

Abstract
Purpose of review
Erythropoietin (EPO) is necessary and sufficient to trigger dynamic transcriptional patterns that drive the differentiation of erythroid precursor cells into mature, enucleated red cells. Because the molecular cloning and Food and Drug Administration approval for the therapeutic use of EPO over 30 years ago, a detailed understanding of how EPO works has advanced substantially. Yet, the precise epigenetic and transcriptional mechanisms by which EPO signaling controls erythroid expression patterns remains poorly understood. This review focuses on the current state of erythroid biology in regards to EPO signaling from human genetics and functional genomics perspectives.
Recent findings
The goal of this review is to provide an integrative view of the gene regulatory underpinnings for erythroid expression patterns that are dynamically shaped during erythroid differentiation. Here, we highlight vignettes connecting recent insights into a genome-wide association study linking an EPO mutation to anemia, a study linking EPO-signaling to signal transducer and activator of transcription 5 (STAT5) chromatin occupancy and enhancers, and studies that examine the molecular mechanisms driving topological chromatin organization in erythroid cells.
Summary
The genetic, epigenetic, and gene regulatory mechanisms underlying how hormone signal transduction influences erythroid gene expression remains only partly understood. A detailed understanding of these molecular pathways and how they intersect with one another will provide the basis for novel strategies to treat anemia and potentially other hematological diseases. As new regulators and signal transducers of EPO-signaling continue to emerge, new clinically relevant targets may be identified that improve the specificity and effectiveness of EPO therapy.
Keywords: epigenetics, erythropoiesis, erythropoietin, functional genomics, signal transducer and activator of transcription 5
INTRODUCTION
The human body is composed of several hundred terminally differentiated cell types. The cellular identity for each of the estimated 10–100 trillion cells in the human body is maintained through stable inheritance of gene expression patterns. The mechanisms underlying differentiation and cell identity have been intensively studied for several decades. Given its experimental tractability, the hematopoietic differentiation system has served as a paradigm for understanding epigenetic and gene regulatory determinants of cell fate. In particular, the production of red cells via erythropoiesis is one of the most well studied branches of the hematopoietic cell lineage.
Erythropoietin (EPO) is the primary hormone regulator that controls erythroid cell maturation, a process that is required for the daily replenishment of nearly 1%(200 billion)of the circulating red blood cells [1]. Terminally differentiating erythroblasts include, in sequence, proerythroblasts (ProEs), basophilic (BasoEs), polychromatophilic (PolyEs), and orthochromatic erythroblasts (OrthoEs) as defined by their morphological staining and immunophenotype characteristics [2]. ProEs depend on EPO for survival and continued differentiation [3]. As the kidneys produce most of the body’s EPO, exogenous EPO therapy effectively manages chronic anemia due to renal failure [4,5]. EPO binding to its cognate receptor activates the Janus Kinase 2 (JAK2), which activates the transcription factor signal transducer and activator of transcription 5 (STAT5) [6]. STAT5, together with other transcription factors TAL1, GATA1, and KLF1 (hereafter, the latter three referred to collectively as ‘master regulators’), bind enhancers to direct erythroid differentiation by orchestrating dynami cgene expression programs that culminate in the massive expression of globin genes and enucleation of red blood cells [7–10]. Enhancers are cis-regulatory elements dispersed throughout the genome that serve to regulate the transcription of genes, often in response to extracellular stimuli or developmental signals. Erythroid enhancers represent a critical link between EPO signal transduction and erythroid transcriptional patterns. Absence of any one of these proteins (EPO, JAK2, STAT5, TAL1, GATA1, or KLF1) in mice results in severe anemia and death by midgestation [11–17].
The goal of this review is to provide an integrative view of the gene regulatory underpinnings for erythroid expression patterns that are dynamically shaped during erythroid differentiation. In particular, this review highlights recent studies involving the interplay of erythroid transcription factor-binding patterns, epigenetics, and chromatin domains (Fig. 1).
FIGURE 1.
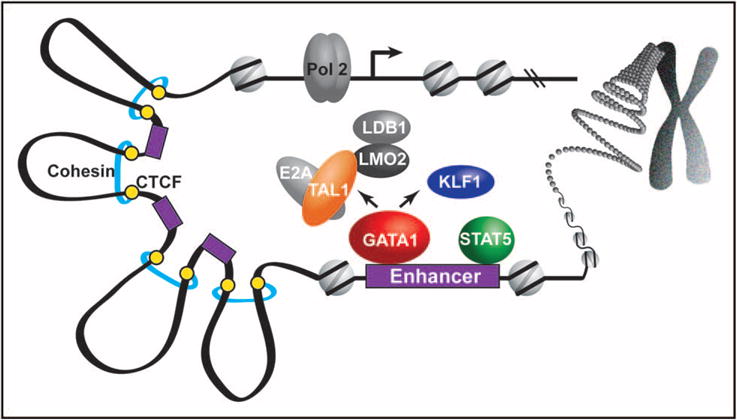
Integrative model illustrating how transcription factors, enhancers, and chromatin looping work together to recruit Pol II to promoters during erythropoiesis. The erythroid master regulator transcription factors (GATA1, KLF1, and TAL1) predominantly bind to enhancers (denoted by purple rectangles) in various assemblages in conjunction with accessory proteins, including E2A, LDB1, and LMO2. E2A is a member of the E protein family of helix-loop-helix transcription factors that forms a heterodimer with TAL1 to bind DNA. With LMO2 tethering LDB1 to erythroid transcription factors, LDB1 facilitates higher order complex assembly and chromatin looping interactions via LDB1 homodimerization [8]. EPO-EPOR-JAK2 signaling directly activates STAT5, which integrates with the master regulators by cooccupying several hundred GATA1 bound enhancers [34■■]. Transcription factors and enhancers interact within the confines of topologically associated chromatin domains that are structurally held together by CTCF and cohesin, as well as other regulatory and structural factors.
Model systems to study erythropoiesis and erythropoietin signaling
From fly to mouse to human, many excellent model cell systems have been developed to study erythropoiesis [2,18]. However, as most model cell systems are cultured in the presence of EPO, among other cytokines, it remains challenging to tease apart EPO-dependent mechanisms of action. Thus, to facilitate the study of EPO-dependent processes, a few model systems have been developed that synchronously proceed through erythropoiesis in response to EPO stimulation [19,20]. In addition, EPO-responsive murine J2E and human UT7 leukemogenic erythroid cell lines provide a nearly unlimited source of cells that are suitable for experiments requiring a large number of cells [21,22]. In some cases, brief serum starvation and EPO add back experiments have been used as a proxy for elucidating EPO-modulated processes [23,24].
Identifying etiological mutations in human genetic studies can also be the source for unique biological insights. For example, a recent report described a rare homozygous mutation in EPO resulting in a condition of severe anemia that was originally diagnosed as Diamond Blackfan Anemia [25■■]. However, genome-wide exome sequencing revealed the EPO R150Q mutation in this patient, suggesting that this case was distinct from Diamond Blackfan Anemia. Remarkably, although the EPO mutant bound to the EPO receptor (EPOR) with near wild-type affinity and activated STAT5, erythroid cell proliferation and differentiation were impaired. The authors proposed a model consistent with the data wherein the anemia-associated EPO mutation alters the kinetics of receptor binding, which results in biased downstream JAK2 signaling response. Importantly, this recent study underscores the value of genetic studies in identifying new and perhaps treatable forms of anemia. Indeed, reorienting ligand-receptor interactions has been proposed as a strategy for developing more potent and selective erythropoietic stimulating agents [22].
Erythropoietin stimulates signal transducer and activator of transcription 5 binding to several hundred genomic locations
In response to environmental stimuli, transcription factors interpret the cellular genome to alter transcriptional output. Erythroid expression patterns are highly dynamic and have been extensively studied by genome-wide expression profiling [1,26–28], providing numerous insights into the molecular pathways that control red blood cell development. Additionally, expression profiling of EPO starvation experiments in ProEs derived from primary fetal liver showed that EPO signaling modulates the expression of several hundred genes involved in cell survival signaling and cell identity [20,24]. Although, the trio of erythroid master regulators (GATA1, KLF1, and TAL1) has been extensively studied and their binding locations known in a number of model systems [8], less is understood regarding how EPO-dependent STAT5 binding throughout the genome is connected with temporal patterns of erythroid gene expression.
During erythropoiesis, EPO signaling primarily activates three signaling pathways: JAK-STAT, phosphatidylinositol-3-kinase, and mitogen-activated protein kinase [29]. In regards to the EPO-JAK-STAT axis, EPO has been suggested to serve as a rheostat, or dimmer switch, for STAT5 signaling in erythroid cells [30]. EPO-stimulated STAT5 activation is necessary and sufficient for erythropoiesis [31]. Yet, a key challenge remains in understanding how STAT5 signaling and chromatin binding are linked to changes in erythroid expression patterns. These data are critical to connecting the molecular dots between EPO stimulation and subsequent STAT5 activation to the erythroid epigenetics and transcription programs. Although STAT5 ChIP-seq data are available in a few cancer cells lines [32,33], a comprehensive set of STAT5-binding locations in an erythroid model have been lacking.
To address this gap in understanding, a recent study identified direct targets of EPO-activated STAT5 during erythropoiesis using complementary functional genomic approaches [34■■]. In a well designed study, a short timeframe enabled identification of direct STAT5-binding targets, whereas nascent transcription profiling captured rapid changes in gene expression in response to EPO stimulation. Within 30 min of EPO stimulation of murine J2E cells, STAT5 occupied over 300 genomic locations that were predominantly promoter distal enhancer regions. STAT5-binding locations that were co-occupied by GATA1 and/or KLF1 tended to be linked to erythroid-related genes, suggesting an integration of EPO-JAK-STAT signaling and the erythroid master regulators. Indeed, nearly half of STAT5-binding locations were co-occupied by GATA1 and/or KLF1. In contrast, housekeeping genes were primarily bound by STAT5 in the absence of GATA1 and KLF1. This raises the question of whether STAT5 and the master regulators (GATA1, KLF1, and TAL1) are each necessary and/or additive for proper expression and timing of erythroid genes. Given that GATA1 and KLF1 are known to assemble into LDB1-mediated complexes [8], how then does STAT5 biochemically fit into this regulatory picture? In summary, a number of important advances were made by this study in regards to how EPO works to promote erythropoiesis, including the identification of EPO-responsive genes, which is relevant to therapeutic treatment with EPO.
Erythropoietin stimulation reprograms the enhancer landscape during erythropoiesis
Transcription factors bind to enhancer regions to drive cell-type-specific gene expression patterns. Although enhancers relevant to erythroid cells have been identified, the mechanism by which EPO alters the dynamics of histone modifications has not been defined. Enhancer elements operate from promoter distal regions of the genome independent of gene orientation [35–37]. Collectively, transcription factors orchestrate cell-type-specific transcription programs by binding to their cognate sequence motifs within specific enhancers and recruiting coregulators and RNA polymerase II (Pol II) to promoters to initiate transcription [38–40]. Enhancer segments are typically co-occupied by combinations of transcription factors that influence gene expression programs, often integrating signals from multiple pathways.
Shaping the epigenome in a given cell type involves coordinate activities of transcription factors, together with nucleosome modifiers and remodelers [41,42]. In particular, enhancer elements are demarcated by histone 3 lysine 4 monomethylation (H3K4me1) and histone 3 lysine 27 acetylation (H3K27ac) [43–45]. This signature distinguishes enhancers from promoters, which are marked by histone 3 lysine 4 tri-methylation (H3K4me3).
Recent reports tracking histone modifications described the erythroid enhancer landscape in human and murine erythroid cells [46–48]. Interestingly, despite the dramatic transcriptional changes that accompany erythropoiesis, previous work found that broad features of chromatin states remain largely unchanged during GATA1-induced differentiation in the murine G1E-ER4 cell line [48]. This study suggested that erythroid enhancers are established in erythroid precursor cells, but precisely when this occurs remains unclear. Although the locations of erythroid enhancers have been determined in murine and human cell systems, how EPO influences the enhancer landscape is currently unknown.
A recent study investigated how EPO modulates the erythroid epigenome by performing epigenetic profiling using an ex-vivo murine cell system that undergoes synchronous erythroid maturation in response to EPO stimulation [49■■]. In this study, the authors identified a repertoire of EPO-modulated enhancers, illuminating a new facet of EPO signaling. EPO stimulation altered the histone mark signatures across several thousand enhancer locations, revealing a cis-regulatory network of EPO-responsive enhancers. In contrast, most of the other identified enhancers remained in an active acetylated state during EPO signaling, suggesting that most erythroid enhancers are established at an earlier precursor stage. Together, these findings defined a cis-regulatory enhancer network for EPO signaling during erythropoiesis, provided a framework for future studies involving the interplay of epigenetics and EPO signaling, and highlighted an underappreciated role for EPO in reprogramming the epigenome.
Erythroid enhancers operate in the context of topologically associated chromatin domains
Enhancers and promoters operate in the context of distinct chromatin neighborhoods. Transcription factors exert their influence on gene expression patterns via enhancer binding and facilitate longrange chromatin contacts between enhancer and promoter regions. These enhancer–promoter interactions are identified at various resolutions, from two specific genomic loci to whole genome. Chromosome conformation capture (3C) technologies identify in-vivo chromatin contacts using a proximity DNA ligation assay [50]. The seminal 3C technique served as the basis for the development of a more diverse tool kit of chromatin contact assays, such as 4C, 5C, Hi-C, ChIA-PET, among others, which have been extensively reviewed [51–54]. Specifically, Hi-C was used to characterize genome-wide topologically associated domains (TADs) in mouse and human that tends to be flanked by the insulator-binding protein CCCTC-Binding factor (CTCF) [55].
In hematology, distant chromatin looping interactions have long been subject of intense study with the experimental paradigm of the β-globin locus and locus control region. Two recent reports applied 3C-based technologies to erythroid cells to understand the molecular mechanisms involved in α-globin gene expression and CTCF-dependent chromatin contacts [56■■,57■■]. Hanssen et al. [56■■] examined whether chromatin topology and transcription of the α-globin locus was dependent on specific CTCF-binding sites. To examine the dynamics and specificity of the α-globin sub-TAD (i.e., sub-TADs typically fall between 40kb and 3Mb), the capture-C assay was applied to murine erythroid and nonerythroid stem cells. Interestingly, although the α-globin locus is flanked by constitutively bound CTCF sites, these sites contact one another specifically in erythroid cells during erythropoiesis. Next, to dissect the regulatory underpinnings of this erythroid tissue-specific sub-TAD, elegant mutational studies were conducted in mice that specifically eliminated convergent CTCF-binding sites flanking the α-globin locus, singlyorinvarious combinations. Disruption of a specific combination of CTFC sites resulted in loss of CTCF binding, as expected, but more importantly altered interactions between the regions of chromatin flanking the α-globin locus. In summary, the authors proposed that the CTCF-bound chromatin positions surrounding the α-globin TAD serve to restrict and guide local enhancer interactions to α-globin promoters.
It is becomingly increasingly clear that in addition to CTCF and cohesin, other factors, such as bromodomain and extraterminal motif protein 2 (BRD2), are critical for establishing and maintaining long-range chromatin contacts [57■■]. Although BRD2 is necessary for the expression of some GATA1-activated genes during erythropoiesis, BRD2 does not colocalize with GATA1 [58], suggesting BRD2 and GATA1 may promote erythroid maturation through distinct mechanisms. To investigate this further, a recent report by Hsu et al. [57■■] examined the role the BRD family of proteins played in establishing transcriptional and architectural chromatin boundaries in erythroid cells. Global protein–DNA interaction profiling showed that BRD2 and CTCF tend to colocalize throughout the genome, and that BRD2 occupancy depends upon CTCF, whereas CTCF occupancy was independent of BRD2. Genome editing followed by the Hi-C assay showed that disrupting CTCF/BRD2-occupied elements impaired the architectural boundaries established by CTCF/BRD2 and resulted in promiscuous enhancer–promoter contacts and inappropriate transcription of nearby genes. Taken together, the authors propose a model wherein CTCF recruits BRD2 as a necessary cofactor to maintain the integrity of transcriptional and architectural boundaries.
CONCLUSION
The glycoprotein hormone EPO stimulates erythropoiesis wherein erythroid precursor cells proceed through an exceedingly complex network of molecular signaling, epigenetic modifications, and transcriptional dynamics that ultimately produce enucleated red cells. Integrating and connecting the molecular dots between EPO signaling and the spatiotemporal transcriptional patterns continues to be a challenging task and many questions remain.
Determining the EPO-induced STAT5-binding locations in the J2E murine erythroid cell line represents an important first step in linking EPO to control of erythroid gene expression [34■■]. However, the extent to which STAT5-binding patterns are conserved from mouse to human remains unclear. Given that altered chromatin occupancy of master regulators has been proposed to underlie the evolutionary divergence in erythroid transcription patterns [27,59], it will be interesting to examine how STAT5 binding-patterns differ among other mouse and human model systems. Although STAT5 co-occupied several hundred locations in mouse J2E cells with GATA1 and KLF1, STAT5 was not enriched at the promoters of the master regulator genes. In contrast, data from an Encyclopedia of DNA Elements study showed that STAT5 was enriched at the GATA1 and TAL1 promoters in the human erythroleukemic K562 cell line [33], which raises the intriguing question of whether EPO-stimulated STAT5 activity regulates the temporal expression of some erythroid master regulator genes in a more physiologically relevant model system for erythropoiesis.
Although transcription factors frequently bind to promoters, their predominant genome-wide distributions reside within distal enhancer regions. In this review, we highlighted a recent report that linked EPO signaling to changes in the enhancer landscape [49■■]. However, more work is necessary to establish functional significance for the several thousand candidate enhancer regions that display EPO responsiveness. Still, establishing a causal link between enhancer activation or repression with a corresponding gene activity is a challenging task, particularly on a genome-wide scale. Further confounding this pairwise enhancer–promoter association is the observation in previous studies that multiple enhancers can together fine tune the expression of a single gene [60,61]. Thus, additional studies are required to establish a regulatory link between enhancer regions and target genes. For example, in a recent study, Hewitt et al. [62■] elegantly dissected the mechanisms underlying the functional relationship between a GATA2-regulated enhancer and a gene involved in red cell regeneration and survival. Although Clustered Regularly Interspaced Short Palindromic Repeats has accelerated efforts to functionally validate enhancers, linking enhancers to their regulatory target genes remains a daunting task. To circumvent this limitation, machine learning methods and computational modeling of cis-regulatory networks can support efforts to understand the rules governing how enhancers associate with target genes and subsequently better focus experimental design.
Given that enhancers frequently skip over the nearest gene to loop to a more distant gene [63], 3C-based assays are necessary to link enhancers to target genes that they potentially regulate. Long-range chromatin interaction assays performed on a genome-wide scale highlighted in this review [56■■,57■■] extend previous locus-specific looping studies in erythroid cells and complement enhancer profiling studies. It is becomingly increasingly clear that in addition to CTCF and cohesin, other factors, such as BRD2 and LDB1, are critical for establishing and maintaining long-range chromatin contacts [64]. For example, a recent study suggested that the Ying Yang 1 transcription factor is a structural regulator of enhancer–promoter loops in a similar manner to CTCF-cohesion-mediated looping [65]. Last, the extent to which EPO signaling influences chromatin looping contacts remains an open question.
Pursuing these avenues of research in an integrative fashion will yield important insights into erythropoiesis and anemia, and more broadly further our understanding of how hormone signaling pathways control gene expression patterns in the context of differentiation systems. As new regulators and signal transducers of EPO-signaling continue to emerge [29], new clinically relevant targets should be identified that improve the specificity and effectiveness of EPO therapy.
KEY POINTS.
An integrative view of transcription factors, epigenetics, and chromatin interactions is critical to understanding the interplay of these areas in erythroid gene regulation.
Key erythroid transcription factors (TAL1, GATA1, KLF1, and STAT5) bind enhancers to direct erythroid differentiation by orchestrating dynamic, cell-type-specific gene expression programs.
EPO stimulation modulates the enhancer repertoire of erythroid precursor cells.
Long-range chromatin contacts during erythropoiesis provide a framework for understanding the intersection of EPO signaling, transcription factor binding, and enhancer dynamics.
Acknowledgments
We would like to thank the hematology community for helpful and inspiring discussions related to this review.
Financial support and sponsorship
This work was supported by the Vanderbilt Molecular Endocrinology Training Program grant 5T32 DK07563.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Kingsley PD, Greenfest-Allen E, Frame JM, et al. Ontogeny of erythroid gene expression. Blood. 2013;121:e5–e13. doi: 10.1182/blood-2012-04-422394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nandakumar SK, Ulirsch JC, Sankaran VG. Advances in understanding erythropoiesis: evolving perspectives. Br J Haematol. 2016;173:206–218. doi: 10.1111/bjh.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koury MJ. Tracking erythroid progenitor cells in times of need and times of plenty. Exp Hematol. 2016;44:653–663. doi: 10.1016/j.exphem.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Koury MJ, Haase VH. Anaemia in kidney disease: harnessing hypoxia responses for therapy. Nat Rev Nephrol. 2015;11:394–410. doi: 10.1038/nrneph.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med. 2015;21:221–230. doi: 10.1038/nm.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. Erythropoiesis: from molecular pathways to system properties. Adv Exp Med Biol. 2014;844:37–58. doi: 10.1007/978-1-4939-2095-2_3. [DOI] [PubMed] [Google Scholar]
- 7.An X, Schulz VP, Mohandas N, Gallagher PG. Human and murine erythropoiesis. Curr Opin Hematol. 2015;22:206–211. doi: 10.1097/MOH.0000000000000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Love PE, Warzecha C, Li L. Ldb1 complexes: the new master regulators of erythroid gene transcription. Trends Genet. 2014;30:1–9. doi: 10.1016/j.tig.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tallack MR, Perkins AC. Three fingers on the switch: Kruppel-like factor 1 regulation of gamma-globin to beta-globin gene switching. Curr Opin Hematol. 2013;20:193–200. doi: 10.1097/MOH.0b013e32835f59ba. [DOI] [PubMed] [Google Scholar]
- 10.Gnanapragasam MN, Bieker JJ. Orchestration of late events in erythropoiesis by KLF1/EKLF. Curr Opin Hematol. 2017;24:183–190. doi: 10.1097/MOH.0000000000000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pevny L, Simon MC, Robertson E, et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature. 1991;349:257–260. doi: 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 12.Nuez B, Michalovich D, Bygrave A, et al. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 13.Robb L, Lyons I, Li R, et al. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci USA. 1995;92:7075–7079. doi: 10.1073/pnas.92.15.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porcher C, Swat W, Rockwell K, et al. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 15.Neubauer H, Cumano A, Muller M, et al. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell. 1998;93:397–409. doi: 10.1016/s0092-8674(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 16.Socolovsky M, Fallon AE, Wang S, et al. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 17.Socolovsky M, Nam H, Fleming MD, et al. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 18.Parker MP, Peterson KR. Mouse models of erythropoiesis and associated diseases. Methods Mol Biol. 2018;1698:37–65. doi: 10.1007/978-1-4939-7428-3_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawyer ST, Koury MJ, Bondurant MC. Large-scale procurement of erythropoietin-responsive erythroid cells: assay for biological activity of erythropoietin. Methods Enzymol. 1987;147:340–352. doi: 10.1016/0076-6879(87)47123-1. [DOI] [PubMed] [Google Scholar]
- 20.Zhang D, Johnson MM, Miller CP, et al. An optimized system for studies of EPO-dependent murine pro-erythroblast development. Exp Hematol. 2001;29:1278–1288. doi: 10.1016/s0301-472x(01)00725-1. [DOI] [PubMed] [Google Scholar]
- 21.Klinken SP, Nicola NA, Johnson GR. In vitro-derived leukemic erythroid cell lines induced by a raf- and myc-containing retrovirus differentiate in response to erythropoietin. Proc Natl Acad Sci USA. 1988;85:8506–8510. doi: 10.1073/pnas.85.22.8506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moraga I, Wernig G, Wilmes S, et al. Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands. Cell. 2015;160:1196–1208. doi: 10.1016/j.cell.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregory T, Yu C, Ma A, et al. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood. 1999;94:87–96. [PubMed] [Google Scholar]
- 24.Singh S, Dev A, Verma R, et al. Defining an EPOR- regulated transcriptome for primary progenitors, including Tnfr-sf13c as a novel mediator of EPO-dependent erythroblast formation. PLoS One. 2012;7:e38530. doi: 10.1371/journal.pone.0038530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■25.Kim AR, Ulirsch JC, Wilmes S, et al. Functional selectivity in cytokine signaling revealed through a pathogenic EPO mutation. Cell. 2017;168:1053–1064. doi: 10.1016/j.cell.2017.02.026. An elegant and thorough genetic study on a pathogenic EPO mutantion that is highlighed in this review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.An X, Schulz VP, Li J, et al. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014;123:3466–3477. doi: 10.1182/blood-2014-01-548305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pishesha N, Thiru P, Shi J, et al. Transcriptional divergence and conservation of human and mouse erythropoiesis. Proc Natl Acad Sci USA. 2014;111:4103–4108. doi: 10.1073/pnas.1401598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L, Lin YH, Sierant MC, et al. Developmental transcriptome analysis of human erythropoiesis. Hum Mol Genet. 2014;23:4528–4542. doi: 10.1093/hmg/ddu167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhrt D, Wojchowski DM. Emerging EPO and EPO receptor regulators and signal transducers. Blood. 2015;125:3536–3541. doi: 10.1182/blood-2014-11-575357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Porpiglia E, Hidalgo D, Koulnis M, et al. Stat5 signaling specifies basal versus stress erythropoietic responses through distinct binary and graded dynamic modalities. PLoS Biol. 2012;10:e1001383. doi: 10.1371/journal.pbio.1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grebien F, Kerenyi MA, Kovacic B, et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood. 2008;111:4511–4522. doi: 10.1182/blood-2007-07-102848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin HY, Willi M, HyunYoo K, et al. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat Genet. 2016;48:904–911. doi: 10.1038/ng.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Zhuang J, Iyer S, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■ 34.Gillinder KR, Tuckey H, Bell CC, et al. Direct targets of pSTAT5 signalling in erythropoiesis. PLoS One. 2017;12:e0180922. doi: 10.1371/journal.pone.0180922. It is highlighted in this review and the first study describing the EPO-dependent genomic targets for STAT5 during erythropoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartman CR, Blobel GA. Perturbing chromatin structure to understand mechanisms of gene expression. Cold Spring Harb Symp Quant Biol. 2015;80:207–212. doi: 10.1101/sqb.2015.80.027359. [DOI] [PubMed] [Google Scholar]
- 37.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 38.Venters BJ, Pugh BF. How eukaryotic genes are transcribed. Crit Rev Biochem Mol Biol. 2009;44:117–141. doi: 10.1080/10409230902858785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15:703–708. doi: 10.1038/nrm3890. [DOI] [PubMed] [Google Scholar]
- 43.Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heintzman ND, Stuart RK, Hon G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 45.Visel A, Blow MJ, Li Z, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, Liu X, Li D, et al. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev Cell. 2016;36:9–23. doi: 10.1016/j.devcel.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su MY, Steiner LA, Bogardus H, et al. Identification of biologically relevant enhancers in human erythroid cells. J Biol Chem. 2013;288:8433–8444. doi: 10.1074/jbc.M112.413260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu W, Cheng Y, Keller CA, et al. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. Genome Res. 2011;21:1659–1671. doi: 10.1101/gr.125088.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■49.Perreault AA, Benton ML, Koury MJ, et al. Epo reprograms the epigenome of erythroid cells. Exp Hematol. 2017;51:47–62. doi: 10.1016/j.exphem.2017.03.004. An epigenetics study highlighted in this review that identified several thousand enhancers that rapidly responded to EPO stimulation, revealing the plasticity of a large number of enhancers during erythropoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 51.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simonis M, Klous P, Splinter E, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 54.Fullwood MJ, Ruan Y. ChIP-based methods for the identification of long-range chromatin interactions. J Cell Biochem. 2009;107:30–39. doi: 10.1002/jcb.22116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■56.Hanssen LLP, Kassouf MT, Oudelaar AM, et al. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat Cell Biol. 2017;19:952–961. doi: 10.1038/ncb3573. A capture-C study of α-globin locus highlighted in this review that showed the requirement of CTCF for proper chromatin contacts and expression of genes nearby the α-globin locus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■■57.Hsu SC, Gilgenast TG, Bartman CR, et al. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol Cell. 2017;66:102–116. doi: 10.1016/j.molcel.2017.02.027. A genome-wide Hi-C study highlighted in this review that demonstrated the importance of BRD2 colocalization with CTCF in the formation of chromatin architecture during erythropoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stonestrom AJ, Hsu HC, Jahn KS, et al. Functions of BET proteins in erythroid gene expression. Blood. 2015;125:2825–2834. doi: 10.1182/blood-2014-10-607309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ulirsch JC, Lacy JN, An X, et al. Altered chromatin occupancy of master regulators underlies evolutionary divergence in the transcriptional landscape of erythroid differentiation. PLoS Genet. 2014;10:e1004890. doi: 10.1371/journal.pgen.1004890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guerrero L, Marco-Ferreres R, Serrano AL, et al. Secondary enhancers synergise with primary enhancers to guarantee fine-tuned muscle gene expression. Dev Biol. 2010;337:16–28. doi: 10.1016/j.ydbio.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 61.Hong JW, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ■62.Hewitt KJ, Katsumura KR, Matson DR, et al. GATA factor-regulated Samd14 enhancer confers red blood cell regeneration and survival in severe anemia. Dev Cell. 2017;42:213–225. doi: 10.1016/j.devcel.2017.07.009. An excellent structure function study of a GATA-regulated enhancer that controls SAMD14 gene expression during erythropoeisis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deng W, Lee J, Wang H, et al. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weintraub AS, Li CH, Zamudio AV, et al. YY1 is a structural regulator of enhancer-promoter loops. Cell. 2017;171:1573–1588. e1528. doi: 10.1016/j.cell.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]