

Abstract
Aggregates of misfolded proteins can compromise the function of the 26S proteasome complex, leaving neurons susceptible to accelerated and impaired protein homeostasis, thereby contributing to the pathogenesis of neurodegeneration. Strategies aimed at enhancing the function of the 26S proteasome via phosphorylation of key subunit epitopes have been effective in reducing protein aggregates in mouse models of disease. We discuss how phosphodiesterase (PDE) inhibitors and G protein-coupled receptor (GPCR)-targeted drugs might be considered as candidate therapeutics, acting on second messenger signal transduction. The range of candidates might address the need forregion-,cell-,oreven cellular compartment-specific modulation. Given the array of clinical and experimental drugs targeting cAMP/cGMP signaling, we propose that proteasome activators targeting secondary messengers might be exploited as novel agents for the treatment or prevention of some neurodegenerative diseases.
The Ubiquitin Proteasome System (UPS) in Neurodegenerative Diseases
Neurodegenerative diseases (see Glossary) such as Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and prion diseases are clinically diverse proteinopathies that share a common pathological feature at autopsy: the presence of aggregates composed of misfolded proteins [1,2]. In humans, disease-specific proteins such as tau, α-synuclein, huntingtin (HTT), and transactive response DNA-binding protein 43 kDa (TDP-43) can accumulate as misfolded, oligomeric, or aggregated forms within cells, and spread between cells from regions of initial vulnerability to distant areas as diseases progress [3]. The most common protein entity associated with aggregates is ubiquitin, implicating the ubiquitin proteasome system (UPS) in processes associated with disease pathogenesis. The UPS is responsible for the degradation of most proteins, and any negative perturbation of the degradation process can have a deleterious effect on general protein quality control. This is particularly detrimental to neurons because they cannot dilute toxic aggregates by mitotic cell division [4]. The UPS enzymatic cascade consists of two discrete and successive steps: first the covalent tagging of the substrate protein by a poly-ubiquitin chain; second, the subsequent degradation of the tagged protein by the 26S proteasome [5]. In eukaryotic cells several enzymes including ubiquitin-activating enzyme (E1 ubiquitin activase), ubiquitin-conjugating enzyme (E2 ubiquitin conjugase), and ubiquitin ligase (E3 ubiquitin ligase) conjugate molecules of ubiquitin to the target lysine on the substrate protein. After multiple rounds of ubiquitination, the poly-ubiquitin chain is formed and the protein is tagged for destruction. Degradation of poly-ubiquitinated substrates is carried out by the 26S proteasome (Box 1) [6].
Box 1. The Mammalian 26A Proteasome.
The 26S proteasome is a large, ~2.5 MDa, multi-catalytic, ATP-dependent protease that degrades proteins into small peptides. It comprises two components: a 19S regulatory particle (RP) and a 20S core particle (CP) that carries out the catalytic activity [5]. The 20S CP is a barrel-shaped structure composed of 28 subunits stacked in four rings with two inner β rings that are flanked by two outer a rings. Three of the β subunits, β1, β2, and β5, contain catalytic sites with different proteolytic specificities. The majority of 20S proteasomes are capped by one or two 19S RPs, composed of 19 different subunits arranged in two distinct subcomplexes termed the base and the lid. The 19S complex is responsible for various biochemical functions, including the recognition of substrates and the binding, deubiquitination, unfolding, and translocation of substrates to the 20S chamber where the protein undergoes irreversible degradation [5,99].
Ubiquitinated conjugates have been shown to decorate paired helical filaments (PHFs) and neurofibrillary tangles (NFTs) composed of the microtubule-binding protein tau in post-mortem brain tissues from patients with AD [7]. Recently, ubiquitin and proteasome components have been identified in protein aggregates in several neurodegenerative diseases [8–12]. In addition to the accumulation of ubiquitinated proteins, decreased proteasome function has been reported in the hippocampal, parietal, and temporal lobe regions of post-mortem AD brain tissue, but not in unaffected areas such as occipital lobe and cerebellum [13]. Subsequent studies of HD [14], PD [15–17], ALS [18,19], and prion disease [11] samples have also provided post-mortem evidence of downregulation of proteasome function. Thus, the current thinking in the field is that altered proteostasis plays a key role in the pathogenesis of proteinopathies [20,21]. Several independent studies suggest that protein aggregates may disrupt protein degradation by physically blocking the gate opening of the 19S regulatory subunit of the 26S proteasome (Box 1). For example, it has been reported that aggregates of prion protein (PrP) can block the opening of 26S and 20S proteasome particles, leading to reduced proteasomal activity in prion-infected brains of transgenic mice [22–24]. Furthermore, incubation of isolated 26S proteasomes with PHF-tau isolated from AD brain, or incubation with recombinant tau aggregates and oligomers [25,26], resulted in inhibition of proteasome activity. Similar results were reported when aggregates of α-synuclein were incubated with purified 26S proteasomes [27,28]. However, we have shown that 26S proteasomes remained defective even after they had been purified from the brain of a mouse model that develops robust tau aggregates [25]. Thus, we speculate that the persistence of proteasome deficiency might be caused by a fundamental change in the quaternary structure of the 26S complex, leading to profound deterioration of its organization. Another possibility is that specific misfolded proteins that resist complete degradation could become an adherent, fibrous ‘blanket’ on proteasome structures, leading to attenuated activity. Future structural analysis and atomic modeling of the 26S proteasomes from healthy and diseased brain tissues will hopefully help elucidate the mechanism(s) by which proteasome impairment occurs in the diseased brain.
We discuss here new research that investigates the mechanism of phosphorylation-dependent activation of proteasome function which can lead to enhanced activity of the proteasome against pathological substrates. We describe a new therapeutic approach based on this mechanism, and propose that it could be developed for the treatment of some neurodegenerative diseases.
Approaches to Enhancing the UPS
Although the UPS has been shown to contribute to disease pathogenesis, interest in this pathway as a therapeutic target has lagged behind other clearance pathways such as macroautophagy. The first studies on the impact of deficient neuronal macroautophagy in mouse models [29,30] led to intense research in the neurodegeneration field, and autophagy inducers were proposed as potential therapeutics. In support, the autophagy-inducer rapamycin was shown to clear aggregates in a mouse model of AD and in Drosophila models of tauopathy and poly-glutamine expansion [31].
Recently, the field has focused on the role of small oligomeric forms of proteins in disease pathogenesis, and attention has been directed at therapeutic approaches involving, for instance, targeting of oligomers or small aggregates by the UPS. One mechanism proposed to enhance substrate degradation is via inhibition of USP14, a proteasome-associated deubiquitinating enzyme. Specifically, USP14 reduces proteolysis in a substrate-specific manner by rapidly trimming substrate-bound ubiquitin chains before the 26S proteasome can initiate degradation of the substrate [32]. As a result, deubiquitinated proteins are released from the 26S proteasome undigested [32]. It is possible to accelerate 26S proteasome-mediated proteolysis using small-molecule USP14 inhibitors which have shown utility in cell cultures and primary neurons [33–35].
Direct Activation of 26S Proteasome Function
A different approach to enhancing the UPS is to target proteasome function directly, as opposed to via ubiquitination pathways. The first study demonstrating the biological significance of enhanced proteasome function showed that post-translational modification of proteasome subunits by phosphorylation positively regulated proteasome-mediated protein degradation in HEK293 cells [36]. Thereafter, reversible proteasome phosphorylation leading to activation of proteasome function was documented in vitro in neurons [37–40] and in cell lines [40–43], as well as in vivo in physiological [42,44–47] and pathological conditions [25,48,49]. Such studies have stimulated a wider search for pharmacological approaches to enhance proteasome function.
For instance, PKA-dependent proteasome phosphorylation has been shown to be a positive regulator of 26S proteasome function in several cell lines [41,43], primary neuronal cells [40,50,51], and also in vivo in mouse models of FTD [25] and HD [49]. Moreover, other kinases have been shown to exert a similar effect on 26S proteasome hydrolytic capacity, including CaMKIIα in primary neuronal cultures [37–39,45], DYRK2 in a breast cancer mouse model and cell lines [48], and PKG in a UPS reporter (GFP-degron) mouse model [44]. However, it is not well understood how kinases that phosphorylate different subunits of the 26S proteasome can have a similar effect on protein breakdown. The data so far show that phosphorylation regulates multiple components of the 26S proteasome complex, and this could impact on several steps in the proteolytic process. For example, PKA activation enhances the activity of ATPase subunits Rpt1–6 [25,41] that are involved in protein engagement of the proteasome as well as in the unfolding and translocation of the protein to the 20S core for degradation (Box 1) [5]. Furthermore, PKA was reported to increase 26S proteasome abundance and stability by inducing the association of the 20S core with the 19S regulatory complex, presumably leading to facilitated protein degradation in cell lines and in vivo in canine hearts [41,42,46,52]. Other observations in primary neuronal cultures have indicated that the translocation and sequestration of 26S proteasomes into dendritic spines depends on proteasome phosphorylation by the kinase CaMKIIα. Indeed, the recruitment of 26S proteasomes to dendritic spines can promote local protein degradation, an important regulatory mechanism for synaptic remodeling and for forming synaptic connections [37–39,45]. In addition, proteasome activity assays have shown that PKA [25,36,40–42,46,49,50], CaMKIIα [38,39,51], DYRK2 [48], and PKG [44] can increase the activity of proteasome peptidases (β1, β2, and β5 subunits). In contrast to phosphorylation acting on proteasome activity directly, phosphorylation might also increase the percentage of proteasomes that are engaged in substrate processing; this speculation is based on a recent cryo-electron microscopy tomography study reporting that in healthy neurons the capacity of the proteasome system is not fully utilized [53]. The study reported the coexistence of multiple conformational states of 26S proteasomes within intact hippocampal neurons, with only 20% of the 26S proteasomes being engaged in substrate processing or degradation, while the remainder were in a substrate-accepting ground state [53]. This latter state is the lowest-energy conformation of the 26S proteasome, in which it is presumed to be ‘idle’ [53]. Therefore, we speculate that phosphorylation of 26S proteasome subunits might increase the ratio of active proteasomes engaged in substrate processing versus ‘idle’ or substrate-accepting ground-state 26S proteasomes. However, the idea that there could be phosphorylation-dependent structural changes in 26S proteasomes remains to be tested.
Nonetheless, these findings suggest a strategy that could be used to treat neurodegenerative diseases, where direct stimulation of proteasome function by phosphorylation of proteasome subunits might restore UPS function, enhance protein clearance, and prevent protein aggregation in cells. Discussed below are two envisaged strategies to activate proteasome-mediated clearance; namely, a non-restricted, cell-wide approach and a compartment-restricted approach.
Stimulating Proteasome-Mediated Protein Degradation via Second Messenger Signaling
The synthesis and degradation of the second messengers cAMP and cGMP are highly regulated processes. After being synthesized by adenylyl cyclase or guanylyl cyclase, respectively, cAMP and cGMP amplify encoded information by activating several effector molecules. The main effector proteins of cAMP and cGMP are PKA and PKG, respectively, which in turn phosphorylate other signaling proteins and transcription factors [54,55]. The PKA holoenzyme structure consists of two regulatory and two catalytic subunits. The catalytic (C) subunits contain the active site, whereas the regulatory (R) subunits keep the C subunits tethered into a tetrameric structure, which is the inactive form of PKA [56]. The regulatory subunits have domains that bind to cAMP, and upon cAMP binding the R subunits release the C subunits, and this triggers their kinase activity. The UPS plays a major role in regulating PKA activity because it swiftly degrades the liberated R subunits [57]. We can speculate that chronic proteasome dysfunction in neurodegenerative disease could lead to increased levels of undigested R subunits and reassociation with the C subunits into an inactive PKA tetramer, which may have an effect on downregulation of the PKA signaling cascade (Box 2). PKG is a homodimeric protein that contains both its regulatory and catalytic elements in the same polypeptide chain [58]. The protein changes conformation upon cGMP binding; however, it is not known if its levels are regulated by UPS-mediated degradation [58].
Box 2. The Negative Regulation Triad in Neurological Synapses.
We speculate that the accumulation of pathological, aggregation-prone proteins in synaptic terminals, either through mislocalization or following trans-synaptic transfer, leads to impaired proteasome activity and synaptic proteolysis. This could lead to downregulation of synaptic cAMP/PKA signaling, which is the key signaling pathway for regulating synaptic plasticity. Synaptic dysfunction is thought to be an early pathological event in AD that precedes synaptic loss [96,97]. The latter is the strongest pathological correlate of cognitive decline [100,101]. Tau, which aggregates and forms neurofibrillary pathology in AD and other tauopathies, is mislocalized from axons to synaptic compartments early in the disease [102,103]. Accumulation of tau in the synaptic compartments can lead to impaired synaptic proteolysis (Figure I). The UPS plays an essential role in the synaptic cAMP/PKA cascade because it regulates the turnover of the R subunits of the PKA holoenzyme and the release of the catalytic C subunits [57]. Reduced degradation of the R subunits can result in increased ratio of the R/C subunits, thus favoring reassociation to form the inactive PKA holoenzyme, which leads to downregulation of PKA signaling – an essential pathway for synaptic plasticity and strengthening (Figure I) [104].
Dysregulation of this intricate triad may be a contributing factor to synaptic dysfunction and loss in AD. Thus, therapies that aim to enhance proteasome function in the synapse may have an impact in restoring the cAMP/PKA signaling cascade, reducing aggregated proteins and preventing cognitive decline.
Figure I. Negative Regulation Between Protein Aggregates, Dysregulated Proteasome Function, and cAMP/PKA Signaling in Humans.
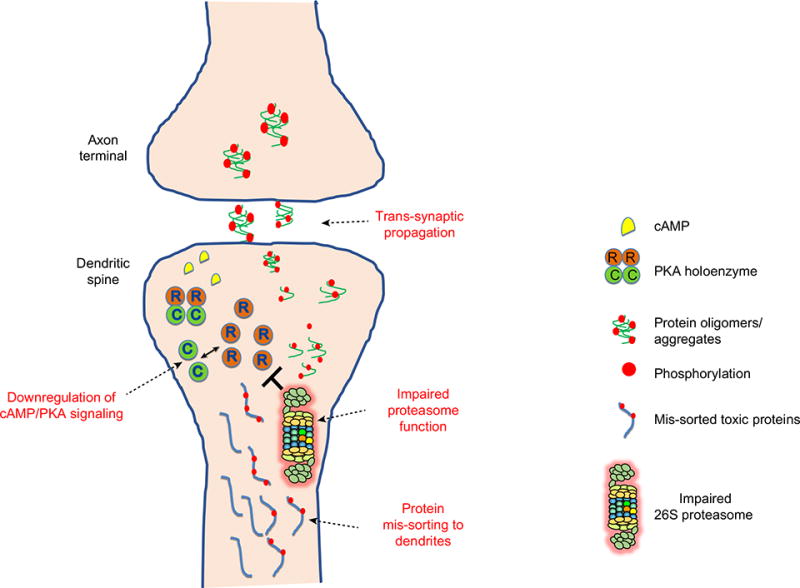
Accumulation of tau in synaptic compartments can occur when tau is mis-sorted from axons to the dendritic compartment and when toxic forms of tau propagate trans-synaptically. The accumulation of oligomers and aggregates is predicted to lead to impaired synaptic proteolysis by physically associating with 26S proteasome complex, thereby impairing proteasome function. The UPS plays an essential role in the synaptic cAMP/PKA cascade because it regulates the turnover of the R subunit of the PKA holoenzyme and the release of the catalytic C subunit. Reduced degradation of the R subunits can result in an increased ratio of the R/C subunits, thus favoring reassociation to inactive PKA holoenzyme and downregulation of PKA signaling.
Recent evidence suggests that stimulation of cAMP second messenger signaling can lead to PKA-mediated phosphorylation of proteasome subunits and enhancement of 26S proteasome function, and this has been shown to facilitate degradation of aggregation-prone proteins in cell lines overexpressing FUS (fused in sarcoma), SOD1 (superoxide dismutase 1), TDP43 (TAR DNA-binding protein 43), or tau [41]. In vivo studies by our group have also shown that inhibiting hydrolysis of cAMP can lead to enhanced proteasome function and attenuation of tauopathy in a mouse model of FTD [25]. This supports the idea that enhancing proteasome function via secondary messengers may potentially help to reduce the burden of aggregates in the brain of patients with neurodegenerative disease.
Activation of Proteasome Function by PDE Inhibitors
The duration and amplitude of the transducing signal induced by second messengers is curtailed by a class of enzymes called cyclic nucleotide phosphodiesterases (PDEs, Box 3) [54,55,59]. Because PDEs can act as negative regulators of cyclic nucleotides, they have been developed as candidate therapies for some neurological disorders, and there are ongoing trials testing their safety and efficacy as cognitive enhancers in AD (clinical trials NCT02491268, NCT02835716, NCT02840279, and NCT02648672) [54], or as neuroprotectant and anti-neuroinflammatory agents in ALS (NCT02714036). PDEs are also being investigated for clinical use in HD [60] and mood disorders [61]. PDEs have been identified as activators of the 26S proteasome [25,41], and this may have broad implications for their potential therapeutic use in neurodegenerative diseases (Figure 1, Key Figure). The first study to show the effect of PDE inhibition on UPS-mediated degradation of disease-related proteins [62,63] demonstrated that administration of either rolipram (a selective PDE4 inhibitor) or purified cell-permeable Uch-L1 (ubiquitin C-terminal hydrolase L1) led to proteasome-mediated degradation in a mouse model of Alzheimer’s-related amyloid deposition [63]. A recent report expanded on earlier studies, demonstrating that PDE4 inhibition by rolipram stimulated PKA, phosphorylated several subunits of 26S proteasome, and rescued impaired proteasome function, leading to facilitated clearance of aggregates of tau ex vivo and in vivo in mice. This led to attenuated tauopathy and reduced cognitive impairment in a mouse model of tauopathy (transgenic line rTg4510) [25] (Figure 1). Furthermore, PDE10 inhibition and activation of PKA in the striatum was shown to have therapeutic effects in a mouse model of HD (line R6/2) because there was a reduction of HTT aggregates [64], increased proteasome function [49], reduced neuropathology [64,65], and alleviation of motor impairment and behavioral deficits [66]. The PDE5 inhibitor, sildenafil, was also reported to activate proteasomes in mouse cardiomyocytes in vivo [44].
Box 3. Human Cyclic Nucleotide Phosphodiesterases.
Human PDEs are structurally related, but they show extensive spatial and functional diversity. There are 11 distinct families of PDEs (PDE1–11) which give rise to almost 100 PDE isoenzyme variants by alternative splicing [59]. PDEs are divided into three broad groups based on their hydrolyzing specificity: cAMP-specific (PDE4, PDE7, and PDE8), cGMP-specific (PDE5, PDE6, and PDE9), and dual-specificity PDEs (PDE1, PDE2, PDE3, PDE10, and PDE11). Almost all PDE isoenzymes are expressed in the CNS, with divergent regional, cellular, and subcellular distributions [55].
Figure 1, Key Figure. Targeting the 26S Proteasome Function To Treat Human Neurodegenerative Diseases.
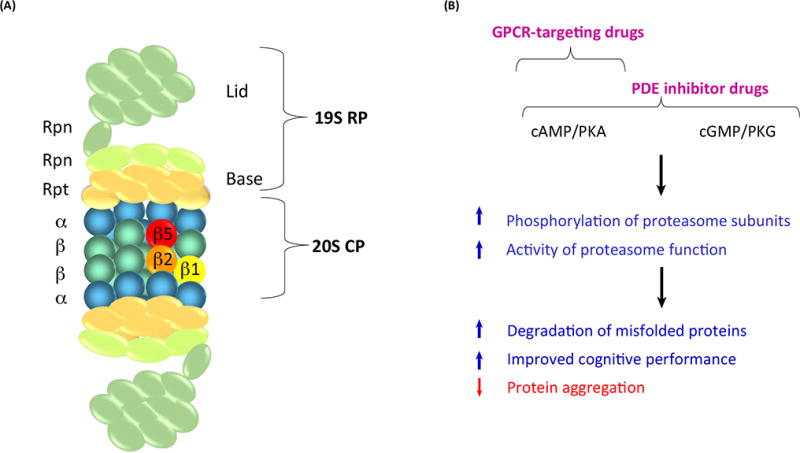
Schematic representation illustrating the mechanism of enhanced proteasome activity. (A) The 26S proteasome is composed of the 20S core particle (CP) and the 19S regulatory particle (RP). The 20S has 28 subunits that form a barrel-shaped structure arranged as four heptameric αββα rings. Three β subunits (β1, β2, and β5) have peptidase activities that hydrolyze proteins into small peptides. Either one or both ends of the 20S proteasome can associate with the 19S regulatory particle to form the 26S proteasome. The 19S RP comprises 19 proteins with different function that have the ability to bind, deubiquitinate, unfold, and translocate polyubiquitinated proteins to the proteolytic chamber of the 20S CP. The 19S RP is further divided into two additional subcomplexes, the ‘base’ and ‘lid’. The base consists of six regulatory particle AAA ATPase subunits (Rpts) as well as four regulatory particle non-ATPase subunits (Rpns). The lid consists of nine different Rpn subunits. (B) A proposed mechanism for enhancing 26S proteasome activity in neurodegenerative diseases is depicted: drugs such as phosphodiesterase (PDE) inhibitors and G protein-coupled receptor (GPCR)-targeted drugs that augment second messenger signaling can lead to phosphorylation of 26S proteasome subunits by PKA or PKG. Phosphorylation of 26S proteasome subunits can stimulate proteasome function and proteasome-mediated protein degradation including toxic misfolded proteins, leading to attenuation of protein aggregation and cognitive decline in patients affected with particular neurodegenerative diseases.
Overall, these studies suggest a new mechanism of action of PDEs, albeit in different models and contexts, whereby PDEs can modulate protein homeostasis through the enhancement of proteasome function. Reports of PDE inhibitor-mediated effects of improved cognition, dampened neuroinflammation, and removal of toxic aggregates in rodent models supports further investigation into their potential use in treating central nervous system (CNS) diseases where proteasome function is impaired. There may be a real opportunity to redirect FDA-approved PDE inhibitors deemed safe for chronic treatment to validate their possible therapeutic use in neurodegenerative diseases. Drugs in this category include the PDE3 inhibitor cilostazol, an antiplatelet drug and vasodilator that works by preventing blood platelets from sticking together, thereby preventing the formation of harmful clots [67], and the PDE5 inhibitor sildenafil which relaxes muscles found in the walls of blood vessels, increasing blood flow to particular areas of the body [68].
Activation of Proteasome Function by GPCR Signaling
GPCRs constitute a large family of membrane proteins that transduce intracellular signals from the extracellular space. Over 90% of GPCRs are expressed in the human brain, with region-and cell type-specific expression patterns [69]. GPCRs regulate signaling pathways involved in a diverse range of events including pain, mood, appetite, vision, the immune system, and cognition. By modulating neurotransmitter release and binding they also play a role in slow synaptic transmission [70,71]. GPCRs are targeted by approximately 30% of FDA-approved drugs [72] and are among the most successful therapeutic targets for diseases including diabetes [73], inflammatory diseases [74], neurodegeneration [75], cardiovascular disease [76], and some psychiatric disorders [77]. Individual GPCRs are structurally similar but they achieve diversity by binding to different isoforms of G proteins (Gs, Gi, Gq/11, G12/13, and Gb/y), thereby activating a multitude of signaling cascades inside cells [72].
GPCR-Mediated Stimulation of Synaptic Proteolysis by the UPS
GPCRs that are coupled to Gs and Gi stimulate or inhibit adenylate cyclase, respectively, and thus regulate cAMP production [75]. Because GPCRs are present in presynaptic and postsynaptic membrane terminals [69], they can regulate cAMP/PKA-mediated UPS protein clearance in synapses. Studies on primary neuronal cultures [37,45,51,78] have shown that calcium-dependent increased neuronal activity can regulate synaptic proteolysis by distributing 26S proteasomes to dendritic spines and by simultaneously upregulating proteasome function via CaMKIIα phosphorylation of the proteasome ATPase subunit Rpt6. Specifically, studies in rats have shown that CaMKIIα-dependent proteasome phosphorylation of Rpt6 is required during long-term memory formation, supporting the idea that enhanced proteolysis by the phosphorylated proteasome is impactful in synapses [45,78].
UPS components are abundant in synaptic terminals, and the UPS is considered to be the canonical pathway for synaptic protein degradation [79,80]. Proteolysis in synaptic terminals has mainly been studied in the context of synaptic plasticity [81,82], where modulation of synaptic strength between presynaptic terminals and postsynaptic dendrites requires swift local protein degradation. Proteolysis has also been studied in the context of physiological synaptic protein degradation [80,83]. In neurodegenerative diseases, where UPS downregulation can be significant, the role of synaptic proteolysis in disease pathogenesis has only recently been recognized. For example, in AD, tau is relocated from its normal location in the axon to the dendritic spines, and tau in this location has been shown to be a target of the UPS [84]. Indeed, synaptoneurosomes derived from patients without AD were found to contain normal non-phosphorylated tau, whereas AD patient-derived synaptoneurosomes were found to accumulate ubiquitinated phospho-tau oligomers, mainly in postsynaptic fractions [85]. Synaptic pathology is also evident in other neurodegenerative diseases; for example, synaptic prion protein deposits [86] have been identified in prion disease brain tissue, and loss of presynaptic terminals and synaptic spine degeneration have been reported in scrapie-infected mice [87,88]. Furthermore, in post-mortem brain tissue samples from patients with PD, α-synuclein-containing aggregates have been observed in the presynaptic compartment of neurons where they are presumed to have a pathological impact [89].
Of relevance to the synaptic location of pathological proteins is the concept of trans-synaptic spread of disease-associated proteins that has been proposed to explain how pathology spreads through the brain in neurodegenerative diseases [90–93]. If toxic proteins that accumulate in synapses drive synaptotoxicity, spread of the disease pathology and progressive worsening of the disease, then therapeutic targets aimed at reducing toxic proteins in pre- or postsynaptic compartments might be able to prevent or attenuate neurodegeneration and slow the cognitive decline associated with progression of the disease.
A novel therapeutic approach aimed at attenuating the spread of pathological proteins between synaptic compartments is the pharmacological modulation of GPCRs present on the membrane of synaptic terminals (Figure 2A). Depending on the GPCR targets, this strategy could be designed to prevent accumulation of proteins in either presynaptic and/or postsynaptic compartments of neurons in affected brain regions. We speculate that compartment-restricted protein degradation via GPCR-mediated cAMP signaling might be regulated by specific anchoring proteins, A-kinase anchor proteins (AKAPs), that bind to and anchor the PKA complex to distinct subcellular compartments, including synapses [94]. Supporting this hypothesis, a report demonstrated in a mouse model of HD (line R6/2) that in vivo chronic stimulation of the A2A receptor resulted in a Gs-coupled response, improved proteasome function by PKA-mediated phosphorylation, and reduced HTT aggregates in striatal synapses [49]. Recently, a study in tau transgenic mice expressing the FTD mutation ΔK280 [95] demonstrated that administration of rolofylline, an antagonist of the Gi-coupled adenosine A1 receptor located on the membrane of synaptic terminals, which can lead to elevated cAMP signaling, restored neuronal activity, and prevented presynaptic impairment and dendritic spine loss [95]. In this study the status of synaptic proteasome function and synaptic tau concentrations were not assessed, but the same improvements in synapses were seen when tau aggregation was inhibited [95]. These findings suggest that enhanced cAMP could mediate the clearance of tau by the UPS [95].
Figure 2. Receptor-Mediated Proteasome Activation: A Putative Strategy To Attenuate Trans-Synaptic Spread in Neurodegenerative Disorders.
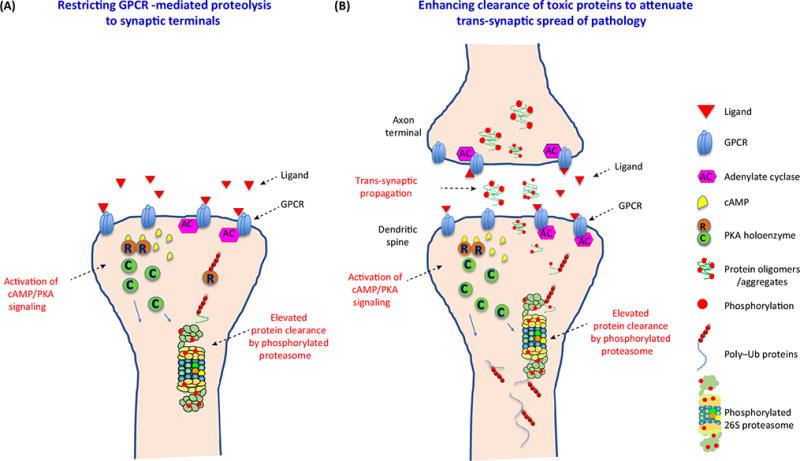
(A) Stimulation of particular GPCRs localized on the membrane of synaptic terminals can result in stimulation of cAMP production by adenylate cyclase (AC). cAMP molecules are able to bind to the R (regulatory) subunits of the PKA tetramer causing tetramer dissociation. In turn, liberated C (catalytic) subunits are now active kinases that can phosphorylate the synaptic 26S proteasome and enhance its function, whereas the R subunits are swiftly degraded by the UPS machinery. (B) GPCR-mediated enhanced synaptic proteolysis by the 26S proteasome can help to clear toxic oligomers that propagate trans-synaptically either before their release in the extracellular space (pre-synaptic compartments) and/or at the site of internalization in postsynaptic compartments. Abbreviations: GPCR, G protein-coupled receptor; Ub, ubiquitin; UPS, ubiquitin proteasome system.
In summary, misfolded, aggregated proteins in synapses that propagate trans-synaptically can impair proteasome function and exacerbate synaptic dysfunction and transmission, which are among the earliest signs of neurodegeneration. In AD, a strong correlation between the extent of synapse loss and the severity of dementia has been documented [96,97]; therefore, intervening with synapse-targeted therapies at an early stage of the illness, including proposed GPCR-targeted drugs, might be able to delay disease progression and prevent overt cognitive decline (Figure 2B).
Concluding Remarks
Drugs that can directly increase 26S proteasome activity can enhance the degradation of pathogenic proteins that have the propensity to aggregate and propagate, and they may therefore be effective therapeutic agents for the treatment of particular neurodegenerative diseases (see Outstanding Questions and Box 4). The proteasome represents a ‘druggable’ target because the first FDA-approved proteasome inhibitor, velcade (bortezomib), an anticancer agent, is now used as a frontline treatment for multiple myeloma and mantle cell lymphoma [98]. Furthermore, the first small-molecule proteasome activators (USP14 inhibitors) [33] are currently in preclinical testing. Because several studies have shown that proteasome activity is phosphorylation-dependent, and that cAMP/cGMP second messenger signaling can stimulate proteasome activity via phosphorylation of proteasome subunits, there are opportunities to develop a wide range of drugs targeting this pathway. Experimental and FDA-approved drugs including PDE inhibitors and GPCR targeting agents are currently available, and we propose that there is merit in testing these classes of drugs, first in preclinical and then in clinical settings (Figure 1). Moreover, the precise, and in many cases restricted, distribution of PDEs and GPCRs makes these targets attractive candidates for the degradation of disease-specific proteins in selected brain regions, cells, or subcellular compartments. This might be uniquely valuable for targeting a drug to a particular disease, or for restricting its action to a vulnerable cell compartment such as a pre- or postsynaptic compartment, which might be particularly effective in combating the spread of pathology through the brain or in alleviating synaptic deficits. We are fortunate to have such a deep reservoir of drugs to choose from to test these potentially game-changing ideas.
Outstanding Questions.
Can misfolded proteins that resist degradation by the UPS cause fundamental changes in the quaternary structure of the 26S complex, leading to persistent deterioration of its subunit organization and its function?
How do multiple kinases that phosphorylate different subunits of the 26S proteasome exert a similar effect on facilitated protein breakdown?
Does site-specific phosphorylation by various kinases result in various degrees of enhanced protein degradation?
Can we design proteasome-targeted therapies to stop transcellular and trans-synaptic propagation of pathogenic proteins by stimulating protein breakdown in specific focal areas of neurons where propagating proteins spread?
Is there feedback regulation between the cAMP/PKA pathway and proteasome-mediated degradation in neurodegenerative diseases? This would imply that modulation of one pathway might impact the other pathway.
Box 4. Clinician’s Corner.
Preventing the aggregation and spread of disease-causing proteins between brain regions during the earliest stage of a neurodegenerative disease might extend the asymptomatic phase and delay (or even halt) disease progression, preventing the devastating cognitive loss that is debilitating for patients and their caregivers.
Enhancing proteasome function by stimulating cAMP or cGMP signaling is a novel putative approach to remove toxic proteins from cells. The mechanism could be exploited for clinical use against neurodegenerative diseases, with the potential to repurpose existing FDA-approved drugs such as PDE inhibitors, cAMP agonists, and GPCR-targeted drugs for rapid clinical impact. The latter represent ~34% of FDA-approved drugs [72] and pharma companies are actively studying small-molecule drugs that can interact with GPCRs as ligands, either as orthosteric or allosteric modulators. The link between GPCRs and AD is relevant because emerging mechanistic insight suggests that this class of receptors may be used to target protein aggregates and treat AD.
Highlights.
Functional changes in the ubiquitin proteasome system may constitute a contributing factor to the pathogenesis of neurodegenerative diseases.
Until recently there has been a lack of pharmacological means to activate proteasome function to clear toxic misfolded proteins.
The notion that the 26S proteasome complex is a passive peptidase of ubiquitin-conjugating machinery has been modified by recent studies demonstrating that activating solely the 26S proteasome can determine the output of protein degradation, especially the output of toxic proteins.
Phosphorylation of proteasome subunits is a positive regulatory mechanism of proteasome-mediated protein degradation.
Drugs that stimulate of cAMP/PKA and cGMP/PKG, such as PDE inhibitors and GPCR-targeted agents can activate the functions of the 26S proteasome in vivo.
Rescuing/activating 26S proteasome function might constitute a putative converging strategy for treating clinically and etiologically diverse neurodegenerative diseases.
Acknowledgments
This work was supported by grants to N.M. (National Institutes of Health, NIH, K01 AG055694, CurePSP, and the Alzheimer’s Association) and K.E.D (NIH RF1 AG056151 and the Cure Alzheimer’s Fund).
Glossary
- A-kinase anchor proteins (AKAPs)
a group of structurally diverse proteins with the common function of binding to the regulatory subunit of PKA and confining the holoenzyme to discrete locations within the cell
- Allosteric modulators
molecules that bind to a site distinct from the orthosteric binding site while still allowing the possibility of orthosteric binding
- Alzheimer’s disease (AD)
a common neurodegenerative disease characterized clinically by progressive deterioration of memory. Pathologically, AD is characterized by extracellular deposits of amyloid-β peptides forming senile plaques and by the intracellular accumulation of hyperphosphorylated conformationally abnormal aggregates of tau into neurofibrillary tangles (NFTs)
- Amyotrophic lateral sclerosis (ALS)
a progressive and fatal adult motor neuron disease characterized by progressive loss of upper and lower motor neurons. In patients, the disease manifests with progressive muscular atrophy, loss of strength, paralysis, and death. ALS is characterized by the aggregation of ubiquitinated proteins in affected motor neurons. Recent studies have identified several new molecular constituents of ALS-linked cellular aggregates, including FUS, TDP-43, OPTN, UBQLN2, and the translational product of intronic repeats in the gene C9ORF72
- Cryo-electron microscopy tomography
an imaging technique in which an electron microscope is used to record a series of 2D images at high resolution (~4 nm) of a biological sample held at cryogenic temperatures. Using computational methods, the 2D images can then be aligned to yield a 3D (tomographic) reconstruction of the sample
- Dendritic spines
small protrusions arising from the dendritic shaft of various types of neurons and receiving inputs from excitatory axons
- Frontotemporal lobe dementias (FTDs)
a family of disorders where the clinical features and the pathology involve populations of neurons in the frontal and/or temporal lobes on one or both sides of the brain. The features include progressive language dysfunction, aphasia, behavioral changes, and deterioration of cognitive function. Neuropathologically, FTD includes three histological subtypes: FTD-tau, FTD-TDP-43, and FTD-FUS
- Huntington’s disease (HD)
a rare neurodegenerative disorder of the central nervous system (CNS) characterized by choreatic movements, behavioral and psychiatric disturbances, and dementia. The disease is caused by an expanded CAG repeat in the huntingtin (HTT) gene and, as a result, the translated protein huntingtin (HTT) contains disease-causing expansions of glutamines (polyQ) that make it prone to misfolding and aggregation into inclusion bodies in neurons
- Neurodegenerative diseases
an umbrella term for a heterogeneous group of diseases affecting the CNS; characterized by the progressive degeneration of the structure and function of brain cells, usually neurons. Common neurodegenerative diseases include AD, FTD, PD, HD, and ALS
- Neurofibrillary tangles (NFTs)
intracellular aggregates of hyperphosphorylated and misfolded tau protein, most commonly known as one of the markers of AD. These aggregates are also found in numerous other diseases known as tauopathies
- Orthosteric binding
the primary, unmodulated binding site on a receptor
- Paired helical filaments (PHFs)
abnormal folding of tau protein leads to the generation of PHFs. In the PHFs of AD, small segments of tau adopt an α-conformation and interact with other tau molecules to form filamentous structures
- Parkinson’s disease (PD)
a chronic and progressive neurodegenerative disorder characterized by motor impairment, autonomic dysfunction, and, in many, psychological and cognitive changes. Pathologically, PD is characterized by the relatively selective loss of dopaminergic neuronal cells in the substantia nigra pars compacta and by the presence of Lewy bodies and Lewy neurites in surviving affected neurons. α-Synuclein is the main component of Lewy bodies and Lewy neurites
- Protein homeostasis
the balancing dynamic network between protein translation, folding, trafficking, and degradation within and outside the cell
- Proteinopathies
neurodegenerative diseases in which specific proteins become structurally abnormal, resist clearance, and aggregate, thereby disrupting the function of cells, tissues, and organs
- Proteotoxicity
impairment of cell function caused by protein misfolding and aggregation
- Prion disease
prions are infectious agents composed of misfolded prion protein (PrPSc) organized in a variety of aggregates. When a prion enters a healthy organism it induces existing properly folded proteins to convert into the misfolded prion form
- Quaternary structure
the 3D arrangement of multiple folded protein subunits in a multisubunit complex such as the 26S proteasome or its components: the 19S regulatory particle and 20S core proteasome
- Repurposing of drugs
the application of an existing therapeutic to a new disease indication. This approach builds upon previous research and prior clinical testing; new therapies could quickly be ready for clinical trials testing a new indication and at a lower cost.
- Synaptic transmission
neurons communicate with each other through two mechanisms: fast and slow synaptic transmission. Fast synaptic transmission occurs in less than 1/1000 of a second and is attributable to the ability of the fast-acting neurotransmitters to open ligand-operated ion channels present in the plasma membrane of postsynaptic cells. Slow synaptic transmission occurs over periods of hundreds of milliseconds to minutes, whereby slow-acting neurotransmitters bind to their receptors and change the level of a second messenger (e.g., cAMP and cGMP). Slow synaptic transmission is mediated by GPCRs
- Synaptic proteolysis
degradation of synaptic proteins within synaptic compartments
- Synaptoneurosomes
isolated synaptic terminals from neurons. Synaptoneurosomes are obtained by mild homogenization of brain tissue under isotonic conditions and subsequent subcellular fractionation using differential and density-gradient centrifugation
- α-Synuclein
a small soluble protein that is present primarily at presynaptic terminals. Its function is not well understood, but studies suggest that it plays a role in maintaining a supply of synaptic vesicles in presynaptic terminals. It forms insoluble fibrils in pathological conditions such as PD, dementia with Lewy bodies, and multiple system atrophy, namely synucleinopathies
- Tau
a microtubule-associated protein that stabilizes neuronal microtubules, promoting axonal outgrowth. It is a natively unfolded protein and is highly soluble, showing little tendency for aggregation. However, in several neurodegenerative diseases tau accumulates into oligomeric or aggregated forms with different conformations such as paired helical filaments and straight or twisted ribbons, depending on the ratio of tau isoforms that are present. Misfolded post-translationally modified tau is a major component of NFTs and neuropil threads. Tau aggregates are characteristic of several neurodegenerative diseases collectively known as tauopathies
- Ubiquitin proteasome system (UPS)
a major system responsible for the degradation of intracellular proteins in eukaryotes. By controlling the levels of key proteins, it regulates almost all cellular activities, including cell-cycle progression, DNA replication and repair, transcription, protein quality control, immune responses, and apoptosis. The UPS includes the ubiquitination system, that marks proteins for degradation, and the proteasome which degrades the ubiquitinated proteins
References
- 1.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 2.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med. 2014;20:130–138. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci. 2017;11:185. doi: 10.3389/fnins.2017.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins GA, Goldberg AL. The logic of the 26S proteasome. Cell. 2017;169:792–806. doi: 10.1016/j.cell.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 2005;12:1191–1197. doi: 10.1038/sj.cdd.4401702. [DOI] [PubMed] [Google Scholar]
- 7.Mori H, et al. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- 8.Blokhuis AM, et al. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:777–794. doi: 10.1007/s00401-013-1125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giasson BI, Lee VM. Are ubiquitination pathways central to Parkinson’s disease? Cell. 2003;114:1–8. doi: 10.1016/s0092-8674(03)00509-9. [DOI] [PubMed] [Google Scholar]
- 10.Bennett EJ, et al. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg AL. On prions, proteasomes, and mad cows. N Engl J Med. 2007;357:1150–1152. doi: 10.1056/NEJMcibr073962. [DOI] [PubMed] [Google Scholar]
- 12.DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 13.Keller JN, et al. Impaired proteasome function in Alzheimer’s disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 14.Seo H, et al. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann Neurol. 2004;56:319–328. doi: 10.1002/ana.20207. [DOI] [PubMed] [Google Scholar]
- 15.Bukhatwa S, et al. A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson’s disease, multiple system atrophy and progressive supranuclear palsy. Brain Res. 2010;1326:174–183. doi: 10.1016/j.brainres.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 16.McNaught KS, et al. Altered proteasomal function in sporadic Parkinson’s disease. Exp Neurol. 2003;179:38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- 17.Alghamdi A, et al. Reduction of RPT6/S8 (a proteasome component) and proteasome activity in the cortex is associated with cognitive impairment in Lewy body dementia. J Alzheimers Dis. 2017;57:373–386. doi: 10.3233/JAD-160946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kabashi E, et al. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:367–371. doi: 10.3109/17482968.2012.686511. [DOI] [PubMed] [Google Scholar]
- 19.Tashiro Y, et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem. 2012;287:42984–42994. doi: 10.1074/jbc.M112.417600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gestwicki JE, Garza D. Protein quality control in neurodegenerative disease. Prog Mol Biol Transl Sci. 2012;107:327–353. doi: 10.1016/B978-0-12-385883-2.00003-5. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka K, Matsuda N. Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim Biophys Acta. 2014;1843:197–204. doi: 10.1016/j.bbamcr.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Deriziotis P, et al. Misfolded PrP impairs the UPS by interaction with the 20S proteasome and inhibition of substrate entry. EMBO J. 2011;30:3065–3077. doi: 10.1038/emboj.2011.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kristiansen M, et al. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26:175–188. doi: 10.1016/j.molcel.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Andre R, Tabrizi SJ. Misfolded PrP and a novel mechanism of proteasome inhibition. Prion. 2012;6:32–36. doi: 10.4161/pri.6.1.18272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myeku N, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP–PKA signaling. Nat Med. 2016;22:46–53. doi: 10.1038/nm.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keck S, et al. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- 27.Snyder H, et al. Aggregated and monomeric alpha-synuclein bind to the S60 proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 28.Lindersson E, et al. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- 29.Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 30.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 31.Berger Z, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 32.Lee BH, et al. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature. 2016;532:398–401. doi: 10.1038/nature17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boselli M, et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem. 2017;292:19209–19225. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Homma T, et al. Ubiquitin-specific protease 14 modulates degradation of cellular prion protein. Sci Rep. 2015;5:11028. doi: 10.1038/srep11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marambaud P, et al. Protein kinase A phosphorylation of the proteasome: a contribution to the alpha-secretase pathway in human cells. J Neurochem. 1996;67:2616–2619. doi: 10.1046/j.1471-4159.1996.67062616.x. [DOI] [PubMed] [Google Scholar]
- 37.Djakovic SN, et al. Regulation of the proteasome by neuronal activity and calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bingol B, et al. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 39.Hamilton AM, et al. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 2012;74:1023–1030. doi: 10.1016/j.neuron.2012.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myeku N, et al. cAMP stimulates the ubiquitin/proteasome pathway in rat spinal cord neurons. Neurosci Lett. 2012;527:126–131. doi: 10.1016/j.neulet.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lokireddy S, et al. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A. 2015;112:E7176–E7185. doi: 10.1073/pnas.1522332112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang F, et al. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem. 2007;282:22460–22471. doi: 10.1074/jbc.M702439200. [DOI] [PubMed] [Google Scholar]
- 43.Lu H, et al. Revealing the dynamics of the 20 S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach. Mol Cell Proteomics. 2008;7:2073–2089. doi: 10.1074/mcp.M800064-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ranek MJ, et al. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128:365–376. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarome TJ, et al. CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front Behav Neurosci. 2013;7:115. doi: 10.3389/fnbeh.2013.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asai M, et al. PKA rapidly enhances proteasome assembly and activity in in vivo canine hearts. J Mol Cell Cardiol. 2009;46:452–462. doi: 10.1016/j.yjmcc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Zong C, et al. Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ Res. 2006;99:372–380. doi: 10.1161/01.RES.0000237389.40000.02. [DOI] [PubMed] [Google Scholar]
- 48.Guo X, et al. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat Cell Biol. 2016;18:202–212. doi: 10.1038/ncb3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin JT, et al. Regulation of feedback between protein kinase A and the proteasome system worsens Huntington’s disease. Mol Cell Biol. 2013;33:1073–1084. doi: 10.1128/MCB.01434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marquez-Lona EM, et al. Phosphorylation of the 19S regulatory particle ATPase subunit, Rpt6, modifies susceptibility to proteotoxic stress and protein aggregation. PLoS One. 2017;12:e0179893. doi: 10.1371/journal.pone.0179893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Djakovic SN, et al. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci. 2012;32:5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo X, et al. UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc Natl Acad Sci U S A. 2011;108:18649–18654. doi: 10.1073/pnas.1113170108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asano S, et al. A molecular census of 26S proteasomes in intact neurons. Science. 2015;347:439–442. doi: 10.1126/science.1261197. [DOI] [PubMed] [Google Scholar]
- 54.Knott EP, et al. Phosphodiesterase inhibitors as a therapeutic approach to neuroprotection and repair. Int J Mol Sci. 2017;18:E696. doi: 10.3390/ijms18040696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kleppisch T. Phosphodiesterases in the central nervous system. Handb Exp Pharmacol. 2009:71–92. doi: 10.1007/978-3-540-68964-5_5. [DOI] [PubMed] [Google Scholar]
- 56.Zhang P, et al. Structure and allostery of the PKA RIIbeta tetrameric holoenzyme. Science. 2012;335:712–716. doi: 10.1126/science.1213979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hegde AN, DiAntonio A. Ubiquitin and the synapse. Nat Rev Neurosci. 2002;3:854–861. doi: 10.1038/nrn961. [DOI] [PubMed] [Google Scholar]
- 58.Osborne BW, et al. Crystal structure of cGMP-dependent protein kinase reveals novel site of interchain communication. Structure. 2011;19:1317–1327. doi: 10.1016/j.str.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Menniti FS, et al. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 60.Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: what’s in the pipeline? Mov Disord. 2014;29:1434–1445. doi: 10.1002/mds.26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt CJ, et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325:681–690. doi: 10.1124/jpet.107.132910. [DOI] [PubMed] [Google Scholar]
- 62.Vitolo OV, et al. Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smith DL, et al. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci U S A. 2009;106:16877–16882. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giampa C, et al. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PLoS One. 2010;5:e13417. doi: 10.1371/journal.pone.0013417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beaumont V, et al. Phosphodiesterase 10A inhibition improves cortico-basal ganglia function in Huntington’s disease models. Neuron. 2016;92:1220–1237. doi: 10.1016/j.neuron.2016.10.064. [DOI] [PubMed] [Google Scholar]
- 66.Harada A, et al. TAK-063, a novel phosphodiesterase 10A inhibitor, protects from striatal neurodegeneration and ameliorates behavioral deficits in the R6/2 mouse model of Huntington’s disease. J Pharmacol Exp Ther. 2017;360:75–83. doi: 10.1124/jpet.116.237388. [DOI] [PubMed] [Google Scholar]
- 67.Bedenis R, et al. Cilostazol for intermittent claudication. Cochrane Database Syst Rev. 2014:CD003748. doi: 10.1002/14651858.CD003748.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwarz ER, et al. The effects of chronic phosphodiesterase-5 inhibitor use on different organ systems. Int J Impot Res. 2007;19:139–148. doi: 10.1038/sj.ijir.3901491. [DOI] [PubMed] [Google Scholar]
- 69.Huang Y, Thathiah A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015;589:1607–1619. doi: 10.1016/j.febslet.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 70.Betke KM, et al. GPCR mediated regulation of synaptic transmission. Prog Neurobiol. 2012;96:304–321. doi: 10.1016/j.pneurobio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerber KJ, et al. Roles for regulator of G protein signaling proteins in synaptic signaling and plasticity. Mol Pharmacol. 2016;89:273–286. doi: 10.1124/mol.115.102210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hauser AS, et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oh DY, Olefsky JM. G protein-coupled receptors as targets for anti-diabetic therapeutics. Nat Rev Drug Discov. 2016;15:161–172. doi: 10.1038/nrd.2015.4. [DOI] [PubMed] [Google Scholar]
- 74.Du C, Xie X. G protein-coupled receptors as therapeutic targets for multiple sclerosis. Cell Res. 2012;22:1108–1128. doi: 10.1038/cr.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang Y, et al. The role of GPCRs in neurodegenerative diseases: avenues for therapeutic intervention. Curr Opin Pharmacol. 2017;32:96–110. doi: 10.1016/j.coph.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 76.Belmonte SL, Blaxall BC. Conducting the G-protein coupled receptor (GPCR) signaling symphony in cardiovascular diseases: new therapeutic approaches. Drug Discov Today Dis Models. 2012;9:e85–e90. doi: 10.1016/j.ddmod.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Komatsu H. Novel therapeutic GPCRs for psychiatric disorders. Int J Mol Sci. 2015;16:14109–14121. doi: 10.3390/ijms160614109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jarome TJ, et al. CaMKII regulates proteasome phosphorylation and activity and promotes memory destabilization following retrieval. Neurobiol Learn Mem. 2016;128:103–109. doi: 10.1016/j.nlm.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 80.Cohen LD, Ziv NE. Recent insights on principles of synaptic protein degradation. F1000Research. 2017;6:675. doi: 10.12688/f1000research.10599.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hegde AN. Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog Neurobiol. 2004;73:311–357. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 82.Hegde AN. Proteolysis, synaptic plasticity and memory. Neurobiol Learn Mem. 2017;138:98–110. doi: 10.1016/j.nlm.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.tom Dieck S, et al. Direct visualization of newly synthesized target proteins in situ. Nat Methods. 2015;12:411–414. doi: 10.1038/nmeth.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tai HC, et al. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kitamoto T, et al. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt–Jakob disease. Am J Pathol. 1992;140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- 87.Jeffrey M, et al. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol Appl Neurobiol. 2000;26:41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 88.Belichenko PV, et al. Dendritic and synaptic alterations of hippocampal pyramidal neurones in scrapie-infected mice. Neuropathol Appl Neurobiol. 2000;26:143–149. doi: 10.1046/j.1365-2990.2000.026002143.x. [DOI] [PubMed] [Google Scholar]
- 89.Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brettschneider J, et al. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16:109–120. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hardy J, Revesz T. The spread of neurodegenerative disease. N Engl J Med. 2012;366:2126–2128. doi: 10.1056/NEJMcibr1202401. [DOI] [PubMed] [Google Scholar]
- 92.Stopschinski BE, Diamond MI. The prion model for progression and diversity of neurodegenerative diseases. Lancet Neurol. 2017;16:323–332. doi: 10.1016/S1474-4422(17)30037-6. [DOI] [PubMed] [Google Scholar]
- 93.Walker LC, et al. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70:304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanderson JL, Dell’Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dennissen FJ, et al. Adenosine A1 receptor antagonist rolofylline alleviates axonopathy caused by human Tau DeltaK280. Proc Natl Acad Sci U S A. 2016;113:11597–11602. doi: 10.1073/pnas.1603119113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scheff SW, et al. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 97.Masliah E, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 98.Raedler L. Velcade (Bortezomib) receives 2 new FDA indications: for retreatment of patients with multiple myeloma and for first-line treatment of patients with mantle-cell lymphoma. Am Health Drug Benefits. 2015;8:135–1340. [PMC free article] [PubMed] [Google Scholar]
- 99.Livneh I, et al. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 2016;26:869–885. doi: 10.1038/cr.2016.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Terry RD, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 101.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 102.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]