemic kidney injury. Untreated MSC, ab‐KIM1‐coated MSC (KIM‐MSC), or vehicle, were injected systemically into the carotid artery of 2‐kidneys, 1‐clip mice 2 weeks after surgery. MSC retention in different organs was explored 24 hours, 48 hours, or 2 weeks after injection. Renal volume, perfusion, and oxygenation were studied 2 weeks after injection using magnetic resonance imaging in vivo, and renal inflammation, apoptosis, capillary density, and fibrosis ex vivo. The ab‐KIM1 coating had little effect on MSC viability or proliferation. The stenotic kidney showed upregulated KIM1 expression, selective homing, and greater retention of KIM‐MSC compared to untreated MSC and compared to other organs. KIM‐MSC‐injected mice improved renal perfusion and capillary density, and attenuated oxidative damage, apoptosis, and fibrosis compared to mice treated with vehicle or with native MSC. In conclusion, MSC coating with ab‐KIM1 increased their retention in the ischemic kidney and enhanced their therapeutic efficacy. This novel method may be useful to selectively target injured kidneys, and supports further development of strategies to enhance cell‐based treatment of ischemic kidney injury. Stem Cells Translational Medicine 2018;7:394–403
Keywords: Mesenchymal stem cell, Cell coating, Kidney injury molecule‐1, Renal artery stenosis
Significance Statement.
Mesenchymal stem cells (MSC) surface coating allows modulating their homing and interaction with target cells. This study coated mouse adipose tissue‐derived MSC with antibodies directed against kidney injury molecule‐1, which is upregulated in injured kidneys, and injected coated cells systemically into mice with renal artery stenosis. The stenotic kidney received coated cells showed selective homing and greater retention of MSC, accompanied with improved renal perfusion, capillary density, attenuated oxidative damage, apoptosis, and fibrosis. This novel method may be useful to selectively target injured kidneys, and supports further development of strategies to enhance cell‐based treatment of ischemic kidney.
Introduction
Mesenchymal stem cells (MSC) are potent vectors in regeneration medicine. Systemically delivered MSC can home to injured sites and play a therapeutic role primarily via paracrine effects. MSC have also been used to treat ischemic kidney diseases and conferred significant improvements in renal functional recovery 1. However, modest retention of exogenously delivered MSC specifically in target organs limits the potential utility and benefits of this approach, as the majority of MSC are initially trapped in the lungs 2, 3. Importantly, increased retention rate of MSC in the infarcted myocardium and inflamed bowel is associated with better therapeutic effects 4, 5. Conceivably, selective MSC retention in the injured kidney may also enhance their therapeutic effects.
Following an ischemic insult, a change in the actin cytoskeleton architecture of microvascular endothelial cells can decrease the cell‐cell adhesion junctions and facilitates MSC transit across the blood vessel wall 6, 7, yet retention of MSC in target organs remains low. Overexpressing cell trafficking‐related CXC chemokine receptor‐4 (CXCR4) by gene‐modification has been used successfully to increase cellular retention rate, but the potential for tumorigenesis associated with genetic manipulation imposes limitations for clinical application 8, 9, 10, 11. Alternatively, coating cells with antibodies alters the cell surface without manipulating genes. For this purpose, an esterified protein, like palpitated protein A/G, has been used to conjugate selected antibodies to the cell surface 12, 13. Thus, palpitated protein‐A (PPA) anchoring anti‐IgG antibodies on the cell surface increased adhesiveness to IgG‐positive B cells in vitro 12. Furthermore, palmitated protein‐G (PPG)‐coated MSC linked to anti‐ICAM‐1 antibodies showed increased binding rate with ICAM‐1‐positive human vascular endothelial cells, and improved efficacy of MSC in inflammatory bowel disease 14, 15. Similarly, chondrogenic progenitor cells coated with antibodies promote binding to injured cartilage extracellular matrix 15. However, similar approaches have not been fully developed to target cell‐based therapy to injured kidneys.
Kidney injury molecule (KIM)‐1 is a transmembrane protein that is expressed minimally in normal kidneys, but upregulated dramatically in damaged kidneys 16. As tubular KIM1 expression is specific to ongoing cell damage, it has been regarded as a marker for kidney injury 17, 18. Compared to the other kidney injury markers, KIM1 rises earlier after injury and is expressed persistently 19, and might therefore serve to target therapeutic interventions specifically to injured kidney. We hypothesized that when conjugated to antibodies directed against KIM1, MSC would show increased retention in injured kidneys, and thereby confer superior therapeutic benefits compared to native MSC.
Methods
All protocols were approved by the Mayo Clinic Institutional Animal Care and Use Committee (A1609‐16). Male 129‐S1 mice (Jackson Lab, Bar Harbor, ME, 11 weeks of age) were studied for 4 weeks after induction of unilateral renal artery stenosis (RAS) or sham surgery. Mice were randomly divided into sham (n = 8), RAS + vehicle (n = 10), RAS + MSC (n = 10), and RAS + KIM‐MSC (n = 10).
RAS was induced by surgical placement of a 0.15 mm diameter arterial cuff, whereas sham surgeries without placement of a cuff were performed in the control group, as previously described 20. This approach leads to a fall in renal volume, blood flow, and function. Blood pressure was measured at baseline, 2, 3, and 4 weeks after surgery by tail‐cuff (Kent Scientific, Torrington, CT).
After 2 weeks, the carotid artery was cannulated via a vascular cut down, and 200 µl phosphate buffered solution (PBS), MSC, or KIM‐MSC (5 × 105 cells in 200 µL PBS) slowly injected. Two weeks later, the mice were scanned with magnetic resonance imaging (MRI) for renal hemodynamics and oxygenation, and subsequently euthanized with CO2. Kidneys and blood samples were collected for in vitro studies. Additionally, the stenotic kidney (STK) and contralateral kidney (CLK) obtained from MSC‐ or KIM‐MSC‐treated mice were used for MSC tracking 1, 2, and 14 days after cell injection (n = 5 each group per time point). Lung and liver were also collected at the 2‐day time point for distribution analysis using fluorescence activated cell sorter (FACS) (FlowSight, Millipore) analysis of dissociated tissues.
To evaluate the effects of KIM1‐MSC on the early phase of RAS, KIM1‐MSC (5 × 105 cells in 200 µl PBS, n = 5) or 200 µl PBS (n = 5) were also injected through the carotid artery immediately after induction of RAS, 24 or 48 hours later, blood samples were collected, and kidneys saved for KIM1 immunofluorescence.
MSC Culture and Coating
Allogeneic MSC were isolated from abdominal adipose tissue (around 1 g) of adult donor mice (129‐S1, 11 weeks) that was digested in collagenase‐H for 45 minutes, filtered, and cultured in Advanced dulbecco's modified eagle medium (DMEM) media supplied with 5% PLTMax (Mill Creek Life Science, Rochester, MN) for about 2 weeks. The third passages of each batch was collected and stored in cell recovery medium in −80°C. Cells from passages 3–7 were collected for phenotyping and transplantation. FACS was used to determine the MSC cell surface markers CD90, CD73, and Sca‐1, and to exclude CD45 and CD34 expression, and an MSC Functional Identification Kit (R&D Systems, Minneapolis, MN) for tri‐lineage differentiation capacity.
Recombinant protein‐G (RPG, Sigma, MO) was derivatized with N‐hydroxysuccinimide ester of palmitic acid (Sigma, MO), and PPG synthesized with palmitation reaction, as previously described 15. Sephadex G‐25 column (Sigma, MO) was used to purify the proteins, and protein concentration measured with bicinchoninic acid (BCA) Assay. Cell coating was conducted as previously described 14. In brief, MSC were washed with serum‐free DMEM and suspended at a density of 5 × 106/ml in DMEM. Varying concentrations of PPG were added to the cell suspension and incubated for 1 hour at 37°C. Cells were washed with DMEM and incubated with 100 µg/ml monoclonal rat anti‐mouse KIM1 antibody (R&D Systems, FAB1817A) for another 1 hour at 37°C. To assess the efficiency and binding rate of the antibody onto the cell surface, DiO cell membrane dye (Molecular Probes, Eugene, OR) was used to label MSC before coating. After incubation with PPG, cells were incubated with the Allophycocyanin (APC)‐conjugated anti‐KIM1 antibody (R&D Systems) following the same procedure and analyzed at the flow cytometry. The percent of APC (red) and DiO (green) positive cells was calculated. To select the PPG concentration appropriate for mouse MSC, the toxicity of different concentrations of PPG was assessed using apoptosis SYTOX staining kit (Invitrogen, Molecular Probes) and quantified by FACS. Cell proliferation rate was assessed using Cell Counting Kit‐8 (Sigma‐Aldrich), by seeding 104 cells in 96 wells and incubating them for 24, 48, or 72 hours, following vendor instructions.
To assess the binding efficiency of KIM1‐MSC to the KIM‐1 protein, a six‐well plate was coated with KIM‐1 protein (0.5, 1.5, and 3 ng/ml, R&D System, MN), then 1.2 × 106 KIM1‐MSC were seeded in each well and incubated at 37°C for 1 hour. Wells were then gently washed with PBS three times, adherent KIM1‐MSC were collected after a short digestion with trypsin, and counted using a cell counter. Nonadhered cells that washed out in PBS were also counted. The efficiency was calculated as the ratio of adhered/seeded cells.
Cell Tracking
Dil cell membrane dye (Molecular, probes) was used to label MSC for cell tracking after injection. In kidney sections (5 µm thick), the expression of endogenous KIM1 was probed with rabbit anti‐mouse KIM1 antibody (Abcam) followed with goat anti‐rabbit second antibody (Alexa Fluor 488). 4′,6‐diamidino‐2‐phenylindole was used to stain nuclei. Total CM‐DiI‐labeled MSC and MSC colocalizing with endogenous KIM1 were counted in at least 8–10 random fields in each section and averaged.
MSC Organ Distribution Analysis
For cell bio‐distribution, organs were weighted, small pieces of each (100 mg) were cut, and then digested with collagenase‐I. Total cells from 100 mg tissue were analyzed by FACs and MSC counted. The numbers of MSCs in the organs were then calculated based on their total weight. Briefly, ice‐cold PBS was used to flush out blood from harvested tissues 48 hours after cell injection. Fresh STK, CLK, lung, and liver were weighted and stored in phosphate buffered saline (Sigma‐Aldrich). Tissues were diced and digested with 0.05 mg/ml Liberase Thermolysin Low (Millipore‐Sigma) and 100 U/ml DNase (Thermo Fischer Scientific). The addition of Roswell Park Memorial Institute (RPMI) media (Sigma‐Aldrich), with 10% fetal bovine serum (FBS), was followed by filtration of the suspension through a cell strainer. The filtrate was then centrifuged at 300 g for 10 minutes, and the pellet resuspended as single cells, analyzed by the FACS for DiI‐positive cells, and their percentage in each organ calculated.
Imaging Protocol and Data Analyses
Two weeks after MSC or vehicle injection, renal volume and hemodynamics were assessed by MRI, as previously described 20. Renal volume was quantified from images acquired using a respiration‐gated three‐dimensional fast imaging. Renal perfusion was measured with arterial spin labeling, and quantified from the flow‐sensitive alternating inversion‐recovery sequence, with rapid acquisition with relaxation enhancement images. Renal oxygenation was assessed with blood oxygen level dependent‐magnetic resonance imaging (MRI). Eight images were reconstructed after zero‐filling the k‐space data to 256 × 256. T2* was quantified by pixelwise mono‐exponential fitting on the averaged magnitude of all eight images over echo times. R2* (1/T2*) was used as an index of blood oxygenation level.
Blood Urea Nitrogen
Blood urea nitrogen (BUN) level was determined using a commercial kit (DetectX Urea Nitrogen kits, Arbor assays, Ann‐Arbor, MI). Briefly, 50 μl of standards or samples were pipetted into a clear microliter plate, an assay diluent was added, and the color‐generating reaction initiated with the Color Reagents. The concentration of BUN was calculated using the delta of the optical density readings at 30 minutes compared to the standard curve.
Renal Injury Pathways
Kidneys were homogenized using a standard technique 21. Specific antibodies against Bax (1:200, Santa Cruz), Bcl‐2 (1:200, Abcam), transforming growth‐factor‐β (TGF‐β) (1:200, Santa Cruz), connective tissue growth factor (CTGF) (1:200, Bio Vision), PAI‐1 (1:200, Abcam), TIMP‐1 (1:200, Santa Cruz) were used with Western blotting protocols. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as loading controls and protein expression quantified with image‐pro plus 6.0 software. The expression of KIM1 in the STK was assessed by immunofluorescence staining. Kidney fibrosis was tested by Masson's Trichrome staining, assessed in 5‐µm sections of each kidney using a computer‐aided image‐analysis program (AxioVision, Carl Zeiss Micro Imaging, Thornwood, NY). Fibrosis area was quantified randomly in 10–15 fields each section. Apoptosis was assessed in renal sections stained with terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL, Promega) and F4/80 (1:200, Abcam) macrophages stained by immunohistochemistry. In 10–15 random fields sampled in each section, positive cells were manually counted. Renal oxidative damage was evaluated by 8‐hydroxy‐2′‐deoxyguanosine (8‐OHDG, 1:200, Abcam) immunohistochemistry, quantified from 10 random fields as percent of positive cells.
Statistical Analysis
Statistical analysis used the JMP software. All data are expressed as mean ± SD or median. Statistical significance was assessed by one‐way analysis of variance followed by unpaired two‐tailed t test for normally distributed data or nonparametric (Wilcoxon and Kruskal‐Wallis) test for no‐normally distributed data. A value of p ≤ .05 was considered significant.
Results
Characterization of Mouse MSC
MSC isolated from abdominal adipose tissue of adult male mice showed capacity for tri‐lineage differentiation into chondrocytes, osteocytes, and adipocytes (Fig. 1A). MSC markers analyzed using flow cytometry were confirmed for the presence of CD90, CD73, and Sca‐1, but negative for the hematopoietic markers CD45 and CD34, as shown in both intensity graphs and representative single‐cell images (Fig. 1B).
Figure 1.
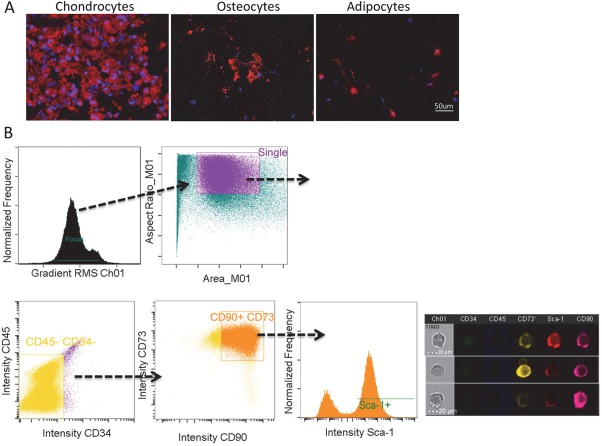
In vitro characterization of mesenchymal stem cells (MSC). (A): Representative images showed that MSC transdifferentiated into chondrocytes (Collagen II), osteocytes (Osteopontin), and adipocytes (FABP4). Scale bar = 50 μm. (B) MSCs were identified using flow cytometry as CD34negCD45negCD90+CD73+Sca‐1+ as showed in histogram and representative single cell images. Scale bar = 20 μm.
Antibodies Directed Against Kidney Injury Molecule‐1 Coating on MSC Has Little Effect on Viability
PPG achieved almost 100% coating rate, compared to under 3% using RPG (control) (Fig. 2A). Increasing concentrations showed little effect on cell proliferation after 24, 48, or 72 hours of incubation, and the number of dead cells by SYTOX dye after coating was not different from uncoated MSC (Fig. 2B, 2C). PPG concentration of 50 µg/ml was used for subsequent experiments 5. After coating with PPG, MSC were labeled with the cell membrane dye CM‐Dio (green) and incubated with APC‐conjugated antibodies directed against kidney injury molecule‐1 (Ab‐KIM1) (red). The successfully KIM‐1 antibody‐coated MSC showed double‐positivity to APC (red) and Dio (green), with coating rate of 100% (Fig. 2D, 2E). Hence, PPG adequately anchored ab‐KIM1 to MSC, with little effects on cell viability. Furthermore, KIM1‐MSC successful dose‐dependent binding to KIM1 protein was confirmed in vitro, reaching 93% at 3 µg of KIM1 (Fig. 2F).
Figure 2.
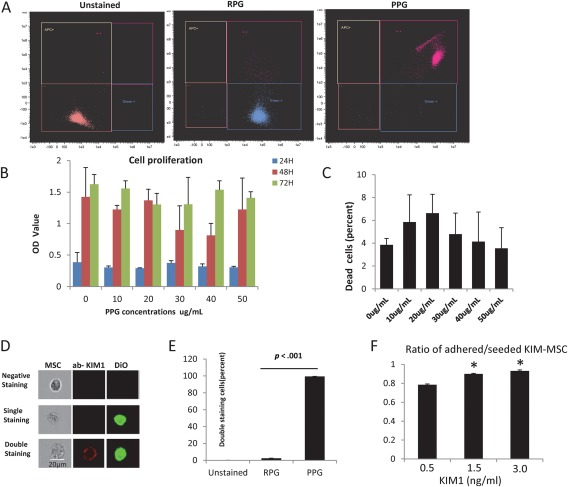
MSC coating with anti‐KIM1 antibody (ab‐KIM1). (A): Flow cytometry analysis showed that there was almost 100% coating rate using PPG, and less than 3% using RPG (control). (B, C): Increasing concentrations showed little effect on cell proliferation after 24, 48, or 72 hours of incubation, and the number of dead cells by SYTOX dye after coating was not different from uncoated MSC. (D, E): The successful KIM‐1 antibody coated MSC (scale bar = 20 μm) showed double positive to allophycocyanin (red) and Dio (green) and the coating rate was 100%. (F): KIM1‐MSC successful dose‐dependent binding to KIM1 protein was confirmed in vitro, reaching 93% at 3 µg of KIM1. Abbreviations: ab‐KIM1, antibodies directed against kidney injury molecule‐1; MSC, mesenchymal stem cells; PPG, palmitated protein‐G; RPG, recombinant protein‐G.
Coating with ab‐KIM1 Increased MSC Delivery to the Mouse STK
KIM1 expression was upregulated in the STK 24 hours after induction of RAS, peaked at 48 hours, and remained upregulated 2 weeks after, but remained minimal in the sham and CLK. Interestingly, injection of KIM‐MSC had no significant effects on KIM1 renal expression (Fig. 3A). CM‐Dil (Red) labeled MSCs were tracked in excised kidneys 24 hours, 48 hours, or 2 weeks after injection (Fig. 3B, 3C). The STK has shown greater homing of MSC compared to the CLK at each time point (p < .01), and almost double the number of KIM‐MSC compared to native MSC. Specifically, the number of Dil‐positive MSC adjacent to endogenous KIM1‐positive tubular cells was also greater in the STK of KIM‐MSC‐treated than in native MSC‐treated mice at each time point, with a peak of cell retention at 48 hours. At 48 hours after injection, a greater MSC fraction was found in STK injected with KIM‐MSC compared with native MSC (p = .01), and KIM‐MSC‐treated mice also showed greater MSC retention in the STK compared to the CLK, lung, and liver, which were not different in mice treated with native MSCs (Fig. 3D). Overall, 8.1% ± 1.7% of total injected native MSC and 14.5% ± 6.5% KIM‐MSC engrafted in the STK.
Figure 3.
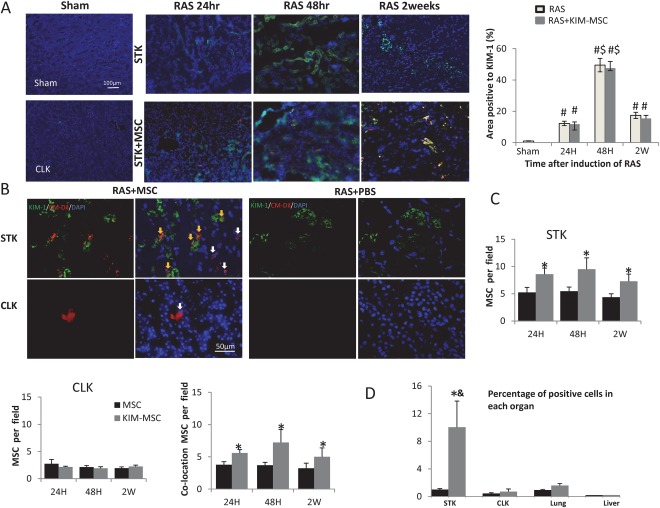
Cell tracking and distribution in mice treated with MSC or KIM‐MSC. (A): KIM1 expression was upregulated in the STK 24 hours after induction of RAS, peaked at 48 hours, remained upregulated 2 weeks after, and was unaffected by the injection of KIM‐MSC. KIM1 expression was minimal in the sham and CLK, scale bar = 100 μm. (B): Representative images of CM‐DiI labeled (red) MSC in KIM1 (green)‐stained STK and CLK 48 hours after delivery into the aorta (yellow arrow shows MSC colocalizing with endogenous KIM1‐positive tubules, scale bar = 50 μm). (C): The STK consistently shows greater homing of MSC compared to the CLK, as well as about twice as many MSC colocalizing with KIM1 in RAS mice injected with KIM‐MSC compared to native MSC. (D): FACS analysis showing the distribution of MSC 48 hours after injection, as a fraction of cellular content in each organ. KIM‐MSC‐treated mice showed the greatest MSC fraction in the STK compared to the CLK, lung, and liver, whereas mice treated with native MSC did not show a difference. #, p < .05 versus Sham, $, p < .05 versus 24 hours, *, p < .05 versus RAS+MSC, &, p < .05 versus RAS+KIM‐MSC CLK, Lung, Liver. Abbreviations: CLK, contralateral kidney; FACS, fluorescence‐activated cell sorting; KIM1, kidney injury molecule‐1; MSC, mesenchymal stem cells; RAS, renal artery stenosis; STK, stenotic kidney.
The Effect of MSC on Renal Function
All mice showed a significant increase of systolic blood pressure (BP) 2, 3, and 4 weeks after RAS compared to sham. Native MSC did not decrease BP in RAS mice, whereas KIM‐MSC tended to slightly lower it at 4 weeks compared to RAS + vehicle, but this difference has not reached statistical significance (p = .1, Fig. 4A). Body weight was lower than sham in all groups 4 weeks after induction of RAS (Fig. 4B), while BUN was elevated 24 hours, 48 hours, and 2 weeks in RAS mice after cell injection, and remained unchanged after either MSC or KIM‐MSC treatment (Fig. 4C). STK volume measured by MRI decreased similarly in all RAS groups 4 weeks after surgery. Conversely, a significant increase in CLK volume in RAS + vehicle was accentuated in RAS + KIM‐MSC (Fig. 4D, 4E).
Figure 4.
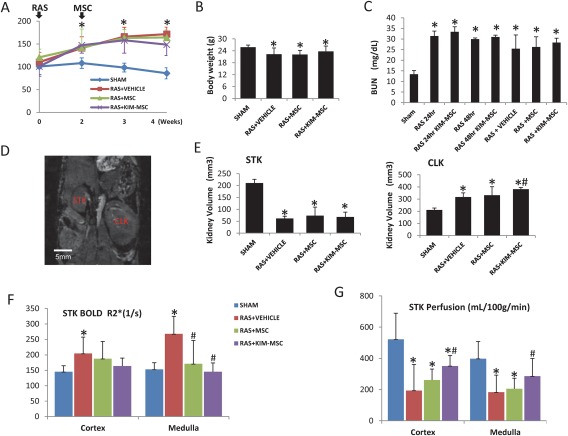
Systolic blood pressure (SBP), body weight, and renal functional parameters. (A): SBP was measured weekly in mice after RAS induction or sham surgery. All RAS mice showed an increase of SBP compared to Sham that remained elevated 4 weeks after surgery (i.e., 2 weeks after cell injection). (B, C): Lower body weight and higher BUN in RAS compared to Sham up to 4 weeks after surgery were unaffected by MSC or KIM‐MSC. BUN was also similarly elevated 24 and 48 hours after cell injection. (D, E): Representative coronal MRI image showing the STK and CLK in a RAS mice, scale bar = 5 mm. STK volume fell 4 weeks after RAS, and was unaffected by MSC or KIM‐MSC. CLK volume increased after RAS, and RAS+KIM‐MSC mice induced a further increase. (F, G): Significant STK cortical and medullary hypoxia in RAS + vehicle was alleviated by both MSC and KIM‐MSC, whereas a decrease in STK cortical and medullary perfusion was improved only by KIM‐MSC. *, p < .05 versus SHAM, #, p < .05 versus RAS + Vehicle. Abbreviations: BUN, blood urea nitrogen; CLK, contralateral kidney; KIM, kidney injury molecule; MSC, mesenchymal stem cells; RAS, renal artery stenosis; STK, stenotic kidney.
Kidney oxygenation assessed by BOLD‐MRI revealed an increase in hypoxic signals in the STK cortex and medulla compared to sham (Fig. 4F). Both MSC and KIM‐MSC similarly alleviated STK hypoxia compared to RAS + vehicle, whereas a decrease in STK cortical and medullary perfusion in RAS was improved only by KIM‐MSC (Fig. 4G). There was no difference in CLK perfusion among the groups (Fig. 4H).
MSC Reduced Renal Macrophage Infiltration, Apoptosis, and Oxidative Stress
F4/80 staining showed macrophage infiltration in the STK in all RAS mice compared to the sham, which was similarly attenuated by MSC and KIM‐MSC (Fig. 5A). TUNEL‐positive cells in the STK increased markedly compared to sham, while MSC‐treated mice exhibited fewer positive cells than RAS + vehicle, which were again further fewer in RAS + KIM‐MSC. RAS‐STK had increased numbers of 8‐OHDG positive cells, which were attenuated in RAS + MSC, and further in RAS + KIM‐MSC. Expression of the pro‐apoptotic protein B‐cell lymphoma‐associated‐X (Bax) was similarly downregulated in RAS + MSC and RAS + KIM‐MSC, whereas Bax/B‐cell lymphoma‐extra‐large ratio that was elevated in RAS + vehicle decreased in RAS + MSC and RAS + KIM‐MSC (Fig. 5B).
Figure 5.
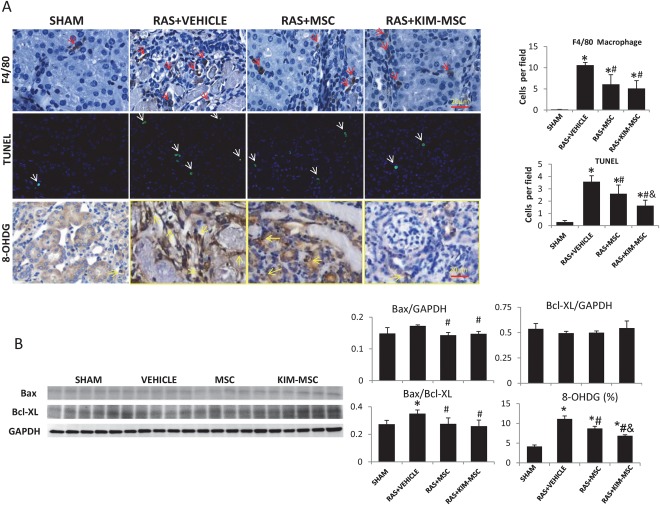
MSC and KIM‐MSC decrease cell apoptosis and macrophage infiltration in stenotic kidneys 2 weeks after injection. (A): Macrophage (red arrows) infiltration increased 4 weeks after induction of untreated RAS (scale bar = 20 μm), but was similarly attenuated 2 weeks after MSC and KIM‐MSC injection. TUNEL‐positive apoptotic signals (white arrows) increased after RAS (scale bar = 20 μm), while RAS +MSC mice exhibited fewer positive cells than the RAS+VEHICLE, which were again fewer in RAS+KIM‐MSC mice. (B): The ratio of Bax/Bcl‐XL significantly increased in RAS+Vehicle, and similarly decreased in RAS+MSC and RAS+KIM‐MSC. *, p < .05 versus SHAM, #, p < .05 versus RAS+Vehicle, &, p < .05 versus RAS+MSC. Abbreviations: 8‐OHDG, 8‐hydroxy‐2′‐deoxyguanosine; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; KIM, kidney injury molecule; MSC, mesenchymal stem cells; RAS, renal artery stenosis.
Pro‐Angiogenic and Antifibrotic Effects of MSC
Four weeks after RAS, CD31 staining showed a decrease of STK microvascular density, which was improved by MSC and slightly further increased by KIM‐MSC (Fig. 6A). The STK also showed marked fibrosis by Masson's trichrome staining. Both MSC and KIM‐MSC mitigated kidney fibrosis compared to RAS + vehicle, yet in RAS + KIM‐MSC renal fibrosis was further blunted compared to RAS + MSC. Compared to Sham, the STK showed upregulated protein expression of TGF‐β, while metalloproteinase inhibitor (TIMP)‐1 and plasminogen activator inhibitor (PAI)‐1 expression remained unchanged. Nevertheless, both MSC and KIM‐MSC downregulated CTGF expression, and RAS + KIM‐MSC also downregulated PAI1, but neither affected TGF‐β expression (Fig. 6B).
Figure 6.
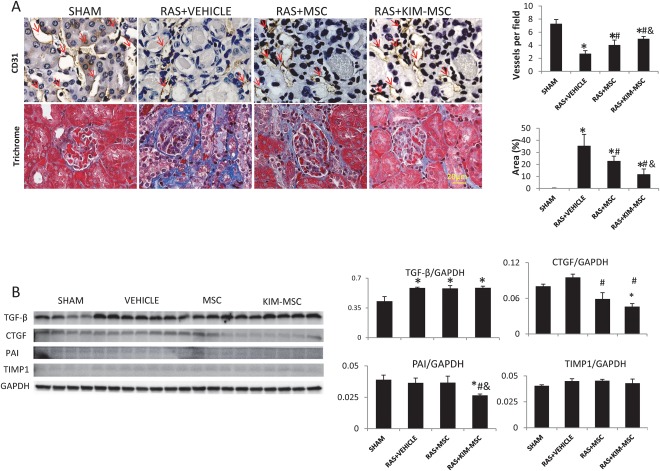
MSC and KIM‐MSC increase microvascular density and alleviate stenotic kidney fibrosis 2 weeks after injection. (A): After 4 weeks of untreated RAS, CD31 staining showing decrease of microvascular density (red arrow) in the STK, which was improved 2 weeks after injection of MSC, and slightly more by KIM‐MSC. Renal fibrosis increased in RAS+Vehicle, but MSC attenuated renal fibrosis compared to RAS+Vehicle, and KIM‐MSC attenuated it further compared to MSC (Scale bar = 20 μm). (B): Pro‐fibrotic TGF‐β protein increased after RAS. MSC and KIM‐MSC did not affect TGF‐β expression, but downregulated STK expression of CTGF, and RAS+KIM‐MSC also downregulated PAI‐1. *, p < .05 versus SHAM, #, p < .05 versus RAS+Vehicle, &, p < .05 versus RAS+MSC. Abbreviations: CTGF, connective tissue growth factor; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase;; KIM, kidney injury molecule; MSC, mesenchymal stem cells; PAI, plasminogen activator inhibitor; RAS, renal artery stenosis; TGF‐β, transforming growth‐factor; TIMP, metalloproteinase inhibitor.
Discussion
In the present study, we tested the feasibility of increasing homing and retention of adipose tissue‐derived MSC to the ischemic kidney by coating them with ab‐KIM1. We found that antibody coating was efficient and had little effect on MSC function. KIM‐MSC preferentially homed to the injured kidney, and were retained adjacent to sites of elevated KIM1 expression, in turn resulting in increased therapeutic efficacy, such as improved renal perfusion and capillary density, and attenuated oxidative damage, apoptosis, and fibrosis. This novel method may thus provide a new tool to increase the utility of cell therapy in ischemic kidney injury.
MSC treatment has been applied in various kidney diseases, including ischemic kidney injury 21. Direct injection of MSC into the renal parenchyma or subcapsular space exerts therapeutic effects 22, 23, but is limited by the technical challenge and potential for tissue damage. On the other hand, following intravenous injections, cells first pass through and lodge in the lungs, diminishing their distribution to the kidneys, resulting in engraftment rates under 3% 24, 25. Intra‐arterial injection of MSC may overcome some of these limitations, but renal retention remains low and amounts to few percent 24, 26. Various methods have been used to overcome the challenge of low engraftment of exogenously delivered cells, including gene transfection, and modifications of culture conditions or protein binding to manipulate the expression of cell surface molecules to promote MSC homing 5, 27, 28. Protein binding allows altering the cell surface but not genome, avoiding tumorigenic risk and thereby expanding the potential utility. Here, we used the protein cell coating method to attach the ab‐KIM1 to the cell surface. This method showed a high binding rate and little effect on cell viability, supporting the feasibility of therapeutic use of these coated MSC.
As a specific injury marker of kidney injury, KIM‐1 is expressed at a very low level in normal kidneys, but markedly upregulated after ischemic or toxic injury. KIM1 is mainly expressed in tubular epithelial cells and persists after ischemic injury, contributing to renal tubulointerstitial inflammation and fibrosis 19. We observed upregulation of KIM1 expression in the STK as early as 24 hours after induction of RAS, which remains consistently elevated by 2 weeks later, positioning KIM1 as an attractive candidate for cell targeting. After carotid artery injection in RAS mice, we found about a twofold higher number of KIM‐MSC in the STK compared with native MSC in each time point, and their colocalization with endogenous KIM1 was correspondingly increased, suggesting more specific targeting of injured sites. Interestingly, the unchanged KIM1 expression suggests that while targeting KIM‐1, KIM‐MSCs ultimately confer their therapeutic effects by restoration of the renal microcirculation and attenuation of renal oxidative stress, apoptosis, and fibrosis, rather than by direct inhibition of endogenous KIM1.
Insufficient blood supply may cause cell death and renal atrophy. Four weeks after RAS, STK volume decreased significantly, whereas the CLK showed a compensatory hypertrophy. In addition, RAS reduces intra‐renal perfusion pressure and renal blood flow, which results in decreased oxygen supply and usage efficiency, and eventually hypoxia 29. In RAS + Vehicle, the STK cortex and medulla both had a significant decrease in oxygenation. We have shown in swine RAS that MSC confer potent antioxidant and pro‐angiogenic effects 30, both of which may increase renal perfusion and oxygenation. Similarly, STK microvascular density in mice increased in RAS + MSC, and further in RAS + KIM‐MSC, suggesting more potent pro‐angiogenic effects. This might have resulted from a larger number of MSC leading to a greater aggregate fall in oxidative stress, which contributes to renal microvascular loss 31. Indeed, 8‐OHDG expression was significantly decreased in STKs treated with KIM‐MSC compared to nonmanipulated MSC. Besides secreting pro‐angiogenic factors, MSC can support new vasculature by acting as pericyte‐like cells 32. KIM‐MSC retained in ischemic kidneys more extensively than untreated MSC, and elicited a superior improvement in cortical and medullary perfusion, although interestingly both tactics improved renal oxygenation to a similar extent. Possibly, a comparable fall in inflammation secondary to MSC and KIM‐MSC might have ameliorated the efficiency of oxygen utilization 33, resulting in similar resolution of hypoxia. Alternatively, these cells might have boosted the renal microenvironment through systemic effects 34.
Inflammation plays an important role in the pathophysiology of ischemic kidney injury, and macrophages are important mediators of this process 35. Both MSC and KIM‐MSC similarly decreased STK macrophage infiltration, in line with their putative anti‐inflammatory properties, and despite the lower engraftment of MSC compare to KIM‐MSC. Possibly, this effect does not require direct contact with inflammatory cells in the ischemic kidney, or has a lower threshold to achieve protection. In fact, a previous study suggested that MSC attenuate renal inflammation through systemic mechanisms 34. Cell apoptosis following persistent renal ischemia involve pro‐apoptotic Bax and anti‐apoptotic Bcl‐XL family proteins 36, the ratio of which increased in RAS + vehicle, consistent with the rise in the number of apoptotic cells. MSC and KIM‐MSC reversed the Bax/Bcl ratio to a similar level, while KIM‐MSC evoked a greater decrease in the number of apoptotic cells, implicating additional mechanisms in their anti‐apoptotic effect. For example, amplified retention of KIM‐MSC in the STK might improve cellular survival through delivery of functional mitochondria via direct cell contact 37, 38.
Incomplete regeneration of injured tubular cells leads to collagen deposition and progression of tissue fibrosis, which is regulated by concerted expression of pro‐ and antifibrotic proteins. STK fibrosis was apparent in RAS + vehicle, accompanied by upregulated expression of pro‐fibrotic TGF‐β. Both MSC and KIM‐MSC decreased fibrosis in RAS without affecting TGF‐β expression, possibly by downregulation of its downstream signaling cascade like CTGF. Furthermore, KIM‐MSC also downregulated PAI‐1 protein, and was more effective than MSC in blunting fibrosis.
Limitations
In addition, a single injection of MSC and KIM‐MSC did not attenuate the rise in BP in RAS mice 2 weeks after treatment, which might require multiple injections 39 or a higher dose of MSC. Membrane dye‐based cell‐tracking does not lend itself to resolving the post‐transplantation fate of MSC, some of which might have lost functionality in situ. Neither did we observe a decrease in renal atrophy and BUN in MSC‐treated mice, possibly because renal perfusion was not fully restored. Further studies will need to investigate the increase in CLK volume in RAS + KIM‐MSC, given the unaltered BP and CLK engraftment, and additional studies are needed to determine the optimal frequency and persistence of the effects of cell delivery.
Conclusion
This study introduces a novel approach to target exogenously administered MSC to the injured kidney. Our data illustrate that targeting MSC to the ischemic kidney by coating ab‐KIM1 is a useful method to increase retention and attenuate kidney injury in murine RAS. This method may form the basis for cell delivery in other diseases, and expand the prospects of cell‐based therapy research.
Author Contributions
X.Z.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; K.J., A.P., K.J., and H.T.: collection and/or assembly of data; Xiangyang Zhu: conception and design, data analysis and interpretation, manuscript writing; L.L.: conception and design, financial support, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
This research was partly supported by NIH Grants DK73608, DK102325, DK104273, HL123160, and DK10081.
Contributor Information
Xiangyang Zhu, Email: zhu.xiangyang@mayo.edu.
Lilach O. Lerman, Email: lerman.lilach@mayo.edu.
References
- 1. Imberti B, Morigi M, Benigni A. Potential of mesenchymal stem cells in the repair of tubular injury. Kidney Int Suppl 2011;1:90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barbash IM, Chouraqui P, Baron J et al. Systemic delivery of bone marrow‐derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation 2003;108:863–868. [DOI] [PubMed] [Google Scholar]
- 3. Kean TJ, Lin P, Caplan AI et al. MSCs: Delivery routes and engraftment, cell‐targeting strategies, and immune modulation. Stem Cells Int 2013;2013:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee RJ, Fang Q, Davol PA et al. Antibody targeting of stem cells to infarcted myocardium. Stem Cells 2007;25:712–717. [DOI] [PubMed] [Google Scholar]
- 5. Ko IK, Kim BG, Awadallah A et al. Targeting improves MSC treatment of inflammatory bowel disease. Mol Ther 2010;18:1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sutton TA. Alteration of microvascular permeability in acute kidney injury. Microvasc Res 2009;77:4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int 2002;62:1539–1549. [DOI] [PubMed] [Google Scholar]
- 8. Cheng Z, Ou L, Zhou X et al. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol Ther 2008;16:571–579. [DOI] [PubMed] [Google Scholar]
- 9. Zhang D, Fan GC, Zhou X et al. Over‐expression of CXCR4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol 2008;44:281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cho SW, Sun HJ, Yang JY et al. Transplantation of mesenchymal stem cells overexpressing RANK‐Fc or CXCR4 prevents bone loss in ovariectomized mice. Mol Ther 2009;17:1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du Z, Wei C, Yan J et al. Mesenchymal stem cells overexpressing C‐X‐C chemokine receptor type 4 improve early liver regeneration of small‐for‐size liver grafts. Liver Transpl 2013;19:215–225. [DOI] [PubMed] [Google Scholar]
- 12. Kim SA, Peacock JS. The use of palmitate‐conjugated protein A for coating cells with artificial receptors which facilitate intercellular interactions. J Immunol Methods 1993;158:57–65. [DOI] [PubMed] [Google Scholar]
- 13. Chen A, Zheng G, Tykocinski ML. Hierarchical costimulator thresholds for distinct immune responses: Application of a novel two‐step Fc fusion protein transfer method. J Immunol 2000;164:705–711. [DOI] [PubMed] [Google Scholar]
- 14. Ko IK, Kean TJ, Dennis JE. Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials 2009;30:3702–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dennis JE, Cohen N, Goldberg VM et al. Targeted delivery of progenitor cells for cartilage repair. J Orthop Res 2004;22:735–741. [DOI] [PubMed] [Google Scholar]
- 16. Ichimura T, Bonventre JV, Bailly V et al. Kidney injury molecule‐1 (KIM‐1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up‐regulated in renal cells after injury. J Biol Chem 1998;273:4135–4142. [DOI] [PubMed] [Google Scholar]
- 17. Ichimura T, Hung CC, Yang SA et al. Kidney injury molecule‐1: A tissue and urinary biomarker for nephrotoxicant‐induced renal injury. Am J Physiol Renal Physiol 2004;286:F552–F563. [DOI] [PubMed] [Google Scholar]
- 18. Vaidya VS, Ramirez V, Ichimura T et al. Urinary kidney injury molecule‐1: A sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol 2006;290:F517–F529. [DOI] [PubMed] [Google Scholar]
- 19. Humphreys BD, Xu F, Sabbisetti V et al. Chronic epithelial kidney injury molecule‐1 expression causes murine kidney fibrosis. J Clin Invest 2013;123:4023–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang K, Ferguson CM, Ebrahimi B et al. Noninvasive assessment of renal fibrosis with magnetization transfer MR imaging: Validation and evaluation in murine renal artery stenosis. Radiology 2016;283:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu XY, Urbieta‐Caceres V, Krier JD et al. Mesenchymal stem cells and endothelial progenitor cells decrease renal injury in experimental swine renal artery stenosis through different mechanisms. Stem Cells 2013;31:117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alfarano C, Roubeix C, Chaaya R et al. Intraparenchymal injection of bone marrow mesenchymal stem cells reduces kidney fibrosis after ischemia‐reperfusion in cyclosporine‐immunosuppressed rats. Cell Transplant 2012;21:2009–2019. [DOI] [PubMed] [Google Scholar]
- 23. Cheng K, Rai P, Plagov A et al. Transplantation of bone marrow‐derived MSCs improves cisplatinum‐induced renal injury through paracrine mechanisms. Exp Mol Pathol 2013;94:466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Devine SM, Cobbs C, Jennings M et al. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood 2003;101:2999–3001. [DOI] [PubMed] [Google Scholar]
- 25. Fischer UM, Harting MT, Jimenez F et al. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first‐pass effect. Stem Cells Dev 2009;18:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao J, Dennis JE, Muzic RF et al. The dynamic in vivo distribution of bone marrow‐derived mesenchymal stem cells after infusion. Cells Tissues Organs 2001;169:12–20. [DOI] [PubMed] [Google Scholar]
- 27. Wang X, Liu C, Li S et al. Hypoxia precondition promotes adipose‐derived mesenchymal stem cells based repair of diabetic erectile dysfunction via augmenting angiogenesis and neuroprotection. PLoS One 2015;10:e0118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wiehe JM, Kaya Z, Homann JM et al. GMP‐adapted overexpression of CXCR4 in human mesenchymal stem cells for cardiac repair. Int J Cardiol 2013;167:2073–2081. [DOI] [PubMed] [Google Scholar]
- 29. Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: Ischemia and beyond. Prog Cardiovasc Dis 2009;52:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eirin A, Zhu XY, Krier JD et al. Adipose tissue‐derived mesenchymal stem cells improve revascularization outcomes to restore renal function in swine atherosclerotic renal artery stenosis. Stem Cells 2012;30:1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhu XY, Chade AR, Rodriguez PM et al. Cortical microvascular remodeling in the stenotic kidney: Role of increased oxidative stress. Arterioscler Thromb Vasc Biol 2004;24:1854–1859. [DOI] [PubMed] [Google Scholar]
- 32. Sanz L, Santos‐Valle P, Alonso‐Camino V et al. Long‐term in vivo imaging of human angiogenesis: Critical role of bone marrow‐derived mesenchymal stem cells for the generation of durable blood vessels. Microvasc Res 2008;75:308–314. [DOI] [PubMed] [Google Scholar]
- 33. Welch WJ, Mendonca M, Aslam S et al. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2K,1C kidney. Hypertension 2003;41:692–696. [DOI] [PubMed] [Google Scholar]
- 34. Hu J, Zhang L, Wang N et al. Mesenchymal stem cells attenuate ischemic acute kidney injury by inducing regulatory T cells through splenocyte interactions. Kidney Int 2013;84:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jo SK, Sung SA, Cho WY et al. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant 2006;21:1231–1239. [DOI] [PubMed] [Google Scholar]
- 36. Borkan SC. The role of BCL‐2 family members in acute kidney injury. Semin Nephrol 2016;36:237–250. [DOI] [PubMed] [Google Scholar]
- 37. Lin HY, Liou CW, Chen SD et al. Mitochondrial transfer from Wharton's jelly‐derived mesenchymal stem cells to mitochondria‐defective cells recaptures impaired mitochondrial function. Mitochondrion 2015;22:31–44. [DOI] [PubMed] [Google Scholar]
- 38. Islam MN, Das SR, Emin MT et al. Mitochondrial transfer from bone‐marrow‐derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 2012;18:759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oliveira‐Sales EB, Maquigussa E, Semedo P et al. Mesenchymal stem cells (MSC) prevented the progression of renovascular hypertension, improved renal function and architecture. PLoS One 2013;8:e78464. [DOI] [PMC free article] [PubMed] [Google Scholar]