

Abstract
Human mesenchymal stem cells (MSCs) have huge potential for regenerative medicine. In particular, the use of pluripotent stem cell‐derived mesenchymal stem cells (PSC‐MSCs) overcomes the hurdle of replicative senescence associated with the in vitro expansion of primary cells and has increased therapeutic benefits in comparison to the use of various adult sources of MSCs in a wide range of animal disease models. On the other hand, fetal MSCs exhibit faster growth kinetics and possess longer telomeres and a wider differentiation potential than adult MSCs. Here, for the first time, we compare the therapeutic potential of PSC‐MSCs (ES‐MSCs from embryonic stem cells) to fetal MSCs (AF‐MSCs from the amniotic fluid), demonstrating that ES‐MSCs have a superior neuroprotective potential over AF‐MSCs in the mouse brain following hypoxia‐ischemia. Further, we demonstrate that nuclear factor (NF)‐κB‐stimulated interleukin (IL)‐13 production contributes to an increased in vitro anti‐inflammatory potential of ES‐MSC‐conditioned medium (CM) over AF‐MSC‐CM, thus suggesting a potential mechanism for this observation. Moreover, we show that induced pluripotent stem cell‐derived MSCs (iMSCs) exhibit many similarities to ES‐MSCs, including enhanced NF‐κB signaling and IL‐13 production in comparison to AF‐MSCs. Future studies should assess whether iMSCs also exhibit similar neuroprotective potential to ES‐MSCs, thus presenting a potential strategy to overcome the ethical issues associated with the use of embryonic stem cells and providing a potential source of cells for autologous use against neonatal hypoxic‐ischemic encephalopathy in humans. Stem Cells Translational Medicine 2018;7:439–449
Keywords: Pluripotent stem cells, Mesenchymal stem cells, Fetal stem cells, Cell transplantation, Embryonic stem cells, Induced pluripotent stem cells
Significance Statement.
Mesenchymal stem cells (MSCs) are currently under assessment in various clinical trials worldwide. Their therapeutic potential is due largely to the factors they secrete. This study shows that embryonic stem cell‐derived MSCs have increased in vitro anti‐inflammatory potential in comparison to amniotic fluid‐derived MSCs and that this translates to an increased in vivo neuroprotective capacity following hypoxic‐ischemic injury of the mouse brain. These findings provide new insight into the molecular mechanisms underlying the therapeutic potential of MSCs. By illuminating these molecular mechanisms, this study may have implications for improving the efficiency of MSC therapies.
Introduction
Mesenchymal stromal/stem cells (MSCs) are multipotent cells found in various tissues throughout development, where they contribute to the repair and regeneration of these tissues. Human fetal MSCs have been isolated from various fetal tissues including first trimester blood, liver, or bone marrow (BM) 1, 2, 3, or from extraembryonic tissues such as the placenta, umbilical cord or amniotic fluid 4, 5. Due to their more primitive phenotype, fetal MSCs present various advantages over their adult counterparts such as longer telomeres, active telomerase and a greater expansion capacity 6. MSCs, as defined by their plastic adherence, tri‐lineage differentiation potential, co‐expression of the cell surface markers CD105, CD90, and CD73 and absence of expression of CD34 and CD45 7, are currently under assessment in numerous clinical trials worldwide targeting a range of disorders from myocardial infarction to stroke 8. However, the specific mechanisms by which they mediate tissue regeneration in a range of disease contexts remain largely elusive.
Increasing evidence suggests that the therapeutic potential of MSCs should not be attributed only to their engraftment and differentiation but also to the release of regenerative paracrine/endocrine factors. These effects of MSCs were first suggested when, after injection, BM‐MSCs exerted therapeutic effects on the infarcted heart despite being detected largely in the lungs and liver in various murine heart disease models 9, 10. Furthermore, MSC‐conditioned medium (CM) has been shown to exert a therapeutic effect in animal models of cardiomyopathy 11 and MSC‐derived exosomes have been shown to ameliorate disorders of the brain, kidney, and joints 12, 13, 14, 15. These studies are paving the way for off‐the‐shelf cell‐free therapies, which would have the benefits of being more well defined and standardized than stem cell treatments.
Standardization of cell‐free therapies will require a thorough understanding of the therapeutic factors that MSCs secrete. One key class of molecules released by MSCs are anti‐inflammatory factors. These include interleukin 1 receptor agonist (IL‐1ra) 16, tumor necrosis factor (TNF)α‐stimulated gene (TSG)6 8, 17, 18, prostaglandin (PG)E2 16, 17, 18, 19, indoleamine 2,3‐dioxygenase (IDO) 20, 21, 22, 23, 24, and IL‐10 19. When in an inflammatory environment, MSCs produce higher levels of these anti‐inflammatory factors, a phenomenon known as “licensing” 25, 26, 27. Mechanistically, this is likely due to activation of nuclear factor (NF)‐κB signaling in MSCs leading to transcription of downstream targets which include anti‐inflammatory cytokines. To support this idea, Dorronsoro et al. 28 have shown that inhibition of NF‐κB through silencing of IκB kinase or the TNFα receptor TNFR1 abolishes the capacity of MSCs to regulate T cell proliferation and various groups have shown that Toll‐like receptor (TLR) engagement leads to enhanced immunosuppressive properties in human BM‐MSCs 18, 29, 30. Moreover, Tomchuck et al. 31 have demonstrated that NF‐κB activation in MSCs leads to increased production of cytokines, chemokines and related TLR gene products such as PGE2.
Human primary MSCs, especially those from aged donors, have a limited proliferation capacity in vitro due to replicative senescence 32, 33, 34 and higher passage MSCs are more likely to trigger an innate immune attack upon transplantation into humans 35. In addition, cells derived from aged donors tend to be more pro‐inflammatory than those from younger donors 36, 37, 38, thereby further restricting the clinical potential of expanded human primary cells. To address these issues, various groups have generated pluripotent stem cell‐derived MSCs (PSC‐MSCs), of which there are two types, embryonic stem cell (ESC)‐derived MSCs (ES‐MSCs) 39, 40, 41 and induced pluripotent stem cell (iPSC)‐derived MSCs (iMSCs). Pluripotent stem cells can be expanded in vitro without undergoing senescence and PSC‐MSCs have been shown to have enhanced therapeutic potential compared with other sources of MSCs in animal models of various diseases including multiple sclerosis 42, cardiomyopathy 11, pulmonary arterial hypertension 43, osteoarthritis 14, and limb ischemia 44, potentially due to their “rejuvenated” phenotype 45. However, some studies report comparable 46, 47, 48 or even decreased 45 immunosuppressive abilities of PSC‐MSCs over other sources of MSCs.
In this study, we compare for the first time the neuroprotective potential of ES‐MSCs and fetal amniotic fluid‐derived (AF)‐MSCs. We show that ES‐MSCs have an increased neuroprotective capacity compared with AF‐MSCs in a perinatal mouse hypoxia‐ischemia model. Further, we demonstrate in vitro that increased NF‐κB activation in ES‐MSCs leads to elevated interleukin (IL)‐13 production, thus providing a potential mechanism for the enhanced neuroprotective capacity of ES‐MSCs over AF‐MSCs. Finally, we compare these two MSC lines to iMSCs, showing that iMSCs also exhibit increased NF‐κB activation, concomitant with elevated IL‐13 production and increased anti‐inflammatory potential in comparison to AF‐MSCs. Together, these findings suggest that PSC‐MSCs are a promising cell source for use in therapeutic strategies targeting neonatal hypoxic‐ischemic encephalopathy (HIE) in humans.
Materials and Methods
Ethics Statements
The healthy donors who provided the amniotic fluid in this study provided written informed consent in accordance with the Declaration of Helsinki. The ethical approval given by the Research Ethics Committees of Hammersmith & Queen Charlotte Hospitals (2001/6234) was in compliance with U.K. national guidelines (Review of the Guidance on the Research Use of Fetuses and Fetal Material [1989], also known as the Polkinghome Guidelines London: Her Majesty's Stationary Office, 1989: Cm762) for the collection of fetal tissue for research.
All animals were handled in strict accordance with good animal practice as defined by the U.K. Home Office Animal Welfare Legislation (PPL 70/8784), The Institutional Licensing Committee of Imperial College London and University College London (UCL) and the Institutional Research Ethics Committee (Institute of Child Health, UCL, London).
Cell Culture
Human AF‐MSCs were isolated as previously described 4. Unless specified, cells were plated at 1 × 104 cells per cm2 in D10 (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM l‐glutamine, 50 IU/ml penicillin, and 50 mg/ml streptomycin [all Sigma‐Aldrich, Dorset, UK]) at 37°C with 5% CO2. Where indicated, 20 ng/ml TNFα (Sigma‐Aldrich), 125 ng/ml IL‐13P (Santa Cruz Biotechnology, CA, USA) or an equal volume of the appropriate vehicle was added to the cells in D10. The human ESC line H1 (WiCell Research Institute, Madison, USA) was cultured in feeder‐free conditioned on Matrigel‐coated plates in mTESR (Stem Cell Technologies, Cambridge, UK).
Cell Proliferation Assay
5 × 104 cells per well were plated in twelve wells of a 12‐well plate and the cells were counted in triplicate 24, 48, 72, and 96 hours after plating.
iPSC Reprogramming of AF‐MSCs
5 × 105 AF‐MSCs were detached using TrypLE (Life Technologies, Paisley, UK), centrifuged at 1,200 rpm for 5 minutes then electroporated as described previously 49 but without the pCXWB‐EBNA1 episomal plasmid. After transfection, cells were plated onto one 0.1% gelatin (Sigma‐Aldrich)‐coated well of a 6‐well plate. Twenty‐four hours post‐transfection, cells were detached and seeded into one 6 cm2 dish precoated with Matrigel (Scientific Laboratory Supplies ltd, Nottingham, UK) in Essential 8 medium (Life Technologies). The medium was then replenished every 2 days until around day 20, when iPSC colonies were passaged manually with a needle into a fresh Matrigel‐coated well of a 6‐well plate in Essential 8 medium. Cells were passaged with 0.5 mM EDTA (Life Technologies) thereafter.
MSC Differentiation of Pluripotent Stem Cells
PSC‐MSC differentiation was performed as described in Chen et al. 50 with some modifications. Three iMSC and three ES‐MSC lines were generated. Briefly, a confluent T25 of ESCs or iPSCs was treated with 10 µM SB431542 for 10 days before the cells were passaged using Tryple Select (Life Technologies) at a 1:1 ratio into StemMACS MSC expansion medium supplemented with CytoMix according to the manufacturer's instructions (both Miltenyi Biotech, Bisley, UK). Cells were subsequently seeded at 2 × 104 cells per cm2 at the first passage, then 1 × 104 cells per cm2 thereafter and analyzed around passage 6.
AF‐MSC Transfection
Cells were electroporated as described above in electroporation buffer containing 2 µg each of either pCMV‐IKK2 or the pCMV‐FLAG control plasmid (both Addgene, Cambridge, UK).
Embryoid Body Formation
iPSCs were detached using EDTA and resuspended in Essential 8 medium containing 10 µM Y‐27632 (Sigma‐Aldrich) in bacterial culture dishes (non‐TC treated). The medium was replaced on day 2, then changed to D10 containing 20% FBS (D20) on days 4 and 6. On day 7, the embryoid bodies were plated onto 0.1% gelatin (Sigma‐Aldrich). The cells were fed every 2 days with D20 until being fixed with 4% PFA on day 14.
Lentiviral Production and Transduction
HEK 293T cells were plated at 80% confluency in one well of a 6‐well plate. They were then transfected using poly polyethylenimine precomplexed with 2.79 µg of the appropriate lentiviral vector along with 0.96 µg VSVg envelope plasmid and 1.79 µg packaging plasmid (pCMVΔ8.74) in OptiMEM (Life Technologies) for 3 hours prior to re‐feeding. Culture medium was then harvested at 48 hours post‐transfection and filtered through a 0.45 µm PVDF filter (Elkay, Basingstoke, UK) on to one well of receiver cells.
NF‐κB Anti‐Inflammatory Assay
HEK 293T cells were transduced with the pLNT‐NF‐κB‐NLuc (NLuc) and pLNT‐SFFV‐VLuc (VLuc) lentiviral plasmids as previously described 49. Reporter cells were then plated in triplicate in 24‐well plates. Twenty‐four hours later, cells were activated with 20 ng/ml TNFα (Sigma‐Aldrich). Twenty‐four hours after activation, 12 ml CM from 6 wells of cells was concentrated in an Amicon Ultra‐15 3K NMWL filter (Millipore, Watford, UK) by centrifugation at 4,000g for 30 minutes. The cells were then counted and the volume of medium added was adjusted according to cell number. The appropriate volume of concentrated medium was added to 1.5 ml D10. 0.5 ml was then added to each well of reporter HEK 293T cells in triplicate. Twenty‐four hours after the addition of CM the medium was replaced with fresh D10 and luciferase assays were performed following a further 24 hours.
RT‐PCR
Total RNA was isolated and reverse transcribed to cDNA as previously described 49. Reverse transcription (RT)‐PCR was then performed using 1 µl cDNA which was amplified for 30 cycles at 54°C using the appropriate primer pair (Table 1). Samples were separated using a 1.5% agarose gel and visualized using a UV transilluminator.
Table 1.
Reverse transcription (RT)‐PCR primers
| Primer target | Forward sequence (5′‐3′) | Reverse sequence (5′‐3′) |
|---|---|---|
| OCT4 | CTGGAGAAGGAGAAGCTGGA | AATAGAACCCCCAGGGTGAG |
| SOX2 | GACCAGCTCGCAGACCTACAT | TACCTCTTCCTCCCACTCCA |
| NANOG | AGCCTCTACTCTTCCTACCACC | TCCAAAGCAGCCTCCAAGTC |
| β‐TUBULIN | ACATCCAGGCTGGTCAGTGT | CCCAGGTTCTAGATCCACCA |
qRT‐PCR
Total RNA was isolated and reverse transcribed to cDNA as previously described 49. Triplicate samples containing the appropriate primers (1 µM, Table 2) were analyzed by quantitative (q)RT‐PCR using SYBR Green PCR Master Mix (Life Technologies) according to the manufacturer's instructions.
Table 2.
Quantitative (q)RT‐PCR primers
| Primer target | Forward sequence (5′‐3′) | Reverse sequence (5′‐3′) |
|---|---|---|
| NF‐κB1 | TGCCAACAGATGGCCCATAC | TGTTCTTTTCACTAGAGGCACCA |
| RelA | GTGGGGACTACGACCTGAATG | GGGGCACGATTGTCAAAGATG |
| IKK2 | CTGGCCTTTGAGTGCATCAC | CGCTAACAACAATGTCCACCT |
| β‐ACTIN | CGGGACCTGACTGACTACC | TGAAGGTAGTTTCGTGGATGC |
Flow Cytometry
Cells were detached as described previously and washed in flow buffer (phosphate‐buffered saline (PBS) + 1% BSA). Cells were then centrifuged at 5,000g for 2 minutes before 1 × 105 cells were resuspended in the appropriate primary antibody (anti‐CD105, anti‐CD90, anti‐CD73, anti‐CD14 [All Miltenyi Biotec], anti‐CD29 [Abcam, Cambridge, UK], or anti‐CD45 [BD Biosciences, Wokingham, UK]) at its optimal dilution (1:10) in flow buffer and incubated for 1 hour at 4°C. For unconjugated antibodies, cells were then washed and resuspended in a 1:10 dilution of FITC‐conjugated donkey anti‐mouse (Jackson ImmunoResearch labs, PA, USA) for 30 minutes at 4°C. Cells were then analyzed using a Becton Dickinson FACScalibur flow cytometer (BD biosciences) using Cell Quest Pro and FlowJo software.
Luciferase Assays
The supernatant was collected from triplicate wells of cells at the appropriate time points and 20 µl was transferred to 20 µl assay buffer (25 mM Tris Phosphate [pH7.8] containing 1% BSA and 30% glycerol, all Sigma‐Aldrich) in a white‐bottomed 96‐well plate (Corning, NY, USA) in technical triplicates. VLuc samples were assayed detecting photonic emissions at 460 nm after addition of 5 nM vargulin (Gold Biotechnology, Olivette, USA) and NLuc photonic emissions at 454 nm after addition of 2 mM coelenterazine (Gold Biotechnology) which had been incubated on ice for 45 minutes with 0.1 M KI and 10 mM EDTA (Sigma‐Aldrich) using a FLUOstar Optima luminometer (BMG Labtech, Ortenberg, Germany). NLuc values were divided by VLuc values before the average fold change over control ± standard deviation was plotted graphically.
p values were calculated using analysis of variance (One‐way ANOVA) followed by Bonferroni's multiple comparison post hoc test.
Immunofluorescent Cell Staining
Immunocytochemistry was performed as previously described 49. Briefly, cells were washed with PBS and fixed in situ using 4% PFA in PBS before being washed, permeabilized with 0.3% Triton in PBS (if required) and blocked for 30 minutes in 2% BSA/0.05% Triton in PBS. Cells were then incubated in the appropriate primary antibody overnight at 4°C before being washed and incubated with the appropriate secondary antibody for 1 hour at room temperature in the dark. Finally, cells were washed, the nuclei were stained with 4',6‐diamidino‐2‐phenylindole (DAPI) and the cells were visualized on a Zeiss Axio Observer A1 fluorescence microscope. Additional antibodies used were anti‐REX1, TRA‐1–60, and DNMT3B (all 1:200, Abcam) and an OCT4A‐specific antibody (Santa Cruz Biotechnology).
Enzyme‐Linked Immunosorbent Assays
Cells were plated at 2 × 104 cells per cm2 in four wells of a 6‐well plate. Seventy‐two hours after seeding, 8 ml CM was removed and concentrated using an Amicon ultra‐4 15ml 3K NMWL filter by centrifugation at 4,000g for 40 minutes. 300 μl of D10 was then added to the concentrated medium and 100 µl of this was added per well of the enzyme‐linked immunosorbent assay (ELISA) plate. The mini ABTS ELISAs (Peprotech, London, UK) were performed as per the manufacturer's instructions.
Western Blot
Western blot was performed as described previously 49 with the following antibodies: anti‐IKK2 and anti‐β‐ACTIN (both 1:1,000, Abcam).
Hypoxic‐Ischemic Induction
All animal experiments and care protocols were carried out according to the U.K. Animals (Scientific Procedures) Act 1986 and approved by the Home Office. The ARRIVE guidelines were followed. All experiments involved C57/Bl6 mice obtained from Charles River (Kent, U.K.). The surgical procedures were performed as previously described 51. Briefly, male and female postnatal day 7 (p7) mice were anesthetized with isofluorane (5% induction followed by 1.5% maintenance) before the left common carotid artery was permanently ligated. The mice were then returned to the dam for 2 hours before being exposed to 8% oxygen/92% nitrogen at 36°C for 1 hour. Immediately after hypoxic‐ischemic (HI) insult, 1 × 105 AF‐MSCs or ES‐MSCs were then injected in 2 µl PBS through a single contralateral injection taking approximately 60 seconds.
Tissue Sample Preparation
Animals were sacrificed at 48 hours post‐injection by intraperitoneal injection of pentobarbitane and perfused as described in Hristova et al. 51 Briefly, following sacrifice, animals were perfused with 30 ml 4% paraformaldehyde (PFA) in PBS. Brains were then isolated, fixed in 4% PFA for 1 hour at 4°C and then cryopreserved for 24 hours in phosphate‐buffered sucrose solution before being frozen in dry ice, cut on a cryostat into 40 µm sections and stored at −80°C until required for analysis.
Immunohistochemistry and Histological Analysis
Five cryosections from each brain (400 µm apart) were rehydrated in H2O and then stained as previously described 51. Infarct volume measurement was performed on cresyl violet‐stained sections. These sections were imaged using a Zeiss Axiplan2 Axiocam HRc camera and imported into Image J, which was then used to determine the area of the following bilateral regions: cortex (CTX), pyriform cortex (PYR), hippocampus (HPP), striatum (STR), thalamus (THAL), and external capsule (EC). The percentage tissue loss in comparison to the control side was then calculated by converting the injured and uninjured areas into mm2 then converting to volume by multiplying by 400 µm. To determine the levels of microglial activation, brain tissue was stained using an anti‐αMb2 integrin antibody (1:5,000, Bio‐Rad, Kidlington, UK) and semi‐quantitative scores were allocated to each brain region (CTX, PYR, HPP, STR, THAL, and EC) by an observer blinded to the treatment of the groups, as previously described 51, 52, to describe microglial appearance, that is, 0 (no activation), 1 (focal microglial activation), 2 (mild phagocytic activation affecting <50% of the region), 3 (phagocytic activation affecting >50% of the region), and 4 (total phagocytic activation). To determine levels of astroglial activation, sections were stained using an anti‐glial fibrillary acidic protein (GFAP) antibody (1:6,000, Dako, Glostrup, Denmark) before three ×20 images per region of both the ipsilateral and contralateral sides were captured. Images were then imported into Image J which was used to obtain the mean and standard deviation for the optical luminosity values (OLVs). The standard deviation was subtracted from the values obtained for the surrounding glass slide. Images were contrast stretched using Adobe Photoshop 6.0.
Statistical Analysis
Statistical analysis was performed using SPSS. One‐way analysis of variance (ANOVA)s followed by Bonferroni's multiple comparison post‐hoc test or Kruskal‐Wallis nonparametric tests were used to obtain p values where appropriate. p < .05 was significant.
Results
Validation of ES‐MSC Identity
We modified the protocol published by Chen et al. 50 to differentiate ESCs into ES‐MSCs. To validate this protocol, we confirmed that the ES‐MSCs had gained a characteristic MSC‐like morphology (Fig. 1Ai) along with the expression of the MSC markers CD105, CD73, CD90 (Fig. 1Aii), and CD29 (Supporting Information Fig. S1A), and lacked the expression of the hematopoietic markers CD45 and CD14 (Supporting Information Fig. S1B, S1C). We also confirmed the loss of pluripotency marker expression (SOX2, OCT4A, and NANOG) in these cells by RT‐PCR (Fig. 1B).
Figure 1.
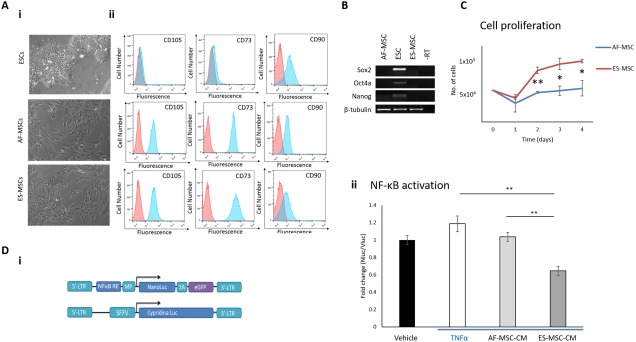
ES‐MSCs have increased anti‐inflammatory potential compared with AF‐MSCs. (Ai): Phase contrast images to show morphology of ESCs, AF‐MSCs, and ES‐MSCs. ×10 magnification. (Aii): Flow cytometry demonstrates the acquisition of MSC marker expression in ES‐MSCs. (B): RT‐PCR shows loss of pluripotency‐associated transcripts in ES‐MSCs. (C): Graph to show the proliferation rate of ES‐MSCs versus AF‐MSCs. (Di): Schematic of pLNT‐NF‐κB‐Nluc and pLNT‐SFFV‐Vluc lentiviral constructs. (Dii): Anti‐inflammatory assay demonstrates that ES‐MSC‐CM decreases NF‐κB activation in reporter cells to a higher degree than AF‐MSC‐CM. *, p < .05; **, p < .01. Abbreviations: AF‐MSC, amniotic fluid‐derived MSC; ESC, embryonic stem cell; ES‐MSC, ESC‐derived MSC; iMSC, iPSC‐derived MSC; iPSC, induced pluripotent stem cell; MSC, mesenchymal stem cell; NF‐κB, nuclear factor‐kappa B; PSC‐MSC, pluripotent stem cell‐derived MSC; TNFα, tumor necrosis factor α.
ES‐MSCs Have Enhanced Neuroprotective Potential in Comparison to AF‐MSCs
Various groups 44, 45, 53 have previously documented the increased proliferative potential of PSC‐MSCs in comparison to BM‐MSCs but we are the first to compare PSC‐MSCs to fetal MSCs. We observed that ES‐MSCs proliferate at an increased rate in comparison to amniotic fluid‐derived MSCs (Fig. 1C).
Numerous groups have demonstrated the improved immunoregulatory potential of PSC‐MSCs in comparison to the parental population 11, 14, 53. In support of these findings, we observed that the media conditioned by ES‐MSCs (ES‐MSC‐CM) had significantly higher anti‐inflammatory potential than that derived from AF‐MSCs in an in vitro NF‐κB reporter assay, using a lentiviral reporter system previously described by us 49 (Fig. 1Di, 1Dii)). Suppression of NF‐κB activation in reporter cells by MSCs or their CM or exosomes has been used by various other groups previously to demonstrate their anti‐inflammatory potential 54, 55, 56. Importantly, neither concentrated D10 medium alone or medium conditioned by ESCs or iPSCs decreased NF‐κB activation reporter cells, thus confirming that the effect is specific to MSC‐CM (Supporting Information Fig. S2A).
To determine whether the increased in vitro anti‐inflammatory potential of PSC‐MSC‐CM translates to an increased therapeutic potential in vivo, we injected the cells into the contralateral side of the brains of neonatal (postnatal day 7) mice subjected to HI insult. Both AF‐MSCs and ES‐MSCs demonstrated the ability to reduce forebrain tissue loss across all examined regions, with the most notable decrease observed in the cortex, pyriform cortex, striatum, and external capsule (Fig. 2Ai). In all examined regions except the thalamus, ES‐MSCs demonstrated a tendency to reduce tissue loss to a higher degree than AF‐MSCs reaching significance in the cortex, pyriform cortex, and overall (Fig. 2Ai). Representative images of the three treated groups (PBS, AF‐MSC, and ES‐MSC) are shown in Figure 2Aii. Both cell types also reduced HI‐induced microglial activation. The reduction achieved by ES‐MSC injection reached significance in all examined regions and overall whereas the reduction achieved by AF‐MSC treatment only reached significance in the striatum (Fig. 2Bi). Notably, ES‐MSCs exhibited a tendency to reduce microglial activation to a higher degree than AF‐MSCs in all examined regions, reaching significance in the thalamus and overall (Fig. 2Bi). Representative images of the levels of microglial activation in all three groups are shown in Figure 2Bii. We observed that MSC injection was able to prevent the transition of microglia from a ramified, inactive morphology to the round, phagocytic phenotype observed in the PBS control group (Fig. 2Biii).
Figure 2.
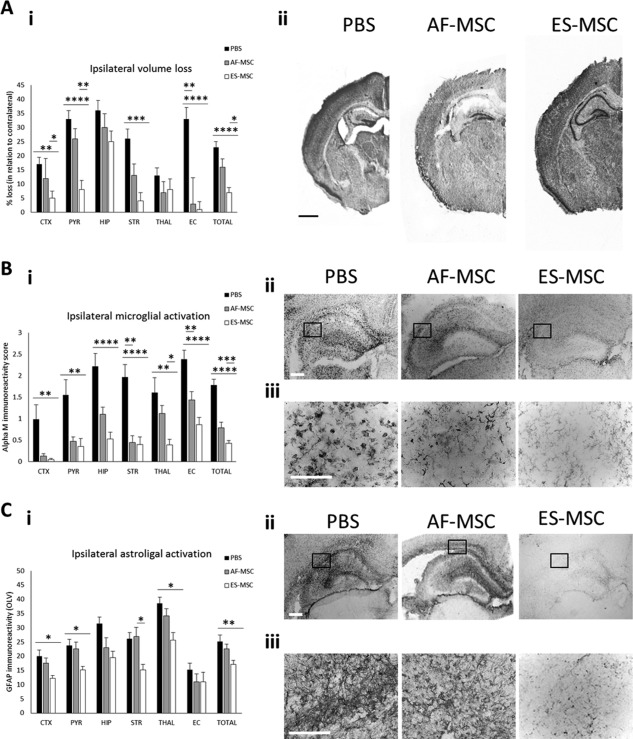
ES‐MSCs have a greater neuroprotective potential than AF‐MSCs following HI injury in the mouse brain. (Ai): Graph and (Aii) phase contrast images to show ipsilateral brain tissue volume loss at 48 hours post‐HI. Scale bar: 900 µm. (Bi): Graph, (Bii) low magnification, and (Biii) high magnification phase contrast images to demonstrate the activation of Alpha M+ microglia. Scale bars: 100 µm. (Ci): Graph, (Cii) low magnification and (Ciii) high magnification phase contrast images to show GFAP immunoreactivity in the ipsilateral side of the brain at 48 hours post‐HI. Scale bars: 100 µm. *, p < .05; **, p < .01; ***, p < .005; ***, p < .001. Abbreviations: AF‐MSC, amniotic fluid‐derived mesenchymal stem cell; ES‐MSC, ESC‐derived mesenchymal stem cell; OLV, optical luminosity values.
A similar effect was observed for astroglial activation assessed through GFAP immunoreactivity. We observed a trend toward reduced levels of GFAP immunoreactivity in response to HI insult in the MSC‐treated groups in all examined regions of the forebrain except the striatum, where only ES‐MSCs decreased astroglial activation (Fig. 2Ci). Again, ES‐MSCs showed a trend toward reducing GFAP immunoreactivity to a higher degree than AF‐MSCs, with this difference reaching significance in the striatum (Fig. 2Ci). No significant differences were observed between treatments in the contralateral side (data not shown). We observed reduced overall GFAP immunoreactivity in response to treatment with either type of MSCs over the PBS control (Fig. 2Cii) and substantially reduced and more punctate staining of GFAP+ astroglial processes in the ES‐MSC treatment group compared with the other two treatment groups (PBS and AF‐MSC) (Fig. 2Ciii).
ES‐MSCs Exhibit Higher Levels of NF‐κB Activation than AF‐MSCs
Since NF‐κB activity has previously been linked to the therapeutic potential of MSCs 28, 29, 30, we then determined whether the increased anti‐inflammatory potential of ES‐MSCs compared with AF‐MSCs was due to increased NF‐κB activation leading to increased production of anti‐inflammatory cytokines. We performed qRT‐PCR for three NF‐κB target genes, NF‐κB1, IKK2, and RelA, in MSCs under basal conditions and observed that the levels of expression of these transcripts were higher in ES‐MSCs compared with AF‐MSCs (Fig. 3A). Additionally, Western blotting revealed an increase in IKK2 protein expression in ES‐MSCs compared with AF‐MSCs (Fig. 3B) and the levels of NF‐κB activation following TNFα stimulation of NF‐κB reporter MSCs were also higher in these cells, as assessed by a luciferase assay (Fig. 3C).
Figure 3.
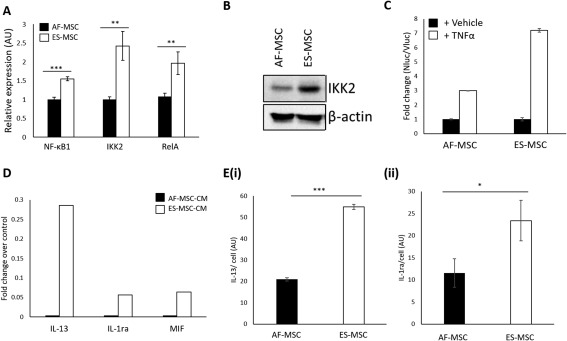
ES‐MSCs exhibit increased NF‐κB activation and anti‐inflammatory cytokine production. (A): qPCR to show NF‐κB target gene expression in ES‐MSCs. (B): Western blot to show IKK2 protein expression in AF‐MSCs and ES‐MSCs. (C): Luciferase assay to show NF‐κB activation in AF‐MSCs and ES‐MSCs in response to TNFα. (D): Cytokine array to show levels of anti‐inflammatory cytokines in ES‐MSC‐CM versus AF‐MSC‐CM. (E): ELISA to show levels of (Ei) IL‐13 and (Eii) IL‐1ra production by ES‐MSCs and AF‐MSCs. *, p < 0.05; **, p < .01; ***, p < .005. Abbreviations: AF‐MSC, amniotic fluid‐derived mesenchymal stem cell; ES‐MSC, ESC‐derived mesenchymal stem cell; CM, conditioned medium; TNFα, tumor necrosis factor α.
To show that these observations were not specific to the origin of the MSCs we also demonstrated increased activation of NF‐κB in fetal BM‐iMSCs compared with the parental fetal BM‐MSC population (Supporting Information Fig. S2B). BM‐iMSC‐CM also showed a tendency toward having increased anti‐inflammatory potential compared with BM‐MSC‐CM (Supporting Information Fig. S2C).
ES‐MSCs Secrete Increased Levels of anti‐Inflammatory Cytokines Compared with AF‐MSCs
We then aimed to identify the anti‐inflammatory cytokines, downstream of NF‐κB, that contribute to the increased anti‐inflammatory potential of ES‐MSC‐CM compared with AF‐MSC‐CM. A cytokine array identified increases in the production of IL‐13, IL‐1ra and macrophage inhibitory factor (MIF) by ES‐MSCs compared with AF‐MSCs (Fig. 3D). Since IL‐13 and IL‐1ra are NF‐κB target genes we investigated the secretion of these cytokines further using ELISAs and confirmed that both IL‐13 and IL‐1ra were released at significantly higher levels by ES‐MSCs than AF‐MSCs (Fig. 3Ei, 3Eii).
NF‐κB‐Induced IL‐13 Production Contributes to the Increased anti‐Inflammatory Potential of ES‐MSCs Compared with AF‐MSCs
To determine whether elevated NF‐κB activation causes increased secretion of IL‐13 and IL‐1ra in MSCs, we used either TNFα stimulation or overexpression of IKK2 to activate NF‐κB signaling in AF‐MSCs. We validated the ability of the IKK2 expression plasmid to increase NF‐κB activation in AF‐MSCs using a luciferase assay (Fig. 4Ai) and qRT‐PCR for NF‐κB target gene expression (Fig. 4Aii). We then used an ELISA to determine whether NF‐κB activation by either TNFα stimulation or overexpression of IKK2 causes increased IL‐13 or IL‐1ra production. We found that increasing NF‐κB activation by either of these methods led to increased levels of IL‐13, but not IL‐1ra, production in AF‐MSCs (Fig. 4Bi, 4Bii).
Figure 4.
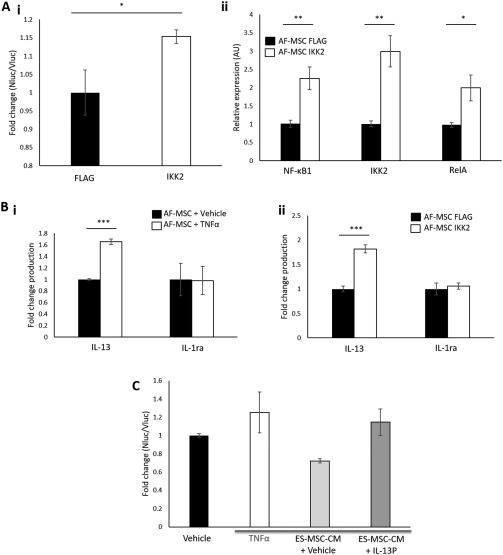
NF‐κB‐induced IL‐13 production contributes to the increased anti‐inflammatory potential of ES‐MSC‐CM compared with AF‐MSC‐CM. (A): Validation of IKK2 activation of NF‐κB by (Ai) luminometry and (Aii) qRT‐PCR. (B): ELISA to show that NF‐κB activation by (Bi) TNFα stimulation and (Bii) IKK2 overexpression causes increased IL‐13 but not IL‐1ra production. (C): Luciferase assay to show that IL‐13 blockade decreases the anti‐inflammatory potential of ES‐MSC‐CM. IKK: inhibitor of κB kinase. *, p < .05, **, p < .01; ***, p < .005. Abbreviations: AF‐MSC, amniotic fluid‐derived mesenchymal stem cell; CM, conditioned medium; ES‐MSC, ESC‐derived mesenchymal stem cell; TNFα, tumor necrosis factor α.
Finally, to determine whether NF‐κB‐induced IL‐13 production contributes to the increased anti‐inflammatory potential of ES‐MSCs compared with AF‐MSCs we blocked IL‐13 production in ES‐MSCs using an inhibitory peptide (IL‐13P). Treatment with the IL‐13P resulted in the decreased anti‐inflammatory potential of PSC‐MSC‐CM (Fig. 4C) thus indicating that, as predicted, NF‐κB‐stimulated IL‐13 production contributes to the increased anti‐inflammatory potential of ES‐MSCs compared with AF‐MSCs. A schematic for the proposed link between NF‐κB activation and IL‐13 production in PSC‐MSCs versus other types of MSCs is shown in the graphical abstract.
NF‐κB‐Stimulated IL‐13 Production Also Contributes to the Increased Anti‐Inflammatory Potential of iMSC‐CM Over AF‐MSC‐CM
We then wanted to determine whether the above findings were also true for the other type of PSC‐MSCs, iMSCs. We first reprogrammed amniotic fluid‐derived (AF)‐MSCs into iPSCs and confirmed the reactivation of the expression of pluripotency markers (Supporting Information Fig. S3A) and their expression of early lineage markers upon embryoid body differentiation (Supporting Information Fig. S3B). We then generated iMSCs using the same protocol used to derive ES‐MSCs and validated their MSC‐like morphology (Supporting Information Fig. S4Ai), cell surface marker expression profile (Supporting Information Fig. S1, S4Aii), and their loss of pluripotency marker expression (Supporting Information Fig. S4B).
Interestingly, similar to ES‐MSCs, iMSCs exhibited an increased proliferation rate in comparison to AF‐MSCs (Supporting Information Fig. S4C). Moreover, iMSC‐CM exhibited increased anti‐inflammatory potential (Fig. 5A), which was concomitant with increased NF‐κB activation (Fig. 5Bi–5Biii) and increased production of anti‐inflammatory cytokines (Fig. 5Ci–5Ciii). Finally, we demonstrated that increased IL‐13 production contributes to the increased anti‐inflammatory potential of iMSC‐CM over AF‐MSC‐CM by showing that an IL‐13 inhibitory peptide significantly reduced the ability of iMSC‐CM to reduce NF‐κB activation in reporter cells (Fig. 5D).
Figure 5.
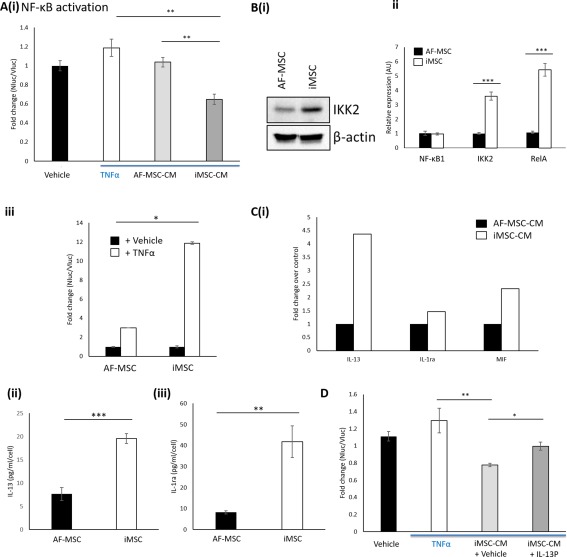
NF‐κB‐induced IL‐13 production contributes to the increased anti‐inflammatory potential of iMSC‐CM compared with AF‐MSC‐CM. (A): Anti‐inflammatory assay demonstrates that iMSC‐CM decreases NF‐κB activation in reporter cells to a higher degree than AF‐MSC‐CM. (Bi): Western blot to show IKK2 protein expression in AF‐MSCs and iMSCs. (Bii): qPCR to show increased NF‐κB activation in iMSCs compared with AF‐MSCs. (Biii): Luciferase assay to show NF‐κB activation in AF‐MSCs and iMSCs in response to TNFα. (Ci): Cytokine array to show levels of anti‐inflammatory cytokines in iMSC‐CM versus AF‐MSC‐CM. (Cii): ELISA to show levels of IL‐13 and (Ciii) IL‐1ra produced by iMSCs and AF‐MSCs. (D): Luciferase assay to show that IL‐13 blockade decreases the anti‐inflammatory potential of iMSC‐CM. *, p < .05; **, p <.01; ***, p < .005. Abbreviations: AF‐MSC, amniotic fluid‐derived mesenchymal stem cell; CM, conditioned medium; ES‐MSC, ESC‐derived mesenchymal stem cell; iMSCs, induced pluripotent stem cell‐derived MSCs; NF‐κB, nuclear factor‐kappa B; TNFα, tumor necrosis factor α.
Discussion
To our knowledge, this is the first direct comparison of the therapeutic potential of PSC‐MSCs and primary fetal MSCs. PSC‐MSCs have been shown to have superior immunomodulatory properties to adult MSCs 43, 53; however, we hypothesized that fetal MSCs may be comparable to PSC‐MSCs due to their more primitive origin. Here we show that, contrary to our hypothesis, PSC‐MSCs proliferate faster than AF‐MSCs and have greater anti‐inflammatory potential than both fetal BM‐MSCs and AF‐MSCs. These results suggest that PSC‐MSCs are the optimal cell source for therapeutic use, even over our current “gold standard,” fetal MSCs. The use of PSC‐MSCs would bypass the challenge of replicative senescence, since pluripotent cells can self‐renew indefinitely in culture, and using the same cell source for multiple therapies would allow a level of standardization that would not be possible with other sources of MSCs.
As mentioned, PSC‐MSCs have superior therapeutic capacity in a wide variety of disease contexts, yet we lack understanding of the molecular mechanisms underlying this phenomenon and of MSC therapeutic mechanisms more generally. Here we show that PSC‐MSCs exhibit increased NF‐κB activation in comparison to AF‐MSCs and that this occurs concomitantly with increased IL‐13 production and anti‐inflammatory capacity, providing for the first time a potential explanation for the increased therapeutic potential of PSC‐MSCs over other sources of MSCs.
Moreover, we demonstrate the first comparison of the two different PSC‐MSC types, iMSCs and ES‐MSCs. We and others 57 show that iMSCs exhibit decreased levels of MSC marker expression in comparison to ES‐MSCs and AF‐MSCs and that iMSCs are a more heterogenous population generally. We have also observed that ES‐MSCs have similar size and granularity to AF‐MSCs than iMSCs do and that ES‐MSCs release higher levels of the anti‐inflammatory cytokines IL‐10 and EGF and lower levels of the pro‐inflammatory cytokine IL‐6 in comparison to iMSCs (data not shown), suggesting that ES‐MSCs are the most appropriate for therapeutic use. Despite this, iMSCs present the advantages of being able to be used autologously, or to be patented, which are not possible in the case of ES‐MSCs.
Our study also shows that first trimester AF‐MSCs have neuroprotective potential following HI injury in the mouse brain. Our HI model mimics prenatal brain injury that can lead to lifelong cognitive disabilities such as cerebral palsy and epilepsy, and the ability of MSCs to prevent this. Our results fit with previous findings that demonstrate the ability of BM‐MSCs and multipotent adult progenitor cells (MAPCs) to exert neuroprotective effects after HI in rodent and larger animal models 58, 59, 60, 61, 62, 63. Moreover, we show for the first time that ES‐MSCs exhibit greater neuroprotective potential than AF‐MSCs in this context.
NF‐κB‐stimulated IL‐13 production may be one of many ways in which MSCs protect the brain after HI injury. For example, NF‐κB is also likely to stimulate the production of additional protective cytokines and small molecules such as IL‐10, PGE2, and IDO either directly or in combination with other factors; we have observed the increased production of IL‐10 by ES‐MSCs in comparison to AF‐MSCs (data not shown). In addition, we and others 42 show that ES‐MSCs express lower levels of IL‐6 in comparison to other MSC types, which may contribute to their increased therapeutic effects since IL‐6 generally acts as a pro‐inflammatory cytokine. We also identify IL‐1ra and MIF as cytokines that are produced at higher levels by PSC‐MSCs than AF‐MSCs. Zhang et al. 11 have previously shown that MIF (and growth/differentiation factor 15) production plays a role in the increased cardioprotective role of iMSC‐CM over BM‐MSC‐CM and IL‐1ra has been shown to be important for the therapeutic effects of MSCs in the lung 64. Lian et al. 44 also demonstrated increased production of stromal cell‐derived factor (SDF)1α, stem cell factor (SCF) and fibroblast growth factor (FGF)2 in iMSCs compared with the parental cell population. It is likely that these cytokines are context‐dependent and that cocktails of factors, rather than one individual factor, are important since the outcomes from early clinical trials with single cytokines for brain and cardiovascular diseases has failed to meet expectations 63, 64. One potential challenge is the lack of stability of growth factors, which could potentially be addressed by the use of extracellular vesicles 12, 13, 15.
Conclusion
By illuminating the molecular mechanisms underlying the improved therapeutic potential of PSC‐MSCs, and therefore of MSCs more generally, our work may have implications for improving the efficiency of MSC therapies. For example, NF‐κB signaling could be manipulated in MSCs using a lentiviral IKK2 overexpression plasmid to improve the therapeutic potential of the extracellular vesicles released by MSCs. The anti‐inflammatory properties of PSC‐MSCs are clearly superior not only to adult MSCs but also to fetal MSCs and it is, therefore, likely that they represent the most promising source for therapeutic use in the future, so long as their long‐term stability is proven and risks of teratoma formation are circumvented.
Author Contributions
K.E.H., P.G., H.H., P.D.C., and P.V.G.: conceptualization; K.E.H., M.H., H.H., and P.V.G.: methodology; K.E.H.: validation; K.E.H. and A.M.R.: formal analysis; K.E.H., M.C., K.D., A.H., A.M.R., F.V., K.‐L. H., and M.H.: investigation; K.E.H.: manuscript writing‐original draft; K.E.H., M.C., K.D., A.M.R., F.V., K.‐L.H., A.H., D.P., P.G., H.H., P.D.C., M.H., and P.V.G.; manuscript writing‐review and editing and acceptance of final manuscript; K.E.H.: visualization; P.V.G.: supervision; P.D.C., D.P., H.H., P.G., M.H., and P.V.G.: funding acquisition.
Disclosure of Potential Conflicts of Interest
D.P. declare consultancy role with Magnus Life Science. The other authors indicated no potential conflicts of interest.
Supporting information
Supporting Information Figure S1
Supporting Information Figure S2
Supporting Information Figure S3
Supporting Information Figure S4
Acknowledgments
We would like to acknowledge Dr. Sindhu Subramanaim for the provision of the cord blood cells and Dr. Lorna Fitzpatrick/Prof. Tristan McKay for the provision of the pCMV‐IKK2 and pCMV‐FLAG plasmids. This study was supported by grants from Great Ormond Street Children's Charity, Sparks Children Medical Research, the Newlife Foundation and by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
Contributor Information
Kate E. Hawkins, Email: k.hawkins@ucl.ac.uk
Pascale V. Guillot, Email: p.guillot@ucl.ac.uk.
References
- 1. Brady K, Dickinson SC, Guillot PV et al. Human fetal and adult bone marrow‐derived mesenchymal stem cells use different signaling pathways for the initiation of chondrogenesis. Stem Cells Dev 2014;23:541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guillot PV, Abass O, Bassett JH et al. Intrauterine transplantation of human fetal mesenchymal stem cells from first‐trimester blood repairs bone and reduces fractures in osteogenesis imperfecta mice. Blood 2008;111:1717–1725. [DOI] [PubMed] [Google Scholar]
- 3. Yang HM, Cho MR, Sung JH et al. The effect of human fetal liver‐derived mesenchymal stem cells on CD34+ hematopoietic stem cell repopulation in NOD/Shi‐scid/IL‐2Ra(null) mice. Transplant Proc 2011;43:2004–2008. [DOI] [PubMed] [Google Scholar]
- 4. Ranzoni AM, Corcelli M, Hau K‐L et al. Counteracting bone fragility with human amniotic mesenchymal stem cells. Sci Rep 2016;6:39656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. In 't Anker PS, Scherjon SA, Kleijburg‐van der Keur C et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells 2004;22:1338–1345. [DOI] [PubMed] [Google Scholar]
- 6. Guillot PV, Gotherstrom C, Chan J et al. Human first‐trimester fetal MSC express pluripotency markers and grow faster and have longer telomeres than adult MSC. Stem Cells 2007;25:646–654. [DOI] [PubMed] [Google Scholar]
- 7. Dominici M, Le Blanc K, Mueller I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8:315–317. [DOI] [PubMed] [Google Scholar]
- 8. Madrigal M, Rao KS, Riordan NH. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med 2014;12:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murry CE, Soonpaa MH, Reinecke H, et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004;428:664–668. [DOI] [PubMed] [Google Scholar]
- 10. Wang C, Cheng L, Xu H et al. Towards whole‐body imaging at the single cell level using ultra‐sensitive stem cell labeling with oligo‐arginine modified upconversion nanoparticles. Biomaterials 2012;33:4872–4881. [DOI] [PubMed] [Google Scholar]
- 11. Zhang Y, Liang X, Liao S et al. Potent paracrine effects of human induced pluripotent stem cell‐derived mesenchymal stem cells attenuate doxorubicin‐induced cardiomyopathy. Sci Rep 2015;5:11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gatti S, Bruno S, Deregibus MC et al. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia‐reperfusion‐induced acute and chronic kidney injury. Nephrol Dial Transplant 2011;26:1474–1483. [21324974] [DOI] [PubMed] [Google Scholar]
- 13. Ophelders DR, Wolfs TG, Jellema RK, et al. Mesenchymal stromal cell‐derived extracellular vesicles protect the fetal brain after hypoxia‐ischemia. Stem Cells Translational Medicine 2016;5:754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu Y, Wang Y, Zhao B, et al. Comparison of exosomes secreted by induced pluripotent stem cell‐derived mesenchymal stem cells and synovial membrane‐derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res Ther 2017;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang G, Zou X, Huang Y et al. Mesenchymal stromal cell‐derived extracellular vesicles protect against acute kidney injury through anti‐oxidation by enhancing Nrf2/ARE activation in rats. Kidney Blood Press Res 2016;41:119–128. [DOI] [PubMed] [Google Scholar]
- 16. Prockop DJ, Youn Oh J. Mesenchymal stem/stromal cells (MSCs): Role as guardians of inflammation. Mol Ther 2012;20:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drago D, Cossetti C, Iraci N et al. The stem cell secretome and its role in brain repair. Biochimie 2013;95:2271–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. English K. Mechanisms of mesenchymal stromal cell immunomodulation. Immunol Cell Biol 2013;91:19–26. [DOI] [PubMed] [Google Scholar]
- 19. Nemeth K, Leelahavanichkul A, Yuen PS et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)‐dependent reprogramming of host macrophages to increase their interleukin‐10 production. Nat Med 2009;15:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meisel R, Zibert A, Laryea M et al. Human bone marrow stromal cells inhibit allogeneic T‐cell responses by indoleamine 2,3‐dioxygenase‐mediated tryptophan degradation. Blood 2004;103:4619–4621. [DOI] [PubMed] [Google Scholar]
- 21. Rong LJ, Chi Y, Yang SG et al. [Effects of interferon‐gamma on biological characteristics and immunomodulatory property of human umbilical cord‐derived mesenchymal stem cells]. Zhongguo shi yan xue ye xue za zhi 2012;20:421–426. [PubMed] [Google Scholar]
- 22. Kang JW, Koo HC, Hwang SY et al. Immunomodulatory effects of human amniotic membrane‐derived mesenchymal stem cells. J Vet Sci 2012;13:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin W, Oh SK, Choo AB et al. Activated T cells modulate immunosuppression by embryonic‐and bone marrow‐derived mesenchymal stromal cells through a feedback mechanism. Cytotherapy 2012;14:274–284. [DOI] [PubMed] [Google Scholar]
- 24. Croitoru‐Lamoury J, Lamoury FMJ, Caristo M et al. Interferon‐gamma regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2,3 dioxygenase (IDO). PLoS One 2011;6:e14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ren G, Zhang L, Zhao X et al. Mesenchymal stem cell‐mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008;2:141–150. [DOI] [PubMed] [Google Scholar]
- 26. Ren G, Su J, Zhang L et al. Species variation in the mechanisms of mesenchymal stem cell‐mediated immunosuppression. Stem Cells 2009;27:1954–1962. [DOI] [PubMed] [Google Scholar]
- 27. Krampera M, Cosmi L, Angeli R et al. Role for interferon‐gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006;24:386–398. [DOI] [PubMed] [Google Scholar]
- 28. Dorronsoro A, Ferrin I, Salcedo JM et al. Human mesenchymal stromal cells modulate T‐cell responses through TNF‐alpha‐mediated activation of NF‐kappaB. Eur J Immunol 2014;44:480–488. [DOI] [PubMed] [Google Scholar]
- 29. Opitz CA, Litzenburger UM, Lutz C et al. Toll‐like receptor engagement enhances the immunosuppressive properties of human bone marrow‐derived mesenchymal stem cells by inducing indoleamine‐2,3‐dioxygenase‐1 via interferon‐beta and protein kinase R. Stem Cells 2009;27:909–919. [DOI] [PubMed] [Google Scholar]
- 30. Yagi H, Soto‐Gutierrez A, Parekkadan B et al. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant 2010;19:667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomchuck SL, Zwezdaryk KJ, Coffelt SB et al. Toll‐like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells 2008;26:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zaim M, Karaman S, Cetin G et al. Donor age and long‐term culture affect differentiation and proliferation of human bone marrow mesenchymal stem cells. Ann Hematol 2012;91:1175–1186. [DOI] [PubMed] [Google Scholar]
- 33. Kretlow JD, Jin YQ, Liu W et al. Donor age and cell passage affects differentiation potential of murine bone marrow‐derived stem cells. BMC Cell Biol 2008;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wagner W, Bork S, Horn P et al. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS one 2009;4:e5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moll G, Rasmusson‐Duprez I, von Bahr L et al. Are therapeutic human mesenchymal stromal cells compatible with human blood?. Stem Cells 2012;30:1565–1574. [DOI] [PubMed] [Google Scholar]
- 36. Kriete A, Mayo KL, Yalamanchili N et al. Cell autonomous expression of inflammatory genes in biologically aged fibroblasts associated with elevated NF‐kappaB activity. Immun Ageing 2008;5:5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Magalhães JP, Curado J, Church GM. Meta‐analysis of age‐related gene expression profiles identifies common signatures of aging. Bioinformatics 2009;25:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age‐associated diseases. J Gerontol Ser A Biol Sci Med Sci 2014;69:S4–S9. [DOI] [PubMed] [Google Scholar]
- 39. Boyd NL, Robbins KR, Dhara SK et al. Human embryonic stem cell‐derived mesoderm‐like epithelium transitions to mesenchymal progenitor cells. Tissue Eng Part A 2009;15:1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Peppo GM, Sjovall P, Lennerås M et al. Osteogenic potential of human mesenchymal stem cells and human embryonic stem cell‐derived mesodermal progenitors: A tissue engineering perspective. Tissue Eng Part A 2010;16:3413–3426. [DOI] [PubMed] [Google Scholar]
- 41. Vodyanik MA, Yu J, Zhang X et al. A mesoderm‐derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell 2010;7:718–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang X, Kimbrel EA, Ijichi K et al. Human ESC‐derived MSCs outperform bone marrow MSCs in the treatment of an EAE model of multiple sclerosis. Stem Cell Reports 2014;3:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Y, Liao S, Yang M, et al. Improved cell survival and paracrine capacity of human embryonic stem cell‐derived mesenchymal stem cells promote therapeutic potential for pulmonary arterial hypertension. Cell Transplant 2012;21:2225–2239. [DOI] [PubMed] [Google Scholar]
- 44. Lian Q, Zhang Y, Zhang J et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation 2010;121:1113–1123. [DOI] [PubMed] [Google Scholar]
- 45. Frobel J, Hemeda H, Lenz M et al. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Reports 2014;3:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fu X, Chen Y, Xie FN et al. Comparison of immunological characteristics of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Tissue Eng Part A 2015;21:616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hao Q, Zhu YG, Monsel A et al. Study of bone marrow and embryonic stem cell‐derived human mesenchymal stem cells for treatment of Escherichia coli endotoxin‐induced acute lung injury in mice. Stem Cells Translational Medicine 2015;4:832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yun YI, Park SY, Lee HJ et al. Comparison of the anti‐inflammatory effects of induced pluripotent stem cell‐derived and bone marrow‐derived mesenchymal stromal cells in a murine model of corneal injury. Cytotherapy 2017;19:28–35. [DOI] [PubMed] [Google Scholar]
- 49. Hawkins KE, Joy S, Delhove JMKM et al. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep 2016;14:1883–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen YS, Pelekanos RA, Ellis RL, et al. Small molecule mesengenic induction of human induced pluripotent stem cells to generate mesenchymal stem/stromal cells. Stem Cells Translational Medicine 2012;1:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hristova M, Rocha‐Ferreira E, Fontana X et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) reduces neonatal hypoxic‐ischaemic brain damage. J Neurochem 2016;136:981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rocha‐Ferreira E, Phillips E, Francesch‐Domenech E et al. The role of different strain backgrounds in bacterial endotoxin‐mediated sensitization to neonatal hypoxic‐ischemic brain damage. Neuroscience 2015;311:292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brown PT, Squire MW, Li WJ. Characterization and evaluation of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Cell Tissue Res 2014;358:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mellows B, Mitchell R, Antonioli M et al. Protein and molecular characterization of a clinically compliant amniotic fluid stem cell‐derived extracellular vesicle fraction capable of accelerating muscle regeneration through enhancement of angiogenesis. Stem Cells Dev 2017;26:1316–1333. [DOI] [PubMed] [Google Scholar]
- 55. Yagi H, Soto‐Gutierrez A, Navarro‐Alvarez N et al. Reactive bone marrow stromal cells attenuate systemic inflammation via sTNFR1. Mol Ther 2010;18:1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang B, Yin Y, Lai RC et al. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev 2014;23:1233–1244. [DOI] [PubMed] [Google Scholar]
- 57. Diederichs S, Tuan RS. Functional comparison of human‐induced pluripotent stem cell‐derived mesenchymal cells and bone marrow‐derived mesenchymal stromal cells from the same donor. Stem Cells Dev 2014;23:1594–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jellema RK, Wolfs TGAM, Lima Passos V et al. Mesenchymal stem cells induce T‐cell tolerance and protect the preterm brain after global hypoxia‐ischemia. PLoS one 2013;8:e73031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee JA, Kim BI, Jo CH et al. Mesenchymal stem‐cell transplantation for hypoxic‐ischemic brain injury in neonatal rat model. Pediatr Res 2010;67:42–46. [DOI] [PubMed] [Google Scholar]
- 60. Perasso L, Cogo CE, Giunti D et al. Systemic administration of mesenchymal stem cells increases neuron survival after global cerebral ischemia in vivo (2VO). Neural Plast 2010;2010:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. van Velthoven CT, van de Looij Y, Kavelaars A et al. Mesenchymal stem cells restore cortical rewiring after neonatal ischemia in mice. Ann Neurol 2012;71:785–796. [DOI] [PubMed] [Google Scholar]
- 62. Yasuhara T, Hara K, Maki M et al. Intravenous grafts recapitulate the neurorestoration afforded by intracerebrally delivered multipotent adult progenitor cells in neonatal hypoxic‐ischemic rats. J Cereb Blood Flow Metab 2008;28:1804–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yasuhara T, Matsukawa N, Yu G et al. Behavioral and histological characterization of intrahippocampal grafts of human bone marrow‐derived multipotent progenitor cells in neonatal rats with hypoxic‐ischemic injury. Cell Transplant 2006;15:231–238. [DOI] [PubMed] [Google Scholar]
- 64. Ortiz LA, Dutreil M, Fattman C et al. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci USA 2007;104:11002–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure S1
Supporting Information Figure S2
Supporting Information Figure S3
Supporting Information Figure S4