

Summary
RNA-editing mechanisms, which induce nucleotide substitution in the RNA, increase transcript and protein diversities. Editing dysregulation has been shown to lead to grave outcomes, and transcriptome-wide aberrant RNA editing has been found in tumors. However, little is known about the involvement of editing in other diseases. Systemic lupus erythematosus (SLE) is a multisystemic autoimmune disease characterized by a loss of tolerance for autoantigens from various tissues and the production of multiple autoantibodies. Here, we show that blood samples from individuals with SLE have abnormally high levels of RNA editing, some of which affect proteins and potentially generate novel autoantigens. We suggest that elevated RNA editing, either by ADARs or APOBECs, may be involved in the pathophysiology of SLE, as well as in other autoimmune diseases, by generating or increasing the autoantigen load, a key requisite for the progression of autoimmunity.
Keywords: systemic lupus erythematosus, SLE, RNA editing, autoimmune disease, immune tolerance, ADAR, APOBEC3, neo-autoantigens, A-to-I editing, C-to-U editing
Graphical Abstract
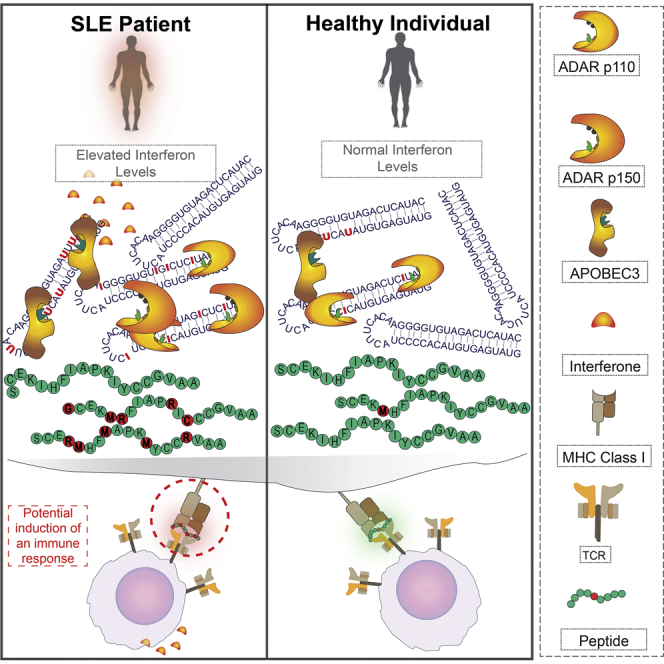
Highlights
-
•
SLE patients have elevated RNA editing in their blood
-
•
In SLE, elevated editing at coding regions can potentially generate autoantigens
-
•
Elevated RNA editing may promote SLE progression by increasing autoantigen load
Roth et al. show that SLE patients have elevated RNA editing, a process that modifies an RNA sequence from the sequence encoded in the genome. This is manifested as both increased numbers and various altered coding sequences, which may promote autoimmune progression by increasing autoantigenic load.
Introduction
Adenosine-to-inosine (A-to-I) editing is a prevalent form of post-transcriptional modification (Bazak et al., 2014a, Ramaswami and Li, 2014), catalyzed by the adenosine deaminase acting on RNA (ADAR) enzymes (Bass, 2002, Nishikura, 2010, Savva et al., 2012). Because inosine is interpreted by most cellular machineries as a guanosine (G), there are several possible outcomes to this deamination (Speyer et al., 1962). Hence, editing can alter splice consensus elements, microRNA seeds, and binding sites, as well as protein coding sequences (Burns et al., 1997, Ekdahl et al., 2012, Higuchi et al., 2000, Rueter et al., 1999). The reaction is catalyzed in humans by the ADAR1 (ADAR) and ADAR2 (ADARB1) enzymes. ADAR2 is confined to the nucleus, whereas ADAR1 has two isoforms—the p110 isoform that is localized in the nucleus and the p150 interferon (IFN)-inducible isoform that shuttles between the nucleus and the cytoplasm (Eckmann et al., 2001, George and Samuel, 1999).
A-to-I RNA editing is a constitutive and finely tuned process that is involved with several systems in the body and requires a delicate balance of activity. On the one hand, insufficient editing has detrimental and even lethal effects, partly due to the accumulation of endogenous double-stranded RNAs (dsRNAs), which triggers an innate immune response (Liddicoat et al., 2015a, Mannion et al., 2014, Pestal et al., 2015). ADAR1 editing deficiency is involved in autoimmune disorders such as Aicardi-Goutières syndrome (AGS) and dyschromatosis symmetrica hereditaria (DSH) (Liu et al., 2006, Rice et al., 2012). Both show some phenotypic similarities to systemic lupus erythematosus (SLE), and AGS patients, like SLE patients, are characterized by elevated type I IFN production (Crow and Manel, 2015, Lee-Kirsch et al., 2014, Rice et al., 2012).
On the other hand, excessive ADAR activity may be harmful. Studies show that elevated levels of ADAR1 usually correlate with higher editing activity, manifested both as higher rates of editing and as editing of non-typical editing sites (Han et al., 2015, Paz-Yaacov et al., 2015). Elevated levels of editing have been shown to have significant effects on the transcriptomic diversity of cancers and, in several cases, even correlate with patient survival (Han et al., 2015, Paz-Yaacov et al., 2015).
The other type of RNA editing in humans is the deamination of cytidine to uridine (C-to-U) by several members of the APOBEC protein family of cytidine deaminases, mainly APOBEC1 and APOBEC3A (Sharma et al., 2015, Smith et al., 2012). Similar to ADAR1, APOBEC3 paralogs are upregulated by IFN (Peng et al., 2006). Overexpression of APOBEC3A was shown to affect thousands of sites that have the potential to recode more than a thousand proteins (Sharma et al., 2017).
SLE is a multisystemic heterogenic autoimmune disease. It is characterized by autoantibodies to a variety of autoantigens derived from various tissues, as well as antinuclear antibodies (Lipsky, 2001, Tsokos, 2011). The causes and pathogenesis of the disease are not fully understood. SLE patients usually have elevated levels of circulating type I IFN and increased expression of both type I and type II IFN-stimulated genes (ISGs) (Chiche et al., 2014, Crow and Manel, 2015, Hagberg and Rönnblom, 2015). Thus, inhibitors of IFN and downstream ISGs are attractive and promising therapeutic targets for treatment (Furie et al., 2017, Hagberg and Rönnblom, 2015, Kirou and Gkrouzman, 2013, Niewold, 2008). Moreover, high artificially induced levels of IFN can cause SLE-like symptoms (Ho et al., 2008, Niewold, 2008). Altered levels of ISGs and RNA modifications have been marked as potential contributors to SLE pathogenesis in several previous studies (Graham and Utz, 2005, Hueber et al., 2004). This includes increased rates of alternative splicing (Ng et al., 2004) and indications of aberrant RNA editing in a few genes, as well as elevated expression of ADAR1 in lymphocytes (Laxminarayana et al., 2002, Laxminarayana et al., 2007, Orlowski et al., 2008). However, these early studies focused only on small subset of editing targets.
Here, by analyzing wide-scale, high-throughput sequencing data (Hung et al., 2015), we show that SLE patients have elevated global RNA-editing levels. In addition, we use computational predictions to demonstrate that this excessive editing can potentially generate autoantigens, which may then be presented on major histocompatibility complex (MHC) molecules and induce an immune response. Editing in coding regions, which alters the amino acid sequence (recoding), may prove immunogenic. Because proteasomal degradation is presumably unaffected by RNA editing, an edited form of a protein could be degraded and eventually be presented by the MHC system. We previously showed that medullary thymic epithelial cells (mTECs), which mediate the presentation of self-epitopes in the thymus, routinely express many edited forms of proteins and thereby forestall the induction of a dangerous immune response against edited peptide-derived autoantigens (Danan-Gotthold et al., 2016). However, possibly due to incomplete presentation and/or confinement of the process to selected edited versions, not all edited forms are presented by the mTECs. As a consequence, T cells specific for edited epitopes may escape negative selection and react to cells that present an editing-derived neo-autoantigen (Danan-Gotthold et al., 2016). Although the main activity of editing is to inhibit the innate immune response to endogenous dsRNA (Liddicoat et al., 2015b, Mannion et al., 2014, Pestal et al., 2015), our results suggest that RNA editing can trigger an immune response directed against editing-originated neo-autoantigens, exacerbating autoimmunity. We therefore suggest it is involved in the etiology and pathogenesis of SLE and possibly other autoimmune diseases.
Results
Enhanced A-to-I Editing in SLE Patients
Many studies of editing in various diseases concentrated on previously detected editing sites. Here, we used RNA sequencing (RNA-seq) data to evaluate changes in global levels of editing, without prior knowledge of specific sites. To compare the RNA-editing levels in SLE patients with those in healthy individuals, we first used two complementary approaches to estimate the global editing rate using RNA-seq samples from Hung et al. (2015) (99 from SLE patients and 18 from healthy individuals) (Table S1): the Alu editing index (AEI), which measures the global rate of editing in Alu repeats (Bazak et al., 2014b), and the global rates of clustered editing sites in each sample, according to the normalized number of hyper-edited (HE) sites (see Experimental Procedures) (Porath et al., 2014). Both types of analysis revealed that global editing was significantly elevated in the blood of SLE patients compared to that of controls (Wilcoxon p value = 4.96e−6 and 8.27e−7 for AEI and HE analyses, respectively). Because higher editing levels may be associated with higher levels of ISGs, we grouped the patients according to the original dataset of interferon signature metric (ISM) division, which measures the presence of the IFN-inducible genes’ expression signature. As expected, ISM-high SLE patients had significantly higher editing levels than controls (Wilcoxon p value = 4.16e−7 and 2.48e−8 for AEI and HE analyses, respectively) (Figures 1A and 1B) and significantly higher editing levels than ISM-low patients (Wilcoxon p value = 1.87e−2 and 1.77e−5 for AEI and HE analyses, respectively) (Figures 1A and 1B). However, even ISM-low patients had significantly higher editing levels than controls (Wilcoxon p value = 3.28e−2 and 3.93e−2 for AEI and HE analyses, respectively) (Figures 1A and 1B). Overall, the editing signal was clean. A-to-G mismatches comprised most mismatches (82.79%), and the ADAR deamination motif (5′ neighbor preference A = U > C > G) (Eggington et al., 2011, Riedmann et al., 2008) was observable (Figures 1C and 1D), indicating that most of the A-to-G substitutions detected in the HE analysis resulted from ADAR editing. Similar results were obtained from another, but smaller, independent dataset from Rai et al. (2016) (Figure S1).
Figure 1.

A-to-I Editing Is Significantly Increased in SLE Patients
Compared cohorts are healthy controls, SLE patients with low levels of ISG (ISM-low, which corresponds to a more quiescent disease), and SLE patients with high levels of ISG (ISM-high), depicted in blue, yellow, and red, respectively. The comparisons were evaluated using the Wilcoxon test.
(A and B) Global levels of editing were assessed by determining (A) the editing in Alu elements (AEI) and (B) the number of hyper-edited (HE) sites (normalized by the number of mapped reads) per sample.
(C and D) Cleanness of the HE signal was assessed by (C) the number A-to-G mismatches compared to all types of mismatches and (D) the neighboring nucleotides frequencies at the sites detected by the HE analysis (which matched the ADAR deamination motif).
(E) Editing levels were assessed using the rates at pre-known sites. The means of the editing rates of ISM-high patients were plotted against controls per site. Sites with significant changes (FDR < 0.1) are depicted in red.
(F and G) Expression levels of (F) ADAR1 and (G) ADAR2 suggest that ADAR1 is responsible for the alterations in editing levels.
As a complement approach, we investigated the level of RNA editing at known sites that have been identified and characterized by previous studies (see Experimental Procedures). Among the sites at which editing levels were found to differ significantly between the controls and the ISM-high patients, most had increased editing levels. This was also the general trend in sites without significant changes (Figure 1E). These results further support the observation that RNA editing is elevated in the blood of ISM-high patients.
To determine whether ADAR1, which is itself an ISG, is responsible for the increased editing levels, we examined its expression levels. As expected, its expression was significantly higher in ISM-high patients compared to controls (2.24-fold change, Wilcoxon p value = 3.77e−6) (Figure 1F). However, in contrast to the results of the global RNA-editing levels, no significant differences were observable between ISM-low patients and controls (Wilcoxon p value = 0.54). This may indicate that ADAR1 expression is regulated directly by IFN, while RNA-editing levels remain relatively high in these patients even without IFN induction. In contrast, the expression of ADAR2 was lower in ISM-high patients compared to controls (0.75-fold change, Wilcoxon p value = 0.035) (Figure 1G). Altogether, our results show significantly elevated ADAR1 expression and editing activity in ISM-high patients.
Enhanced C-to-U Editing in SLE Patients
Proteins from the additional deaminase family, the APOBEC, were also significantly upregulated in ISM-high patients compared to controls (Figure 2A; Table S2). This may lead to the accumulation of C-to-U mismatches, which can serve as a potential source for neo-autoantigens. A dataset of putative C-to-U editing sites in monocytes and macrophages has been published (Sharma et al., 2015). Of the sites, 252 were detected in the examined dataset (96.5% of the expressed sites), and most of them (75%) were edited in more than 20% of the samples. Out of these, 26 had significantly elevated editing rates in ISM-high patients (false discovery rate [FDR] < 0.1), and a similar global trend was observable (Figure 2B). Because the editing probably occurs only in specific cell types (e.g., monocytes and macrophages), very low editing rates were observed in the total cell population (90% of the sites had a mean rate of 0%–2%). To further assess the C-to-U editing levels in SLE patients and healthy individuals, we measured the global rates of HE C-to-U clusters in each sample (see Experimental Procedures). As was the case for the A-to-I editing, ISM-high patients had significantly more clustered C-to-U sites per sample (Wilcoxon p value = 4.87e−3) (Figure 2C), and their numbers correlated well with APOBEC3A levels (r = 0.70) (Figure 2D) indicating that this is the main deaminase driving this elevation.
Figure 2.

Elevated C-to-U Editing in SLE Patients
ISM-high patients, ISM-low patients, and controls are depicted in red, yellow, and blue, respectively. The paired comparisons were evaluated using the Wilcoxon test.
(A) Expression levels of APOBEC3A in ISM-high patients, ISM-low patients, and controls.
(B and C) Elevated C-to-U editing in SLE patients was assessed by (B) the mean C-to-U mismatch rates per site at pre-known sites of ISM-high patients, plotted against the controls (sites with significant changes [FDR < 0.1] are depicted in red) and (C) the number of HE C-to-U sites (normalized by the number of mapped reads) per sample.
(D) Correlation between APOBEC3A expression levels and normalized number of HE C-to-U sites.
Elevated Levels of Recoding Events in SLE Patients
Only a small portion of A-to-I RNA editing results in the recoding of proteins. To identify recoding sites associated with SLE, we systematically searched for differentially edited sites with a non-synonymous outcome. Because the current approaches for de novo detection of recoding sites without a matched DNA sequences from the same individual perform poorly, we limited the analyses to high-credibility sites found within the HE regions or to the previously verified ones.
We first examined putative recoding sites detected by the HE analysis, in which the potentially editable adenosine is found on the transcribed strand. Excluding known SNPs and sites within highly polymorphic genomic regions, we identified 624 putative recoding sites. As expected, most of these sites (95%) were edited in SLE samples, and their prevalence was higher than in controls (an average of 1.100 sites per million mapped reads in SLE patients versus 0.967 in controls, Wilcoxon p value = 0.018) (Figure 3A). The obtained signal was noisy (Figure S2A), indicating that a portion of these sites are not bona fide ADAR targets. Nevertheless, the neighbor preferences obtained fairly agree with the ADAR motif (Figure S2B), supporting the contribution of genuine editing by ADAR. Many of these sites were stochastically edited (545 of 624 sites were edited in fewer than five samples). We therefore compared the relative number of samples that expressed the edited version of the transcript between the groups. Two such sites, in the genes IFITM1 and ODF3B, were found to be edited in a higher percentage of ISM-high samples compared to controls (Fisher’s exact test, FDR < 0.05), probably because of elevated expression levels in SLE patients (FDR < 1e−5). This elevated expression results in increased detectability by our analysis (and presumably in higher numbers of the recoded versions of peptide), which should also reflect higher visibility of them to the immune system. Several edited sites were found within the SRSF4 and VIM genes, which are known targets for autoantibodies of SLE (Sherer et al., 2004).
Figure 3.

A-to-I Editing in Non-synonymous Sites Is Significantly Elevated, Resulting in the Potential Generation of Epitopes
ISM-high patients, ISM-low patients, and controls are depicted in red, yellow, and blue, respectively. The paired comparisons were evaluated using the Wilcoxon test.
(A) Normalized number of recoding sites within HE regions (normalized by the number of mapped reads) per sample.
(B and C) Editing levels of (B) SH3BP2 (at chr4:2,835,556) and (C) ARL6IP4 (SRp25, at chr12:123,466,262).
(D) Normalized number of sites per sample creating edited peptides that have superior HLA affinity over the non-edited forms.
We also examined the editing rates at recoding sites identified in other studies (see Experimental Procedures). Two sites were found to have higher editing levels in ISM-high patients (Wilcoxon, FDR < 0.05) (Figures 3B and 3C), with no significant expression difference in SLE patients and controls in the genes SH3BP2 and ARL6IP4 (at chr4:2,835,556 and chr12:123,466,262, respectively).
These results support the hypothesis that the high global editing levels in SLE patients can give rise to a variety and higher levels of the edited versions of proteins.
RNA Editing Results in Potential MHC Class I Epitopes
To investigate any putative immunogenicity caused by elevated editing in the patients, we evaluated the potential of MHC class I epitopes to be derived from peptides originating from edited transcripts, using histocompatibility leukocyte antigen (HLA) allele predictions for each sample (Szolek et al., 2014). We measured the editing rates of recoding editing events that generate peptides with a predicted high affinity for MHC class I molecules (see Experimental Procedures). SLE patients had significantly higher rates at such sites than controls (Wilcoxon p value = 1.59e−14 and 3.56e−5 for ISM-high and ISM-low patients, respectively), and ISM-high patients had higher rates than ISM-low patients (Wilcoxon p value = 1.65e−3). We focused on a subset of these sites for which editing enhances the binding affinity of the peptides (see Experimental Procedures). ISM-high patients had significantly higher rates at these sites than controls (Wilcoxon p value = 9.13e−7) (Figure 3D) and ISM-low patients (Wilcoxon p value = 1.86e−3). It is evident that these patients have higher editing rates, even at a fraction of the recoding sites, whose resulting proteins have higher affinities for the HLA alleles.
We also assessed the number of possible epitopes from each peptide, both before and after editing (see Experimental Procedures), which was mostly not statistically different, suggesting that the edited forms of the peptides have the same potential to be derived to epitopes as the non-edited forms.
Discussion
The results of this study suggest the possibility of a connection between autoimmunity and excessive RNA editing. We surmise that the latter may facilitate the generation of autoantigens in peripheral tissues. Because these autoantigens might not necessarily be expressed in the thymus, reactive T cells may escape the negative selection and recognize these recoded proteins as non-self. The elevated global editing activity in SLE patients, one of the manifestations of the inflammatory condition, results in increased variety and higher levels of edited forms of proteins. Similarly, conditions that lead to editing alterations can result in new recoding events (Daniel et al., 2012). These have the potential to be processed into autoepitopes that may then be subsequently presented on the MHC molecules, thus stimulating an autoimmune response (Figure 4). Our results therefore enrich knowledge about the recently discovered role of ADAR1 and RNA editing in regulating the innate immune system and support the connection between imbalance of RNA editing and immune dysfunction.
Figure 4.

Pro-inflammatory Editing Feedback
(A) As part of an inflammatory response, interferons are released into the intercellular environment.
(B) Uptake of the interferons triggers a response via the JAK-STAT signaling pathway. Among the many genes affected, the ADAR1 p150 isoform is produced.
(C) This upregulation of ADAR1 leads to higher editing levels, due both to the increasing rates at normally edited sites and to the editing of sites that are not edited under normal conditions (left, non-inflamed cell; right, inflamed cell).
(D) Elevated editing levels lead to increased numbers and a variety of recoded proteins.
(E) Some recoded proteins might be degraded into peptides that will eventually become epitopes, resulting in presentation of recoded epitopes that are usually not found on cell surfaces.
(F) These edited epitopes might trigger an immune response (thus becoming neo-autoantigens), contributing to the inflammatory state and potentially initiating a positive feedback loop.
We hypothesize that the elevated editing may be involved in positive feedback, aggravating autoimmunity (Figure 4). Inflammatory cytokines produced by an immune response triggered by elevated editing levels may maintain or even further increase the editing levels by stimulating the IFN-induced deaminases, resulting in the production of still more, potentially immunogenic, editing-recoded peptides. In addition, the edited epitope not only may prove immunogenic but also may initiate a process of epitope spreading (Lehmann et al., 1992, Sercarz, 1998, Vanderlugt and Miller, 2002). Moreover, SLE has several characteristics that may promote the immunogenicity of RNA editing, such as the upregulation of epitope presentation pathway by IFNs and the accumulation of dead cell debris (Boehm et al., 1997, Lipsky, 2001, Rusinova et al., 2013, Schroder et al., 2004).
Although the results presented here are promising and demonstrate the potential of editing to create neo-autoantigens, there are several limitations to this type of research, leading to incomplete detection of the editing-derived proteome and assessment of its immunogenicity. First, from an analytical perspective, while the HE analysis enables the discovery of recoding editing sites, the HE reads comprise only a minority (∼1%) of the total reads, and presumably a small fraction of the total editing. Moreover, the recoding sites detected here have a relatively low signal-to-noise ratio, so the credibility of each site is not high enough, even though most of them are genuine and altogether the sites are credible. Second, from a biological perspective, several limitations arise. The RNA was extracted mainly from living cells; thus, it is possible that editing sites generating particularly immunogenic peptides may be underrepresented in the data because of their elimination by a fiercer immune reaction against such cells. Another potential drawback is introduced by the RNA-seq data used for the analysis being derived from whole blood, which contains heterogeneous cell types, which may dampen the signal and add noise. Thus, it is probable that the number of recoding sites detected here is an underestimation of the actual number of recoding editing sites in SLE patients.
In summary, we have shown significantly elevated RNA editing in SLE patients and revealed its potential to give rise to neo-autoantigens, implying a role for RNA editing in the etiology and progress of SLE. These findings provide another link between RNA editing and autoimmune diseases.
Experimental Procedures
The Supplemental Experimental Procedures includes more details of the applied analyses.
Datasets
A whole-blood (Paxgene) RNA-seq dataset of 18 healthy individuals and 99 active SLE patients (GEO: GSE72509) from Hung et al. (2015) and a smaller dataset of 12 SLE patients and 4 controls (GEO: GSE80183) from Rai et al. (2016) were downloaded. Details are provided in Table S1.
The reads were aligned to the human genome (hg19) using STAR 2.4.2 (Dobin et al., 2013).
HE
We used a recently described pipeline (Porath et al., 2014) that enables the measurement of editing of heavily edited reads, which standard schemes fail to align correctly (Carmi et al., 2011)
AEI
To measure the editing in Alu elements, we used a previously described method (Bazak et al., 2014b). Due to the magnitude of the effect and the specific occurrence of editing in Alu elements (such as editing in clusters), this measurement results in a clean signal with a minimal false-positive rate (Bazak et al., 2014a).
Known Sites
RNA editing levels for a list of pre-known editing sites were calculated using REDIToolKnown, which is part of the REDItools package (Picardi and Pesole, 2013). The list of editing sites was compiled from previous studies (Khermesh et al., 2016, Pinto et al., 2014, Ramaswami et al., 2012).
Expression Analysis
The DESeq package (Anders and Huber, 2010) in R was used for analyzing differential gene expression in all control and SLE patient samples.
Statistical Analysis
The statistical analysis was done using R (R Project for Statistical Computing, http://www.r-project.org/). Unless otherwise specified, the statistically significant difference (5% confidence level) among two groups was tested using the Wilcoxon rank test. The tests were performed using the default parameters and in a two-sided manner. Where appropriate, p values were corrected for multiple testing.
Recoding Editing Sites Analyses
To provide the peptide sequence for the analyses and find recoding, HE and pre-known sites filtered for SNPs and HLA genes were annotated using wANNOVAR (Chang and Wang, 2012, Wang et al., 2010). HLA alleles for each sample were derived using OptiType (Szolek et al., 2014). For each sample, recoding sites (detected by the HE analysis) inside peptides that have at least one window (see Supplemental Experimental Procedures) with strong affinity to the sample-predicted HLA alleles were selected according to the NetMHCPan4.0 (Jurtz et al., 2017) predictions. A recoding event was considered to have an affinity-enhancing effect if the edited version of the peptide, for all windows on average, had a higher affinity (of at least 10%). The number of epitopes per peptide chain was calculated as previously described in Agranovich et al. (2013).
Acknowledgments
We thank Eli Eisenberg, Amos Schaffer, Binyamin A. Knisbacher, Lea Shallev, and various Levanon lab members for the helpful discussions. We thank Ziv Paz and George Tsokos for the stimulating discussions at the early stages of the project. We thank Tamar Roth-Fenster for assisting in editing and drafting the manuscript. M.D.-G. is grateful to the Azrieli Foundation for the award of an Azrieli Fellowship. E.Y.L. was supported by the European Research Council (311257) and the Israel Science Foundation (1380/14). C.J.C. was supported by the Israel Science Foundation (1422/15).
Author Contributions
E.Y.L., S.H.R., and M.D.-G. designed the research. S.H.R. and M.D.-G. developed and preformed the computational analyses under the guidance of E.Y.L. M.B.-I. and Y.L. preformed the epitope counting analysis. E.Y.L., S.H.R., and M.D.-G. wrote the manuscript with input from C.J.C. and G.R. All authors read and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: April 3, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.036.
Supplemental Information
References
- Agranovich A., Maman Y., Louzoun Y. Viral proteome size and CD8+ T cell epitope density are correlated: the effect of complexity on selection. Infect. Genet. Evol. 2013;20:71–77. doi: 10.1016/j.meegid.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F.J., Rechavi G., Li J.B., Eisenberg E., Levanon E.Y. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L., Levanon E.Y., Eisenberg E. Genome-wide analysis of Alu editability. Nucleic Acids Res. 2014;42:6876–6884. doi: 10.1093/nar/gku414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U., Klamp T., Groot M., Howard J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Burns C.M., Chu H., Rueter S.M., Hutchinson L.K., Canton H., Sanders-Bush E., Emeson R.B. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- Carmi S., Borukhov I., Levanon E.Y. Identification of widespread ultra-edited human RNAs. PLoS Genet. 2011;7:e1002317. doi: 10.1371/journal.pgen.1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X., Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012;49:433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiche L., Jourde-Chiche N., Whalen E., Presnell S., Gersuk V., Dang K., Anguiano E., Quinn C., Burtey S., Berland Y. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014;66:1583–1595. doi: 10.1002/art.38628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow Y.J., Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- Danan-Gotthold M., Guyon C., Giraud M., Levanon E.Y., Abramson J. Extensive RNA editing and splicing increase immune self-representation diversity in medullary thymic epithelial cells. Genome Biol. 2016;17:219. doi: 10.1186/s13059-016-1079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel C., Venø M.T., Ekdahl Y., Kjems J., Öhman M. A distant cis acting intronic element induces site-selective RNA editing. Nucleic Acids Res. 2012;40:9876–9886. doi: 10.1093/nar/gks691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann C.R., Neunteufl A., Pfaffstetter L., Jantsch M.F. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol. Biol. Cell. 2001;12:1911–1924. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington J.M., Greene T., Bass B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl Y., Farahani H.S., Behm M., Lagergren J., Öhman M. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res. 2012;22:1477–1487. doi: 10.1101/gr.131912.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furie R., Khamashta M., Merrill J.T., Werth V.P., Kalunian K., Brohawn P., Illei G.G., Drappa J., Wang L., Yoo S., CD1013 Study Investigators Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017;69:376–386. doi: 10.1002/art.39962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George C.X., Samuel C.E. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- Graham K.L., Utz P.J. Sources of autoantigens in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2005;17:513–517. doi: 10.1097/01.bor.0000171215.87993.6b. [DOI] [PubMed] [Google Scholar]
- Hagberg N., Rönnblom L. Systemic Lupus Erythematosus--A Disease with A Dysregulated Type I Interferon System. Scand. J. Immunol. 2015;82:199–207. doi: 10.1111/sji.12330. [DOI] [PubMed] [Google Scholar]
- Han L., Diao L., Yu S., Xu X., Li J., Zhang R., Yang Y., Werner H.M.J., Eterovic A.K., Yuan Y. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M., Maas S., Single F.N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- Ho V., Mclean A., Terry S. Severe systemic lupus erythematosus induced by antiviral treatment for hepatitis C. J. Clin. Rheumatol. 2008;14:166–168. doi: 10.1097/RHU.0b013e3181775e80. [DOI] [PubMed] [Google Scholar]
- Hueber W., Zeng D., Strober S., Utz P.J. Interferon-alpha-inducible proteins are novel autoantigens in murine lupus. Arthritis Rheum. 2004;50:3239–3249. doi: 10.1002/art.20508. [DOI] [PubMed] [Google Scholar]
- Hung T., Pratt G.A., Sundararaman B., Townsend M.J., Chaivorapol C., Bhangale T., Graham R.R., Ortmann W., Criswell L.A., Yeo G.W., Behrens T.W. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science. 2015;350:455–459. doi: 10.1126/science.aac7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V., Paul S., Massimo A., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khermesh K., D’Erchia A.M., Barak M., Annese A., Wachtel C., Levanon E.Y., Picardi E., Eisenberg E. Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer’s disease. RNA. 2016;22:290–302. doi: 10.1261/rna.054627.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirou K.A., Gkrouzman E. Anti-interferon alpha treatment in SLE. Clin. Immunol. 2013;148:303–312. doi: 10.1016/j.clim.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Laxminarayana D., Khan I.U., Kammer G. Transcript mutations of the alpha regulatory subunit of protein kinase A and up-regulation of the RNA-editing gene transcript in lupus T lymphocytes. Lancet. 2002;360:842–849. doi: 10.1016/s0140-6736(02)09966-x. [DOI] [PubMed] [Google Scholar]
- Laxminarayana D., O’Rourke K.S., Maas S., Olorenshaw I. Altered editing in RNA editing adenosine deaminase ADAR2 gene transcripts of systemic lupus erythematosus T lymphocytes. Immunology. 2007;121:359–369. doi: 10.1111/j.1365-2567.2007.02582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Kirsch M.A., Wolf C., Günther C. Aicardi-Goutières syndrome: a model disease for systemic autoimmunity. Clin. Exp. Immunol. 2014;175:17–24. doi: 10.1111/cei.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann P.V., Forsthuber T., Miller A., Sercarz E.E. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- Liddicoat B.J., Piskol R., Chalk A.M., Ramaswami G., Higuchi M., Hartner J.C., Li J.B., Seeburg P.H., Walkley C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–1120. doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat B.J., Chalk A.M., Walkley C.R. ADAR1, inosine and the immune sensing system: distinguishing self from non-self. Wiley Interdiscip. Rev. RNA. 2015;7:157–172. doi: 10.1002/wrna.1322. [DOI] [PubMed] [Google Scholar]
- Lipsky P.E. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat. Immunol. 2001;2:764–766. doi: 10.1038/ni0901-764. [DOI] [PubMed] [Google Scholar]
- Liu Q., Jiang L., Liu W.-L., Kang X.-J., Ao Y., Sun M., Luo Y., Song Y., Lo W.H.Y., Zhang X. Two novel mutations and evidence for haploinsufficiency of the ADAR gene in dyschromatosis symmetrica hereditaria. Br. J. Dermatol. 2006;154:636–642. doi: 10.1111/j.1365-2133.2006.07133.x. [DOI] [PubMed] [Google Scholar]
- Mannion N.M., Greenwood S.M., Young R., Cox S., Brindle J., Read D., Nellåker C., Vesely C., Ponting C.P., McLaughlin P.J. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng B., Yang F., Huston D.P., Yan Y., Yang Y., Xiong Z., Peterson L.E., Wang H., Yang X.-F. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 2004;114:1463–1470. doi: 10.1016/j.jaci.2004.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewold T.B. Interferon alpha-induced lupus: proof of principle. J. Clin. Rheumatol. 2008;14:131–132. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski R.J., O’Rourke K.S., Olorenshaw I., Hawkins G.A., Maas S., Laxminarayana D. Altered editing in cyclic nucleotide phosphodiesterase 8A1 gene transcripts of systemic lupus erythematosus T lymphocytes. Immunology. 2008;125:408–419. doi: 10.1111/j.1365-2567.2008.02850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N., Bazak L., Buchumenski I., Porath H.T., Danan-Gotthold M., Knisbacher B.A., Eisenberg E., Levanon E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015;13:267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- Peng G., Lei K.J., Jin W., Greenwell-Wild T., Wahl S.M. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J. Exp. Med. 2006;203:41–46. doi: 10.1084/jem.20051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K., Funk C.C., Snyder J.M., Price N.D., Treuting P.M., Stetson D.B. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E., Pesole G. REDItools: high-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–1814. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- Pinto Y., Cohen H.Y., Levanon E.Y. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 2014;15:R5. doi: 10.1186/gb-2014-15-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath H.T., Carmi S., Levanon E.Y. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 2014;5:4726. doi: 10.1038/ncomms5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R., Chauhan S.K., Singh V.V., Rai M., Rai G. RNA-seq Analysis Reveals Unique Transcriptome Signatures in Systemic Lupus Erythematosus Patients with Distinct Autoantibody Specificities. PLoS ONE. 2016;11:e0166312. doi: 10.1371/journal.pone.0166312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G., Li J.B. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014;42:D109–D113. doi: 10.1093/nar/gkt996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G., Lin W., Piskol R., Tan M.H., Davis C., Li J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods. 2012;9:579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G.I., Kasher P.R., Forte G.M.A., Mannion N.M., Greenwood S.M., Szynkiewicz M., Dickerson J.E., Bhaskar S.S., Zampini M., Briggs T.A. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann E.M., Schopoff S., Hartner J.C., Jantsch M.F. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA. 2008;14:1110–1118. doi: 10.1261/rna.923308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter S.M., Dawson T.R., Emeson R.B. Regulation of alternative splicing by RNA editing. Nature. 1999;399:75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- Rusinova I., Forster S., Yu S., Kannan A., Masse M., Cumming H., Chapman R., Hertzog P.J. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41:D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva Y.A., Rieder L.E., Reenan R.A. The ADAR protein family. Genome Biol. 2012;13:252. doi: 10.1186/gb-2012-13-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Sercarz E.E. Immune focusing vs diversification and their connection to immune regulation. Immunol. Rev. 1998;164:5–10. doi: 10.1111/j.1600-065x.1998.tb01202.x. [DOI] [PubMed] [Google Scholar]
- Sharma S., Patnaik S.K., Taggart R.T., Kannisto E.D., Enriquez S.M., Gollnick P., Baysal B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015;6:6881. doi: 10.1038/ncomms7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Patnaik S.K., Kemer Z., Baysal B.E. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol. 2017;14:603–610. doi: 10.1080/15476286.2016.1184387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer Y., Gorstein A., Fritzler M.J., Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin. Arthritis Rheum. 2004;34:501–537. doi: 10.1016/j.semarthrit.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Smith H.C., Bennett R.P., Kizilyer A., McDougall W.M., Prohaska K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012;23:258–268. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speyer J.F., Lengyel P., Basilio C., Ochoa S. Synthetic polynucleotides and the amino acid code. II. Proc. Natl. Acad. Sci. USA. 1962;48:63–68. doi: 10.1073/pnas.48.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolek A., Schubert B., Mohr C., Sturm M., Feldhahn M., Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–3316. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokos G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- Vanderlugt C.L., Miller S.D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.