

Abstract
“Candidatus Fokinia solitaria” is an obligate intracellular endosymbiont of a unicellular eukaryote, a ciliate of the genus Paramecium. Here, we present the genome sequence of this bacterium and subsequent analysis. Phylogenomic analysis confirmed the previously reported positioning of the symbiont within the “Candidatus Midichloriaceae” family (order Rickettsiales), as well as its high sequence divergence from other members of the family, indicative of fast sequence evolution. Consistently with this high evolutionary rate, a comparative genomic analysis revealed that the genome of this symbiont is the smallest of the Rickettsiales to date. The reduced genome does not present flagellar genes, nor the pathway for the biosynthesis of lipopolysaccharides (present in all the other so far sequenced members of the family “Candidatus Midichloriaceae”) or genes for the Krebs cycle (present, although not always complete, in Rickettsiales). These results indicate an evolutionary trend toward a stronger dependence on the host, in comparison with other members of the family. Two alternative scenarios are compatible with our results; “Candidatus Fokinia solitaria” could be either a recently evolved, vertically transmitted mutualist, or a parasite with a high host-specificity.
Keywords: genome reduction, bacterial symbiont, Paramecium, Midichloriaceae, blobology
Introduction
Bacterial endosymbionts that live within eukaryotic cells establish highly diverse relationships with their hosts, ranging from mutualism to parasitism. Furthermore, obligate intracellular endosymbionts, which have become host-dependent, tend to experience reductive genome evolution (McCutcheon and Moran 2011). Such phenomenon is observed in bacteria with very different behaviors, from the infectious pathogen Chlamydia (Nunes and Gomes 2014) to the noninfectious, vertically transmitted mutualist Buchnera (Shigenobu et al. 2000). In the case of mutualist bacteria, especially for those that provide metabolites necessary for the hosts’ survival, genomes can undergo extreme reduction, pushing toward the limit of becoming an organelle—for example, “Candidatus Tremblaya princeps” genome is ∼139 kb, and the mtDNA of Reclinomonas americana is ∼69 kb (Sagan 1967; Schwartz and Dayhoff 1978; McCutcheon and Moran 2007, 2011). On the other side, genomic shrinkage can occur also in infectious parasites or pathogens, and, in this case, it can be considered a signature of host specificity (Moran 2002; Dagan et al. 2006; Bäumler and Fang 2013).
The order Rickettsiales encompasses intracellular bacteria that infect highly diverse hosts. Rickettsiales, as most intracellular bacteria, possess small genomes, however they do not show extreme reduction. Their genomes are mainly in the range of 1–1.5 Mb (Darby et al. 2007; Gillespie et al. 2012). Most Rickettsiales show the capability to transfer horizontally, either as a part of their life cycle (Vaughan et al. 2012; Dumler and Walker 2015; Schulz et al. 2016; Senra et al. 2016), or in their recent evolutionary history (Epis et al. 2008; Gillespie et al. 2012; Castelli et al. 2016). Some Rickettsiales are pathogenic to humans or other vertebrates (Dumler and Walker 2015; Thomas et al. 2016), while others have a strong interdependence with their host (Comandatore et al. 2013; Nikoh et al. 2014). However, often the relationships between Rickettsiales and their hosts are not easily classified or are yet to be explained.
“Candidatus (Ca.) Midichloriaceae” (hereafter Midichloriaceae) is the most recently described family of the order Rickettsiales (according to the definition by Szokoli et al. 2016a). Its members are widespread and live in association with diverse hosts, including amoebae, ticks, corals, flagellates, fish, and mammals (Montagna et al. 2013; Senra et al. 2016; Szokoli et al. 2016b). However, only three genomes, derived from two species, of the family are available on databases (Sassera et al. 2011; Wang and Wu 2014a; Schulz et al. 2016). “Ca. Fokinia solitaria” (hereafter, F. solitaria), the first representative of a novel genus within the family, was recently detected in the ciliate Paramecium sp. collected from a wastewater treatment plant in Rio de Janeiro, Brazil (Szokoli et al. 2016b). Ciliates are unicellular eukaryotes that frequently harbor endosymbiotic bacteria (Fokin 2004; Schweikert et al. 2013), including several Rickettsiales (Castelli et al. 2016). Endosymbionts of ciliates may entertain a wide range of relationships with their hosts, including necessary mutualists (Vannini et al. 2012) and parasites (Kaltz and Koella 2003). Nevertheless, in most cases the effect on host fitness has not been yet elucidated, although in some conditions an apparent benefit for the host was observed (Soldo and Godoy 1973; Kusch et al. 2002; Bella et al. 2016). To gain insights on F. solitaria mechanisms of interaction with the host and on the evolutionary and ecological patterns of Midichloriaceae, we sequenced the complete genome of this organism. Here, we present a set of genomic analyses to compare the novel sequence with other members of the family Midichloriaceae and the order Rickettsiales.
Materials and Methods
Total DNA was extracted from a culture of Paramecium sp. strain Rio ETE_ALG 3VII using a modified CTAB procedure (Gustincich, et al. 1991). Prior to DNA extraction, cells were starved for 2 days and filtered through eight layers of sterile gauze before cell-harvesting via gentle centrifugation (Szokoli et al. 2016b). After DNA quality check, sequencing was performed using Illumina HiSeq 2500, to generate 14,783,394 150-nt paired-end reads. Reads were assembled using SPAdes v3.9.0 (Bankevich et al. 2012; Nurk et al. 2013), and the resulting preliminary assembly was subjected to the blobology pipeline (Kumar et al. 2013), to select the putative F. solitaria reads based on contig coverage, GC%, and taxonomy. The contigs belonging to the 100–1,000× coverage, estimated using Bowtie2 (Langmead and Salzberg 2012), and >30 GC% on the blobology plot were thus selected (supplementary fig. S1, Supplementary Material online). In parallel, contigs outside this range were scanned to detect potential signatures suggesting they could belong to the F. solitaria genome, performing a BLAST search using a database of Rickettsiales genomes, and manually inspecting contigs showing significant hits (e-value <10−5). In addition, the extracted DNA was subjected to Pacific Biosciences Sequel sequencing, obtaining 12,243,617 reads. All PacBio reads and the selected Illumina reads were used to perform a second assembly step using the software Unicycler (Wick et al. 2017). The softwares Bandage (Wick et al. 2015) and BLAST were then used to manually perform bioinformatic joins on the result of this second assembly. The obtained genome was annotated using Prokka (Seemann, 2014) followed by extensive manual curation.
A data set of 13 Rickettsiales and 4 outgroup organisms was constructed (supplementary table S7, Supplementary Material online), selecting all other available Midichloriaceae genomes (n = 3), a similar number of Anaplasmataceae (n = 5) and Rickettsiaceae (n = 4), more closely related (n = 2) and more distant (n = 2) outgroups. The data set was used to infer the phylogenetic position of F. solitaria through two different approaches. In the phylogenomic approach, we identified conserved orthologs using OrthoMCL (Li et al. 2003) (inflation value: 1.1; %identity cutoff: 30). Subsequently, the orthologous protein sequences were aligned using Muscle (Edgar 2004), polished with Gblocks (Talavera and Castresana 2007) and used to build a phylogenomic tree using the software RAxML (Stamatakis 2015), with 100 bootstraps (LG + I+G + F model inferred by using ProtTest 3.4.2; Darriba et al. 2011, sorting models according to AIC and BIC). In the conserved genes data set approach, protein sequences of 24 highly conserved genes, previously selected to give a stable phylogenetic signal were utilized (Lang et al. 2013), aligned with Muscle and used to build a phylogenomic tree using RAxML, with 100 bootstraps (LG + I+G + F model inferred by using ProtTest 3.4.2, sorting models according to AIC and BIC).
COGs of the available Midichloriaceae genomes were annotated using the NCBI COG pipeline (Galperin et al. 2015). The identified COGs were compared and graphically reported in a Venn diagram using the R software (R Core Team 2015).
Metabolic pathway analysis was performed using the BioCyc and Pathway Tools suites (Karp et al. 2015; Caspi et al. 2016). Presence of insertion sequences, prophages, secretion systems, and secreted proteins was predicted using, respectively, ISsaga (Varani et al. 2011), PHAST (Zhou et al. 2011), TXSScan (MacSyFinder-based; Abby et al. 2014), SignalP (Emanuelsson et al. 2007), and TMHMM (Krogh et al. 2001) (with default parameters) and comparing the obtained results with the manually curated annotation.
Results and Discussion
The complete genome of F. solitaria (fig. 1) is contained in a single circular chromosome. With a size of 837,348 bp, this is the smallest reported genome in the order Rickettsiales to date (January 2018), slightly shorter than Neorickettsia sennetsu (859,006 bp) (Dunning Hotopp et al. 2006). Other partial genomes, shorter than the genome of F. solitaria, are published, but such sequences were estimated by the authors to be incomplete (Martijn et al. 2015; Tully et al. 2016). The main genomic characteristics of F. solitaria, including GC content and average gene length, are encompassed within the range of Rickettsiales diversity. The genome contains just two insertion sequences and a paucity of phagic sequences, again characteristics similar to many other Rickettsiales genomes (table 1 and supplementary table S1, Supplementary Material online). On the other hand, although F. solitaria presents the smallest genome in the Rickettsiales order, it does not show features reported for highly reduced genomes, such as very high coding density (often >92%) or overlapping genes (Mira et al. 2001; McCutcheon and Moran 2011). Therefore, F. solitaria should be considered at a similar stage of genome reduction as the other Rickettsiales, and not at the more extreme stage of reduction found in other endosymbiont lineages.
Fig. 1.
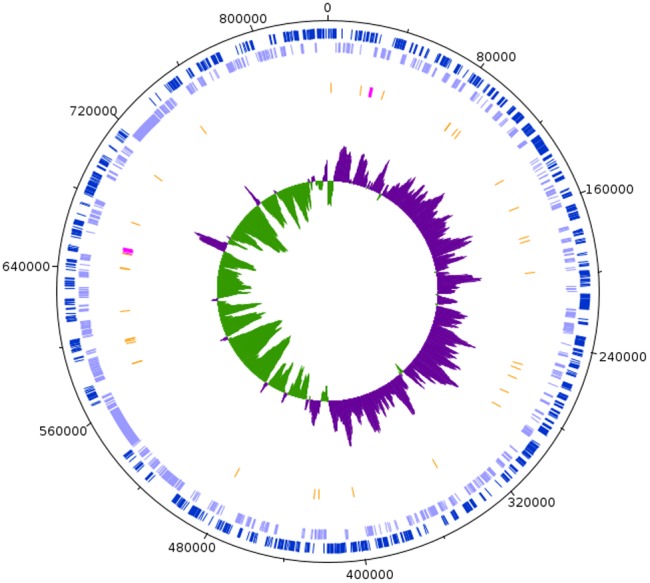
—Graphical representation of the genome of Fokinia solitaria. Circles, from the outermost to the innermost show, respectively, coding sequences (CDSs) on the plus strand in light blue; CDSs on the minus strand in green; tRNAs in orange and rRNAs in fuchsia; the GC skew, positive in green, negative in purple. Position 1 was arbitrarily set as the start of the dnaA gene.
Table 1.
Genomic Characteristics of Selected Members of the Order Rickettsiales
| Organism | GC % | Genome Size (Mb) | Predicted IS Number | Coding Percentage | Family |
|---|---|---|---|---|---|
| Fokinia solitaria | 35.8 | 0.83 | 2 | 88.54 | Midichloriaceae |
| Neorickettsia sennetsu | 41.1 | 0.86 | 1 | 85.20 | Anaplasmataceae |
| Neorickettsia risticii | 41.3 | 0.88 | 1 | 85.08 | Anaplasmataceae |
| Neorickettsia helminthoeca | 41.7 | 0.88 | 1 | 87.49 | Anaplasmataceae |
| Wolbachia wOv | 32.1 | 0.96 | 7 | 72.42 | Anaplasmataceae |
| Rickettsia prowazekii | 29 | 1.11 | 6 | 77.12 | Rickettsiaceae |
| Rickettsia typhi | 28.9 | 1.11 | 0 | 75.95 | Rickettsiaceae |
| Ehrlichia chaffeensis | 30.1 | 1.18 | 0 | 77.84 | Anaplasmataceae |
| Midichloria mitochondrii | 36.6 | 1.18 | 109 | 79.17 | Midichloriaceae |
| Wolbachia wMel | 35.2 | 1.24 | 138 | 87.41 | Anaplasmataceae |
| Rickettsia rickettsii | 32.5 | 1.27 | 7 | 82.86 | Rickettsiaceae |
| Wolbachia wNo | 34 | 1.30 | 136 | 88.31 | Anaplasmataceae |
| Rickettsia australis | 32.3 | 1.30 | 34 | 83.40 | Rickettsiaceae |
| Anaplasma marginale | 49.8 | 1.47 | 0 | 70.15 | Anaplasmataceae |
| Anaplasma phagocytophilum | 41.6 | 1.50 | 0 | 69.62 | Anaplasmataceae |
| Rickettsia bellii | 31.6 | 1.52 | 47 | 85.93 | Rickettsiaceae |
| Jidaibacter acanthamoeba UWC8 | 34.8 | 1.62 | 19 | 89.21 | Midichloriaceae |
| Rickettsia endos. Ixodes scapularis | 33.3 | 1.82 | 547 | 90.56 | Rickettsiaceae |
| Orientia tsutsugamushi | 30.4 | 2.13 | 537 | 79.15 | Rickettsiaceae |
| Jidaibacter acanthamoeba UWC36 | 33.7 | 2.37 | 0 | 80.31 | Midichloriaceae |
Note.—Organisms ordered according to the increasing size of the genome. Fokinia solitaria is reported in bold. Minimum and maximum values of each column are underlined.
Phylogenomic analyses were performed from a data set of 13 Rickettsiales genomes and 4 outgroup genomes (2 Holosporales, 1 Rhodospirillales, and 1 Caulobacterales). Two sets of genes were retrieved, one encompassing 75 orthologs present in all taxa, another including 24 highly conserved proteins (Lang et al. 2013). The two resulting trees present identical topologies (fig. 2 and supplementary fig. S1, Supplementary Material online). They are in agreement with previous 16S rRNA gene phylogenies (Montagna et al. 2013; Senra et al. 2016; Szokoli et al. 2016b) and to recent phylogenomic studies (Driscoll et al. 2013; Wang and Wu 2014b, 2015) in placing the Midichloriaceae as sister group of the Anaplasmataceae, and in positioning F. solitaria within the Midichloriaceae as early diverging respect to the Midichloria and Jidaibacter clade. The branch leading to F. solitaria is long in both trees, again consistent with the previously published 16S rRNA gene trees. This result provides additional evidence to strengthen the hypothesis that F. solitaria could be subjected to an accelerated rate of molecular evolution in relation to other Rickettsiales (Szokoli et al. 2016b). It is tempting to speculate a correlation between the hypothesized fast evolving nature of F. solitaria and its reduced genome size, possibly indicating an instance of the general trend of fast evolution and high genome reduction of bacterial symbionts, caused by genetic drift.
Fig. 2.

—Phylogenomic tree showing the relationships between selected members of the order Rickettsiales, and four outgroup genomes. Tree was obtained from an alignment of 75 orthologous proteins analyzed with RAxML with 100 bootstraps. Bootstrap values are shown above each node. Scale bar stands for estimated sequence divergence.
An ortholog search to compare the genome of F. solitaria with that of the three other available Midichloriaceae genomes revealed that F. solitaria possesses 32 COGs that are not present in other genomes of the family (Venn diagram representation in fig. 3). Some of these COGs are related to functions that are putatively connected with host interaction, but no clear complete pathway or structure was found (see supplementary table S2, Supplementary Material online). A total of 435 COGs are shared by all the considered Midichloriaceae (supplementary table S3, Supplementary Material online), while 208 orthologs are conserved in the three other available Midichloriaceae genomes, but absent in F. solitaria (supplementary table S4, Supplementary Material online).
Fig. 3.
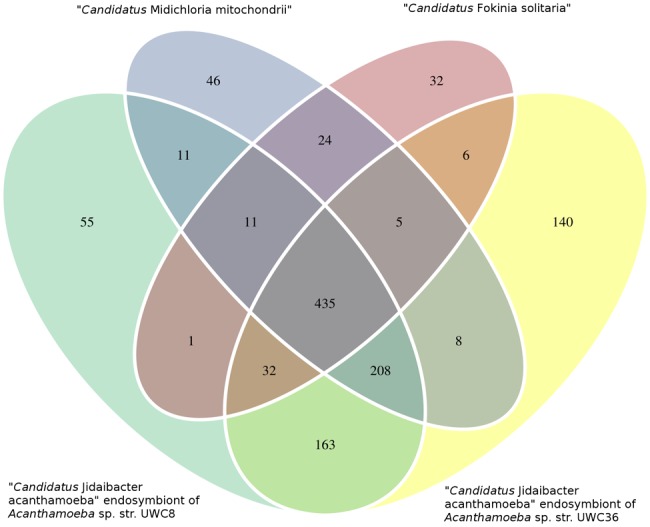
—Venn diagram representation of the distribution of Clusters of Orthologous Groups (COGs) in the four available genomes of Midichloriaceae bacteria. Intersections indicate the numbers of COGs shared by two or more genomes.
A detailed inspection of the ortholog comparison, and of the F. solitaria genome repertoire, indicates that its predicted metabolic capabilities are a reduced version of other Midichloriaceae, consistent with the small genome size. From the energy metabolism point of view, the main difference is the complete absence of the Krebs cycle. This is, to our knowledge, a unique case among all Rickettsiales, even though other members of the order possess an incomplete Krebs cycle (Min et al. 2008). Nevertheless, some capability to perform oxidative phosphorylation is retained (NADH: quinone dehydrogenase, cytochrome bd ubiquinol oxidase, and ATP synthase complexes are present, but cytochrome c reductase and oxidase complexes are not), likely exploiting NADH derived from glycolysis. Genes for biosynthetic pathways are also scarce: just two amino acids (alanine and glycine), some cofactors (biotin, folate, ubiquinone, Fe-S clusters, and, partially, coenzyme A), and no nucleotides can be produced. Additionally, only partial methylerythrol phosphate pathway for isoprenoid biosynthesis is present, while the pentose phosphate pathway is likely absent (only one gene of the entire pathway appears to be present). The presence of two ATP/ADP translocases probably enables F. solitaria to scavenge energy and nucleotides directly from the host. Other Midichloriaceae are overall richer in such pathways, especially in TCA cycle, cytochrome c oxido-reductases, nonoxidative pentose phosphate pathway, amino acids (five more) (Sassera et al. 2011; Wang and Wu 2014a; Schulz et al. 2016). For what concerns cell structures, the genome sequence indicates that F. solitaria is able to synthesize membrane phospholipids and cell wall peptidoglycan. The capability of producing acetyl-CoA, thanks to the pyruvate dehydrogenase complex, is probably useful only for these biosynthetic pathways (since the Krebs cycle is missing), a situation possibly comparable to other endosymbionts of Paramecium (Dohra et al. 2014). On the other hand, lipopolysaccharide (LPS) biosynthesis appears to be fully missing, differently from other Midichloriaceae, but similarly to all members of the family Anaplasmataceae, where this trait is supposed to confer a specific advantage, probably helping to evade host defense mechanisms (Lin and Rikihisa 2007). This condition is, on the other hand, also found in mutualists such as Buchnera (Shigenobu et al. 2000), and could suggest a convergence in pathway loss in strongly reduced genomes.
A rich set of membrane transporters appears to be present in the genome of F. solitaria (such as ATP-binding cassette and major facilitator superfamily), and could complement metabolic deficiencies enabling direct uptake of small molecules from the host. The presence of multiple secretion systems (supplementary table S5, Supplementary Material online) and putative secreted proteins (supplementary table S6, Supplementary Material online), including an ankyrin repeat protein, likely enables F. solitaria to establish and regulate interactions with the host (Al-Khodor et al. 2010). Surprisingly, many of these annotated proteins are typically intracellular. On the other hand, among rickettsial species, there is increasing evidence for the surface localization of proteins that typically function in the bacterial cytoplasm or periplasm, suggesting “moonlighting” functions on cell surfaces of divergent rickettsial species (Gillespie et al. 2015).
Genes coding for flagellar proteins were not detected in the genome. While this result is in accordance with the electron microscopy observations of F. solitaria, it represents a difference with the other members of the family Midichloriaceae. All the three other available genomes, and even the partial genome of the symbiont of Trichoplax adhaerens (Driscoll et al. 2013) code for flagellar genes, even though to our knowledge no direct detection of flagellar structures has been reported for these organisms. Moreover, genes that are typically involved in host cell invasion in several bacteria, such as pili or adhesins, are not present in the F. solitaria genome, a feature consistent with the available genome reports of the other Midichloriaceae.
In summary, the small genome of F. solitaria shows the absence of important genes in comparison with the other Midichloriaceae (i.e., Krebs cycle, cytochrome c reductase and oxidase, pentose posphate pathway, LPS, flagella, synthesis of five amino acids), and the phylogenomic analyses confirm a fast-evolving genome. These results clearly indicate a stronger dependence on the host compared with other so far sequenced Midichloriaceae. It must be noted that some members of the two other Rickettsiales families also display reduction of metabolic capability in a variable and lineage-specific fashion, for example, Rickettsia spp. and Orientia tsutsugamushi are extremely scarce in biosynthetic pathways (Driscoll et al. 2017). In comparison to other metabolically depleted Rickettsiales genomes, we can observe in F. solitaria a different pathway specificity, which might be possibly related to differences in host organisms and interaction. However, the effect of F. solitaria on the host and the dynamics of transmission are unclear. The relatively higher genome reduction could be compatible with a recently evolved, vertically transmitted mutualist or, by contrast, with a parasite with some degree of host specificity. Some genomic traits, such as the absence of LPS biosynthesis and flagella, may support both hypotheses, while others are in favor of either of the two interpretations. For example, synthesized cofactors could provide a beneficial effect to the host Paramecium. By contrast, the previously observed autophagosomal lysis of F. solitaria by its host (Szokoli et al. 2016b) may indicate a parasitic behavior of the bacterium, which is supported by the presence of genes coding for ATP/ADP translocases. Further comparative and experimental analyses may help to elucidate the behavior of F. solitaria in Paramecium.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was in part supported by PRIN 2012 to C.B. and G.P., by European Commission FP7-PEOPLE-2009-IRSES project CINAR PATHOBACTER (project number 247658) to G.P., and by HFSP Grant RGY0075-2017 to D.S. The authors thank Margherita Bersani, Alessandra Cafiso, Franziska Szokoli, Francesco Comandatore, and Stefano Gaiarsa for technical help.
Literature Cited
- Abby SS, Néron B, Ménager H, Touchon M, Rocha EPC.. 2014. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9(10):e110726.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodor S, Price CT, Kalia A, Kwaik YA.. 2010. Ankyrin-repeat containing proteins of microbes: a conserved structure with functional diversity. Trends Microbiol. 18(3):132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, et al. , 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumler A, Fang FC.. 2013. Host specificity of bacterial pathogens. Cold Spring Harb Perspect Med. 3(12):a010041.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bella C, et al. , 2016. Fitness impact of obligate intranuclear bacterial symbionts depends on host growth phase. Front Microbiol. 7:2084.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, et al. , 2016. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44(D1):D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli M, Sassera D, Petroni G.. 2016. Biodiversity of ‘non-model’ Rickettsiales and their association with aquatic organisms In: Thomas S, editor. Rickettsiales. Cham, Switzerland: Springer International Publishing. p. 59–91. [Google Scholar]
- Comandatore F, et al. , 2013. Phylogenomics and analysis of shared genes suggest a single transition to mutualism in Wolbachia of nematodes. Genome Biol Evol. 5(9):1668–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan T, Blekhman R, Graur D.. 2006. The ‘domino theory’ of gene death: gradual and mass gene extinction events in three lineages of obligate symbiotic bacterial pathogens. Mol Biol Evol. 23(2):310–316. [DOI] [PubMed] [Google Scholar]
- Darby AC, Cho N-H, Fuxelius H-H, Westberg J, Andersson SGE.. 2007. Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23(10):511–520. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27(8):1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohra H, Tanaka K, Suzuki T, Fujishima M, Suzuki H.. 2014. Draft genome sequences of three Holospora species (Holospora obtusa, Holospora undulata, and Holospora elegans), endonuclear symbiotic bacteria of the ciliate Paramecium caudatum. FEMS Microbiol Lett. 359(1):16–18. [DOI] [PubMed] [Google Scholar]
- Driscoll T, Gillespie JJ, Nordberg EK, Azad AF, Sobral BW.. 2013. Bacterial DNA sifted from the Trichoplax adhaerens (Animalia: Placozoa) genome project reveals a putative rickettsial endosymbiont. Genome Biol Evol. 5(4):621–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll TP, et al. , 2017. Wholly Rickettsia! Reconstructed metabolic profile of the quintessential bacterial parasite of eukaryotic cells. mBio 8:e00859-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler JS, Walker DH. 2015. Rickettsiales. In: Whitman et al., editors. Bergey’s manual of systematics of archaea and bacteria. Bergey’s Manual Trust. [Google Scholar]
- Dunning Hotopp JC, et al. , 2006. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2(2):e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H.. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2(4):953–971. [DOI] [PubMed] [Google Scholar]
- Epis S, et al. , 2008. Midichloria mitochondrii is widespread in hard ticks (Ixodidae) and resides in the mitochondria of phylogenetically diverse species. Parasitology 135(4):485–494. [DOI] [PubMed] [Google Scholar]
- Fokin SI. 2004. Bacterial endocytobionts of ciliophora and their interactions with the host cell. Int Rev Cytol. 236:181–249. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Makarova KS, Wolf YI, Koonin EV.. 2015. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43(D1):D261–D269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie JJ, Nordberg EK, Sobral BWS, Azad AF. 2012. Phylogeny and comparative genomics: the shifting landscape in the genomics era. In: Palmer GH, Azad AF, editors. Intracellular pathogens II: Rickettsiales. Washington (DC): American Society of Microbiology Press. p. 84–141. [Google Scholar]
- Gillespie JJ, et al. , 2015. Secretome of obligate intracellular Rickettsia. FEMS Microbiol Rev. 39(1):47–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustincich S, Manfioletti G, Del Sal G, Schneider C, Carninci P.. 1991. A fast method for high-quality genomic DNA extraction from whole human blood. BioTechniques 11(3):298–302. [PubMed] [Google Scholar]
- Kaltz O, Koella JC.. 2003. Host growth conditions regulate the plasticity of horizontal and vertical transmission in Holospora undulata, a bacterial parasite of the protozoan Paramecium caudatum. Evolution 57(7):1535–1542. [DOI] [PubMed] [Google Scholar]
- Karp PD, et al. , 2015. Computational metabolomics operations at BioCyc.org. Metabolites 5(2):291–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, Von Heijne G, Sonnhammer EL.. 2001. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 305(3):567–580. [DOI] [PubMed] [Google Scholar]
- Kumar S, Jones M, Koutsovoulos G, Clarke M, Blaxter M.. 2013. Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots. Front Genet. 4:237.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch J, et al. , 2002. Competitive advantages of Caedibacter-infected Paramecia. Protist 153(1):47–58. [DOI] [PubMed] [Google Scholar]
- Lang JM, Darling AE, Eisen JA.. 2013. Phylogeny of bacterial and archaeal genomes using conserved genes: supertrees and supermatrices. PLoS One 8(4):e62510.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS.. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13(9):2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Rikihisa Y.. 2007. Degradation of p22phox and inhibition of superoxide generation by Ehrlichia chaffeensis in human monocytes. Cell Microbiol. 9(4):861–874. [DOI] [PubMed] [Google Scholar]
- Martijn J, et al. , 2015. Single-cell genomics of a rare environmental alphaproteobacterium provides unique insights into Rickettsiaceae evolution. ISME J. 9(11):2373–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA.. 2011. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 10(1):13–26. [DOI] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA.. 2007. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc Natl Acad Sci U S A. 104(49):19392–19397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min CK, et al. , 2008. Genome-based construction of the metabolic pathways of Orientia tsutsugamushi and comparative analysis within the Rickettsiales Order. Comp Funct Genomics 2008:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA.. 2001. Deletional bias and the evolution of bacterial genomes. Trends Genet. 17(10):589–596. [DOI] [PubMed] [Google Scholar]
- Montagna M, et al. , 2013. ‘Candidatus Midichloriaceae’ fam. nov. (Rickettsiales), an ecologically widespread clade of intracellular alphaproteobacteria. Appl Environ Microbiol. 79(10):3241–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA. 2002. Microbial minimalism. Cell 108(5):583–586. [DOI] [PubMed] [Google Scholar]
- Nikoh N, et al. , 2014. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc Natl Acad Sci U S A. 111(28):10257–10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes A, Gomes JP.. 2014. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect Genet Evol. 23:49–64. [DOI] [PubMed] [Google Scholar]
- Nurk S, et al. , 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 20(10):714–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team 2015. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Sagan L. 1967. On the origin of mitosing cells. J Theor Biol. 14(3):225–IN6. [DOI] [PubMed] [Google Scholar]
- Sassera D, et al. , 2011. Phylogenomic evidence for the presence of a flagellum and cbb(3) oxidase in the free-living mitochondrial ancestor. Mol Biol Evol. 28(12):3285–3296. [DOI] [PubMed] [Google Scholar]
- Schulz F, et al. , 2016. A Rickettsiales symbiont of amoebae with ancient features. Environ Microbiol. 18(8):2326–2342. [DOI] [PubMed] [Google Scholar]
- Schwartz RM, Dayhoff MO.. 1978. Origins of prokaryotes, eukaryotes, mitochondria, and chloroplasts. Science 199(4327):395–403. [DOI] [PubMed] [Google Scholar]
- Schweikert M, Fujishima M, Görtz H-D. 2013. Symbiotic associations between ciliates and prokaryotes. In: Rosemberg E, et al., editors. The prokaryotes. Berlin, Germany: Springer-Verlag, p. 427–463. [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. [DOI] [PubMed] [Google Scholar]
- Senra MVX, et al. , 2016. A house for two–double bacterial infection in Euplotes woodruffi Sq1 (Ciliophora, Euplotia) sampled in Southeastern Brazil. Microb Ecol. 71(2):505–517. [DOI] [PubMed] [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H.. 2000. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature 407(6800):81–86. [DOI] [PubMed] [Google Scholar]
- Soldo AT, Godoy GA.. 1973. Molecular complexity of Paramecium symbiont lambda deoxyribonucleic acid: evidence for the presence of a multicopy genome. J Mol Biol. 73(1):93–108. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2015. Using RAxML to infer phylogenies. Curr Protoc Bioinformatics 51:6.14.1–14. [DOI] [PubMed] [Google Scholar]
- Szokoli F, Castelli M et al. , 2016. a. Disentangling the taxonomy of Rickettsiales and description of two novel symbionts (“Candidatus Bealeia paramacronuclearis” and “Candidatus Fokinia cryptica”) sharing the cytoplasm of the ciliate protist Paramecium biaurelia. Appl Environ Microbiol. 82(24):7236–7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szokoli F, Sabaneyeva E et al. , 2016. b. ‘ Candidatus Fokinia solitaria’, a novel ‘stand-alone’ symbiotic lineage of Midichloriaceae (Rickettsiales). PLoS One 11(1):e0145743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Castresana J.. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56(4):564–577. [DOI] [PubMed] [Google Scholar]
- Thomas S, Alexander W, Gilligan FJ, Rikihisa Y. 2016. The Importance of Rickettsiales Infections. In: Thomas S, editor. Rickettsiales. Cham, Switzerland: Springer International Publishing. p. 3–21. [Google Scholar]
- Tully BJ, Sachdeva R, Graham ED, Heidelberg JF.. 2016. 290 metagenome-assembled genomes from the Mediterranean Sea: a resource for marine microbiology. Peer J. 5:e3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini C, Ferrantini F, Ristori A, Verni F, Petroni G.. 2012. Betaproteobacterial symbionts of the ciliate Euplotes: origin and tangled evolutionary path of an obligate microbial association. Environ Microbiol. 14(9):2553–2563. [DOI] [PubMed] [Google Scholar]
- Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M.. 2011. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12(3):R30.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan JA, Tkach VV, Greiman SE.. 2012. Neorickettsial endosymbionts of the digenea: diversity, transmission and distribution. Adv Parasitol. 79:253–297. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu M.. 2015. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci Rep. 5(7949). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wu M.. 2014. a. Complete genome sequence of the endosymbiont of Acanthamoeba Strain UWC8, an amoeba endosymbiont belonging to the ‘Candidatus Midichloriaceae’ family in Rickettsiales. Genome Announc. 2(4):e00791-14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wu M.. 2014. b. Phylogenomic reconstruction indicates mitochondrial ancestor was an energy parasite. PLoS One 9(10):e110685.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Schultz MB, Zobel J, Holt KE.. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31(20):3350–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, Gorrie CL, Holt KE.. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 13(6):e1005595.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS.. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39(Web Server issue):W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.