

Abstract
β-lactamases are enzymes which are commonly produced by bacteria and which degrade the β-lactam ring of β-lactam antibiotics, namely penicillins, cephalosporins, carbapenems, and monobactams, and inactivate these antibiotics. We performed a rational and comprehensive investigation of β-lactamases in different biological databases. In this study, we constructed hidden Markov model profiles as well as the ancestral sequence of four classes of β-lactamases (A, B, C, and D), which were used to identify potential β-lactamases from environmental metagenomic (1206), human microbiome metagenomic (6417), human microbiome reference genome (1310), and NCBI’s nonredundant databases (44101). Our analysis revealed the existence of putative β-lactamases in the metagenomic databases, which appeared to be similar to the four different molecular classes (A–D). This is the first report on the large-scale phylogenetic diversity of new members of β-lactamases, and our results revealed that metagenomic database dark-matter contains β-lactamase-like antibiotic resistance genes.
Keywords: β-lactam antibiotics, β-lactamase, ancestral sequence reconstruction, metagenome, antibiotic resistance, HMM profile
Introduction
β-lactamases are widely distributed in the bacterial domain (Hall and Barlow 2004; Wright 2007) and are responsible for resistance towards β-lactam antibiotics, such as penicillins, cephalosporins, carbapenems, and monobactams. The number of unique, naturally occurring β-lactamases exceeds 1,300 (Bush 2013). Currently, β-lactamase enzymes are classified according to two methods: 1) molecular classification (Ambler 1980) and 2) functional classification (Bush 1989, 2013). Molecular classification is based on conserved and unique amino acid motifs, which divide β-lactamases into classes A, B, C, and D (Ambler 1980). Classes A, C, and D are serine β-lactamases, which have serine at their active site, whereas class B includes zinc (Zn) dependent metalloenzymes (metallo-β-lactamase) that show a hydrolytic mechanism which is different to that of serine β-lactamases (Bush et al. 1995). The metallo-β-lactamases hydrolyze β-lactam rings using Zn metal ions (Philippon et al. 1998). Functional classification is based on substrate profile inhibitor and divides β-lactamases into functional groups, namely 2a, 2 b, and 2be (Bush 1989, 2013). For example, class A sequences have been classified into multiple functional groups, such as group 2 b which includes penicillinases, groups 2c which include penicillinases and some cephalosporinases, group 2e which includes cephalosporinases, group 2be which includes third-generation cephalosporins extended-spectrum β-lactamases, group 2br includes β-lactamase inhibitor resistance, and group 2f includes serine carbapenemases (Bush 2013). The molecular class C β-lactamases (cephalosporinases) includes the largest serine β-lactamases in comparison with class A and D (Bush 2013). Class D β-lactamases hydrolyze oxacillin and have a distinct structural architecture in comparison with other serine β-lactamases (Paetzel et al. 2000). β-lactamases were proposed to exist in nature before the discovery of modern antibiotics (D’Costa et al. 2011). For instance, serine β-lactamase enzymes were estimated to originate over two billion years ago (Hall and Barlow 2004). However, the increased use of β-lactam antibiotics in recent decades has led to the evolution of many new β-lactamase generations, which are believed to have spread antibiotic resistance (Wilke et al. 2005). The spread of genes and mutations is also believed to have conferred resistance to β-lactam antibiotics and increased the evolution of β-lactamases (Heritage et al. 1999).
Metagenomic research has been a powerful tool for the discovery of novel biocatalysts and other valuable biomolecules (Iqbal et al. 2012; Rabausch et al. 2013). However, in the context of β-lactamase enzymes, environmental, and human metagenomic databases have not yet been adequately explored. To date, little is known about the environmental reservoirs of antibiotic resistance genes and even less is known about the potential sources of β-lactamases. Identifying the source of resistant genes and tracking their evolutionary history will promote efforts to combat antibiotic resistance among human pathogens. With this in mind, the current study was conducted to identify novel β-lactamases parsed in different biological databases. We then addressed the question of the existence of β-lactamases in metagenomic databases, followed by the analysis of their phylogenetic diversity. To identify new putative β-lactamases, we performed large-scale metagenome mining with the help of reconstructed putative ancestral sequences and hidden Markov model (HMM)-based profiles of extant enzymes. These two methods were able to identify numerous β-lactamases from biological databases.
Materials and Methods
Figure 1 shows an overview of the current analysis pipeline.
Fig. 1.

—Schematic view of the protocol used to identify putative β-lactamases from biological databases. Putative β-lactamase sequences were identified using the HMM profile search and reconstructed ancestral sequence-based BLAST analysis.
Retrieval of β-Lactamase Sequences
Protein sequences of β-lactamase enzymes were obtained from Antibiotic Resistance Gene-ANNOTation (ARG-ANNOT) database (Gupta et al. 2014). A total of 1,155 sequences (bacterial sequences) were retrieved, which were then classified into four Ambler molecular classes using available literature and resources in the public domain. These sequences were categorized as 620 in Class A, 174 in Class B, 151 in Class C, and 210 in Class D.
Homologous Sequence Detection from Biological Databases
In order to identify of homologous sequences from sequence databases, we used two methods; a BLAST search analysis with the reconstructed ancestral sequences and an HMM profile search. Ancestral sequences and HMM profiles were generated for each class of β-lactamases (A–D) separately. We used ancestral sequences and HMM profiles as a probe/query for homologous sequence similarity searches in biological sequence databases. In supplementary file S1, Supplementary Material online, we show that ancestral sequences are more efficient than extant enzymes, and the BLAST search analysis of extant enzymes retrieved significant hits which were completely covered by BLAST hits of the ancestral sequence. We selected potential homologous sequences under the following BLAST settings: ≥30% sequence identity, ≥50% query length coverage, and an e-value cut-off of 1e-10.
HMM profiles of extant β-lactamases were generated using HMMER3 (Eddy et al. 2011) software. A profile is a description of the consensus of a multiple sequence alignment and uses a position-specific scoring system to capture information about the degree of conservation at different position in the alignment. The maximum-likelihood multiple sequence alignment of each class of β-lactamases were subjected to HMMER3 using the “hmmbuild” command. The HMM profiles of four classes of β-lactamases were then queried against biological sequence databases (for a homologous sequence search) using the “hmmsearch” command. An e-value of 1e-10 was adopted during the HMM profile search.
To identify novel β-lactamases, four biological databases, environmental protein sequence database (env_nr), the human microbiome project reference genome (HMP-HMRGD), the human microbiome project metagenomic database (HMP-HMASM), and the NCBI nonredundant (nr) database were considered. At the time of analysis (September 2015), there were 6,891,928 protein sequences in the env_nr database, 78,625,685 protein sequences in the NCBI nr database, 2,537,354 protein sequences in the HMP reference genome database, and 75,199,622 (predicted ORF) protein sequences in the HMP metagenome database.
To detect homologous sequences in the human microbiome database, 50 million assembled metagenomic scaffolds were downloaded. These sequences were associated with 16 body sites (anterior nares, attached keratinized gingiva, buccal mucosa, hard palate, left retroauricular crease, midvagina, palatine tonsils, posterior fornix, right retroauricular crease, saliva, stool, subgingival plaque, supragingival plaque, throat, tongue dorsum, and vaginal introitus). Potential protein coding regions (ORFs) were predicted using the getorf command in EMBOSS (Rice et al. 2000). ORFs with internal stop codons and encoding proteins shorter than 100 amino acids were removed. The remaining protein sequences (75,199,622) were used for subsequent analysis.
Identifying the Domain Composition of Putative β-Lactamases
All the candidate sequences identified through the HMM profile search and ancestral sequences were queried against the Pfam release 30.0 web interface (Bateman et al. 1999) and the NCBI’s Conserved Domain Database (Marchler-Bauer 2011; Marchler-Bauer et al. 2015). The Pfam and CDD were executed with the default setting on their respective websites.
BLAST Search Analysis against PATRIC Database
All the selected putative β-lactamases were queried in a protein BLAST search analysis against the PATRIC (Pathosystems Resource Integration Center) database (Wattam et al. 2014) to detect of antibiotic resistance genes. A total of 90,597 sequences were obtained from the PATRIC database. We selected potential sequences under the following BLAST settings: ≥30% sequence identity, ≥50% query length coverage, and an e-value cut-off of 1e-10.
The protein BLAST search analysis of all identified putative sequences against the PATRIC database indicates that all these sequences are antibiotic resistance genes (supplementary table S1, Supplementary Material online).
Comprehensive Phylogenetic Tree Building
To explore the phylogenomic diversity of β-lactamases, we first generated a phylogenetic tree of extant β-lactamases. The phylogenetic tree of extant enzymes of class A (620 sequences), B (174 sequences), C (151 sequences), and D (210 sequences) grouped these sequences into clades: a-1 to a-5, b-1 to b-4, c-1 to c-2, and d-1 to d-4 known clades, respectively (supplementary file S1 and figs. S1–S4, Supplementary Material online). We considered a total of 126, 30, 27, and 33 sequences from classes A–D as being representative for further large-scale phylogenetic tree analysis.
Next, we performed a multiple sequence alignment of putative homologous sequences (identified through an HMM profile search and ancestral sequence BLAST search analysis) and extant β-lactamases using the MUSCLE algorithm. Multiple sequence alignment trimming was performed using the trimAL, which removes poorly aligned regions (Capella et al. 2009). Approximate maximum-likelihood phylogenetic trees were constructed using the WAG substitution model in FastTree (Price et al. 2010) and visualized with FigTree-v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/; last accessed July 5, 2017).
Results
Homologous Sequence Identification and Phylogenetic Tree Construction
For the comprehensive identification of homologous sequences, we executed three types of analysis: a BLAST search analysis with the extant β-lactamases, a BLAST search analysis with the reconstructed ancestral sequences, and an HMM profile search in four biological databases (details can be found in Materials and Methods). In the present study, we found numerous putative β-lactamases through the HMM profile search, as compared with the extant and ancestral sequence-based BLAST search analyses. Moreover, we noticed that most (>95%) of the significant putative homologues (detected by the ancestral and extant BLAST) were also identified by the HMM profile search. Therefore, in this study, we mainly discuss the results obtained from the HMM profile search, and present ancestral sequence-based results when novel hits were obtained.
Class A
The HMM profile search of class A β-lactamases in four biological databases retrieved a total of 13,720 (env_nr: 298, HMP metagenome: 2812, HMP reference genome: 261, NCBI nr: 10349) significant putative β-lactamase-like sequences, which contain β-lactamase functional characteristics (functional β-lactamase domain, and characteristic amino acid motifs) (supplementary table S1, Supplementary Material online). The presence of the β-lactamase domain suggests that these are possibly functional β-lactamases.
In contrast, the BLAST search analysis of reconstructed ancestral sequences was able to identify a total of 10,182 (env_nr: 160, HMP metagenome: 1886, HMP reference genome: 143, NCBI nr: 7993) significant putative β-lactamase-like sequences (supplementary table S1, Supplementary Material online). Of these sequences, 97% (9,889) were detected through the HMM profile search and only 3% (293) from the NCBI nr database were found to be unique and not detected by the HMM profile search. This finding indicates that the HMM profile search was able to detect a greater number of sequences in comparison to the ancestral sequence-based BLAST search. Next, we removed the redundant sequences from both results and selected the representative sequences for further multiple sequence alignment and phylogenetic tree construction. The multiple sequence alignment of extant and newly identified sequences showed conserved motifs S69XXK, S131DN, and K235XG (supplementary file S2, Supplementary Material online). Here, “X” represents sequences that can accommodate multiple substitutions.
To understand the relationship between extant and novel sequences, a large-scale phylogenetic tree was constructed to decipher the overall affiliations of putative new and extant β-lactamases. The midpoint rooted phylogenetic tree of class A contains representative extant and putative new sequences, which delineated two major novel clades (labeled as A-1 and A-2) (fig. 2). Since these clades are clustered separately without any extant enzymes, they were considered as “novel clades”.
Fig. 2.
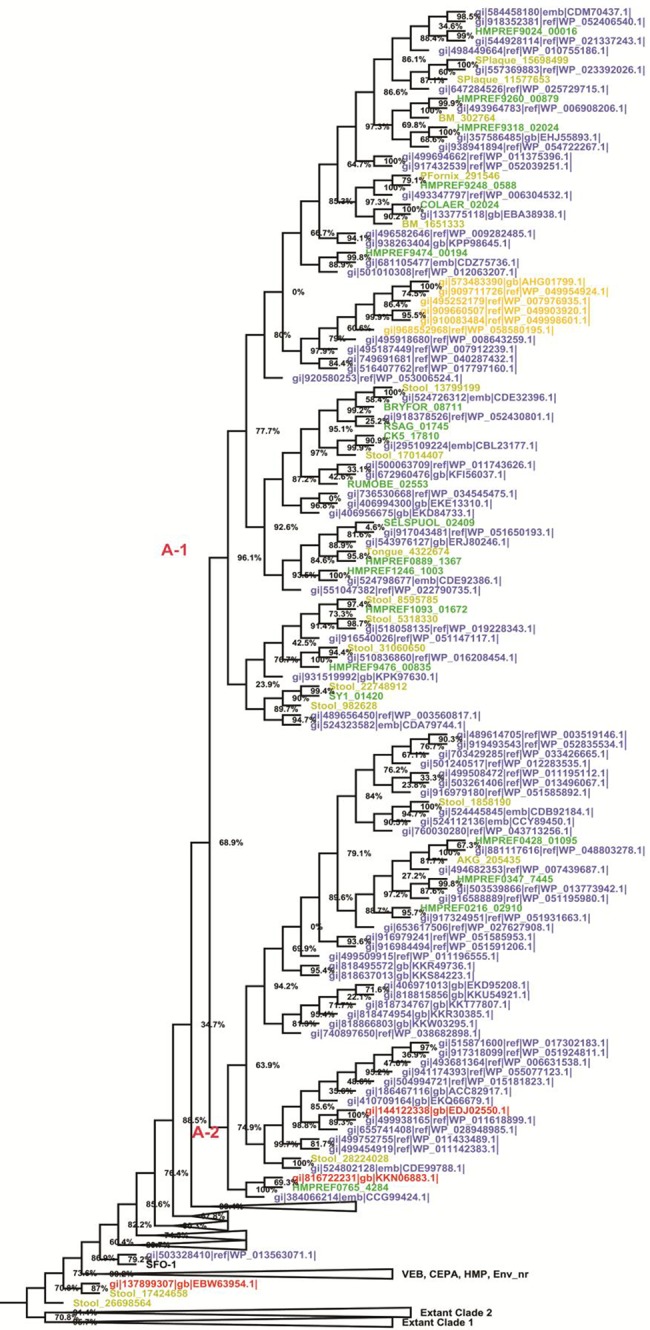
—Midpoint rooted phylogenetic tree of putative class A β-lactamase enzymes. The tree was constructed using the approximate maximum-likelihood method in FastTree and visualized in FigTree. The numbers represent node support values. Because of a vast number of hits, only representative sequences from each database are shown (see main text). The coloring scheme of the leaves indicates sequences derived from distinct databases, that is, red, env_nr; blue, nr; dark green, HMP-Ref; olive, HMP-metagenomic; and black, ARG-ANNOT. Orange indicates archaeal sequences.
The bacterial β-lactamase sequences were considered for the construction of ancestral and HMM profiles. In this report, the HMM profile search analysis detected remote homologous sequences from archaea domains (such as Haladaptatus paucihalophilus, Halobacteriaceae archaeon SB9, Halococcus agarilyticus, Halococcus sediminicola, Halostagnicola larsenii, and Halostagnicola larsenii JCM 1346). This finding indicates that the HMM profile search analysis detected homologues which were distantly related to archaea. Other than archaea, all sequences are related to bacterial domain and metagenomic. The metagenomic sequences are not annotated in public databases, therefore, their source of organisms are unknown.
Class B
The metallo-β-lactamase class B HMM profile search detected a total of 7,931 (env_nr: 393, HMP metagenome: 994, HMP reference genome: 266, NCBI nr: 6,278) significant putative sequences which contains functional β-lactamase domains. Similarly, the other method, the ancestral sequence, detected a total of 1,953 putative sequences. Of these ancestral results, 93.75% (1831) of sequences had already been detected by the HMM profile search, while 6.25% (122) of the sequences were new and were absent in the HMM profile search result. We selected all these sequences (detected by both methods) and removed redundancies. We then, selected the representative sequences (through the CD hit clustering method) and constructed a phylogenetic tree with their multiple sequence alignments, which revealed four novel clades, B-1 to B-4 (fig. 3). The multiples sequence alignment shows H131,133,222,291 conserved amino acid residues (supplementary file S3, Supplementary Material online).
Fig. 3.
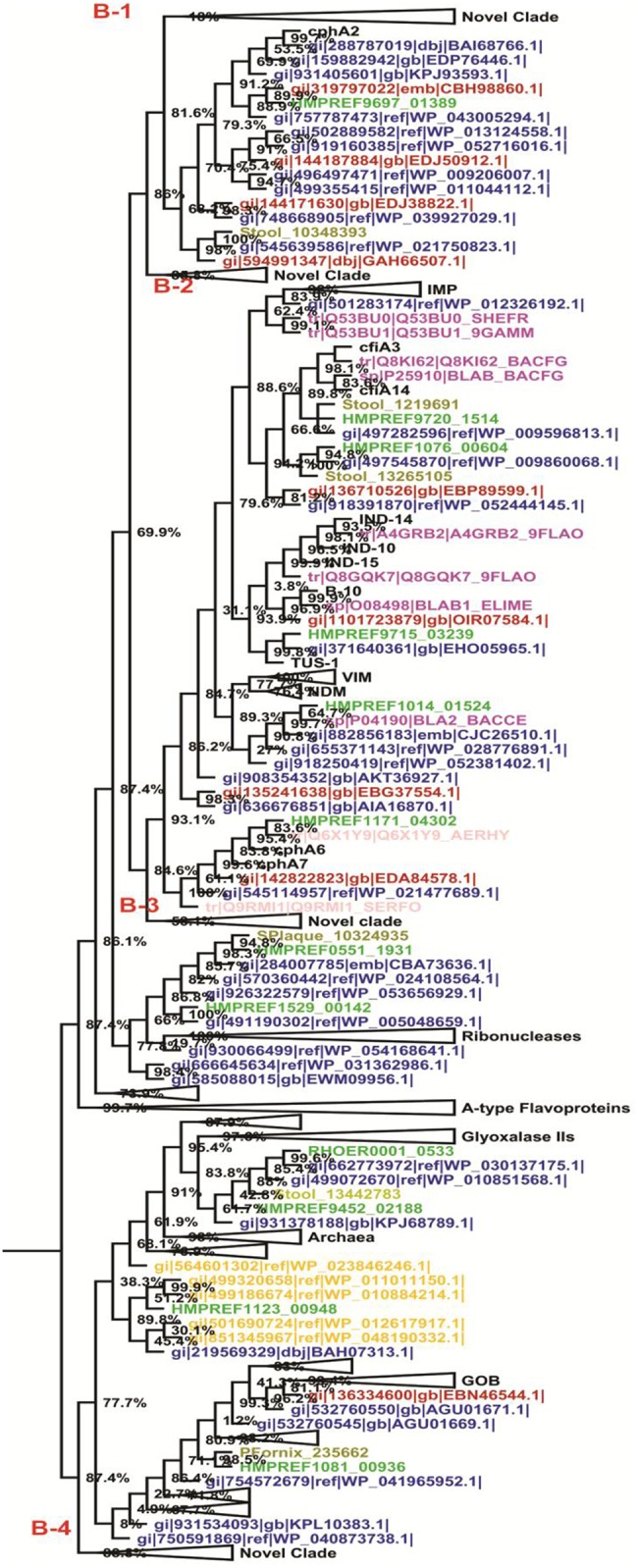
—Midpoint rooted phylogenetic tree of putative class B β-lactamase enzymes. Color scheme and annotations are as in figure 2.
The Class B HMM profile detected putative β-lactamase-like sequences from archaea (such as Bathyarchaeota archaeon, Candidatus Methanosphaerula, Candidatus Nitrosoarchaeum, Candidatus Nitrosopumilus, Ferroglobus placidus, Halalkalicoccus jeotgali, Halobacteriaceae archaeon, Methanobacterium sp., Methanocella conradii, Methanocella paludicola SANAE, Methanohalobium evestigatum, Methanolobus psychrophilus, Methanolobus tindarius, Methanomethylovorans hollandica, Methanosaeta concilii, Methanosaeta sp. SDB, Methanosphaera, Methanotorris igneus, Natrinema versiforme, Natronorubrum bangense, Natronorubrum tibetense, Palaeococcus pacificus, Picrophilus torridus, Pyrococcus furiosus, Pyrococcus shinkaii, Sulfolobus acidocaldarius, Sulfolobus islandicus, Sulfolobus solfataricus, Thaumarchaeota archaeon, Thermococcus barophilus, Thermococcus sibiricus, Vulcanisaeta distribute, Vulcanisaeta moutnovskia Prokofeva 2006, and Vulcanisaeta souniana). This finding suggested that the HMM profile of bacterial sequences was able to detect homologous sequences which were remote from the archaea domain. Other than archaea, all sequences are related to bacteria and metagenomes. The metagenomic sequences are not annotated in public databases, therefore, their source of organisms are unknown.
A number of other proteins contain the metallo-β-lactamase domain. These proteins include glyoxalase IIs, A-type flavoproteins, and ribonucleases (Alderson et al. 2014). Therefore, in the present phylogenetic diversity analysis (fig. 3), we selected some sequences from this diverse set of enzymes, along with extant and newly identified putative β-lactamase-like sequences. The above-mentioned “novel clades” do not contain any extant β-lactamase and a diverse set of enzymes (glyoxalase IIs, A-type flavoproteins, and ribonucleases).
Class C
The queried reconstructed the ancestral sequence of extant enzymes in a BLAST search and was able to identify a total of 4,287 (env_nr: 113, HMP metagenome: 25, HMP reference genome: 75, NCBI nr: 4,074) significant putative β-lactamases. Of these sequences, only 77 (1.79%) sequences has not been identified though the HMM search analysis, and these sequences has been identified through the ancestral BLAST search analysis.
In contrast, the HMM profile search identified a greater number of sequences compared with ancestral sequence search. Through the HMM profile search, we were able to detect a total of 26,719 (env_nr: 490, HMP metagenome: 2,204, HMP reference genome: 667, NCBI nr: 23,358) significant sequences which contain the β-lactamase domain. This finding indicates that HMM profile search analysis is more sensitive than the ancestral BLAST search analysis. We then combined all sequences from the above analysis for clustering, followed by a multiple sequence alignment and phylogenetic tree analysis. The multiple sequence alignment of these sequences shows conserved S111XXK and Y209XN amino acid motifs (supplementary file S4, Supplementary Material online). Here, “X” represents sequences that can accommodate multiple substitutions.
The phylogenetic tree presents eight novel clades; C-1 to C-8 (fig. 4), which contain archaea, bacterial, and metagenomic sequences. The archaea sequence (detected by the HMM profile search) belonged to Aeropyrum camini JCM 12091, Methanocorpusculum labreanum, Methanolacinia paynteri, Methanoregula formicica, Methanosarcina sp., Methanospirillum hungatei, Natrialba chahannaoensis, Natrialba hulunbeirensis, Natrialba magadii, Natronobacterium gregoryi, Natronorubrum bangense, Natronorubrum tibetense, Pyrobaculum aerophilum, Pyrobaculum ferrireducens, Pyrococcus abyssi, Pyrococcus sp. NA2, Thaumarchaeota archaeon, Thermoplasmatales archaeon, Thermoproteus sp., Vulcanisaeta distribute, Vulcanisaeta moutnovskia, and Vulcanisaeta sp. (listed in supplementary table S1, Supplementary Material online). This finding indicates that the HMM profile of bacterial sequences was able to detect homologues which are remote from Archaea. Other than archaea, all sequences are related to bacteria and metagenomes. The metagenomic sequences are not annotated in public databases and, therefore, their sources of organisms are unknown.
Fig. 4.
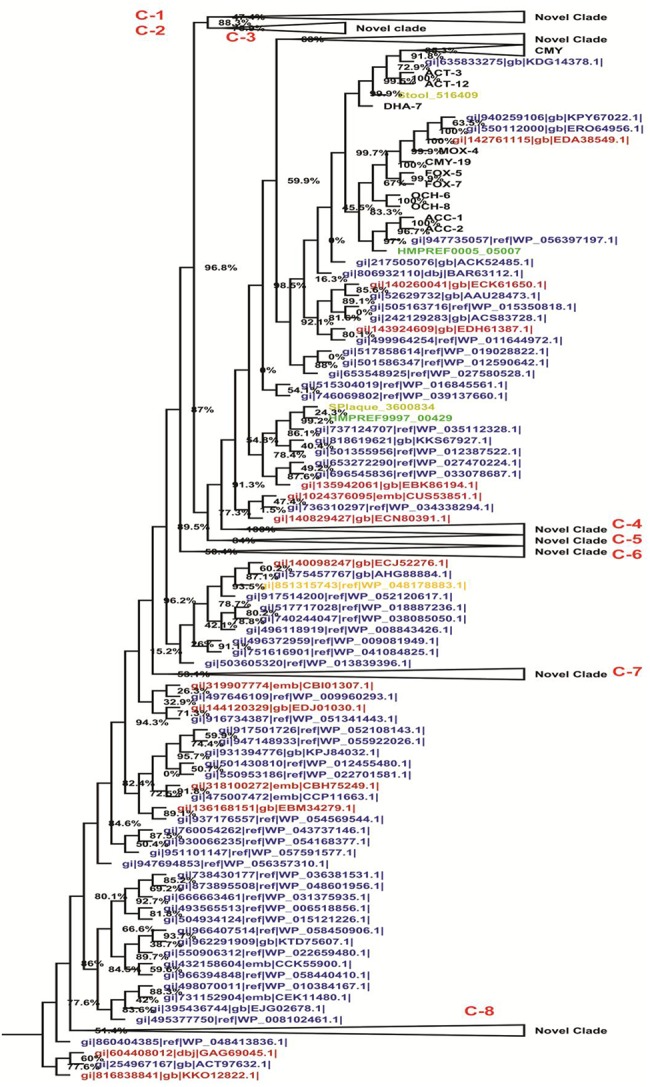
—Midpoint rooted phylogenetic tree of putative class C β-lactamase enzymes. Color scheme and annotations are as in figure 2.
Class D
The HMM profile of class D β-lactamase identified a total of 4,127 (env_nr: 22, HMP metagenome: 404, HMP reference genome: 115, NCBI nr: 3,586) significant putative homologous sequences which contain the β-lactamase domain (supplementary table S1, Supplementary Material online). In contrast, the BLAST search analysis with the ancestral sequence revealed a total of 4,170 (env_nr: 22, HMP metagenome: 404, HMP reference genome: 115, NCBI nr: 3629) significant sequences. In the present analysis, the BLAST search of ancestral sequences identified more sequences (43 sequences) than the HMM profile search analysis. All these sequences contain the β-lactamase domain, which indicates that these sequences are probably functional β-lactamases. All the identified sequences in this class were related to the bacterial domain and metagenomic, but we did not obtain any sequences from the archaea domain. Metagenomic sequences have not been annotated in the public domain and, therefore, it was not possible to identify their source organism. The representative sequences were selected for multiple sequence alignment followed by the construction of a phylogenetic tree which reveals the diversity of class D β-lactamases. The multiple sequence alignment of these sequences shows conserved S58XXK, K194XG amino acid motifs. Here, “X” represents sequences that can accommodate multiple substitutions (supplementary file S5, Supplementary Material online).
The phylogenetic tree of class D contains extant as well as newly identified representative sequences forming two novel clades D-1 and D-2 (fig. 5). These novel clades belong to the Oceanicaulis sp., Bacillus pumilus, Hungatella hathewayi WAL-18680, Eubacterium sp., Staphylococcus aureus, Clostridium sp., and Lachnospiraceae bacteria. The class D HMM profile search was unable to detect any sequences from the archaeal domain.
Fig. 5.—
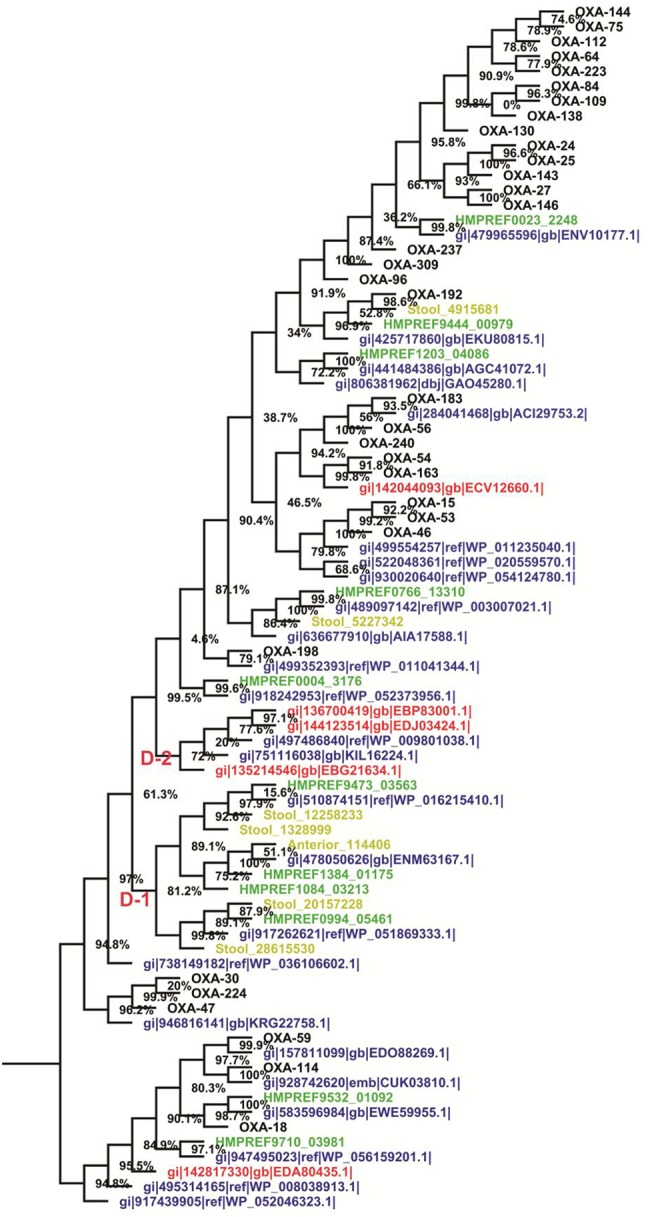
Midpoint rooted phylogenetic tree of putative class D β-lactamase enzymes. Color scheme and annotations are as in figure 2.
Discussion
The objective of this study was to identify putative β-lactamases from biological databases (the HMP reference genome database, the HMP metagenome database, the environmental metagenome database, and NCBI’s nr database), and to describe their phylogenetic diversity. With this in mind, we built HMM profiles and most probable ancestral sequences of extant enzymes, which were considered to be more efficient in recognizing remotely homologous sequences. We then predicted β-lactamase domains and conserved amino acid residues/motifs in putative sequences, which suggested their probable functional involvement in the catalytic mechanism.
We constructed the putative ancestral sequences and HMM profile of four classes (class A–D) of extant β-lactamases to identify homologous sequences from distinct databases. It has previously been shown that ancestral sequences have similar and extended functional characteristics compared with modern enzymes (Risso et al. 2013). For instance, the resurrected Precambrian β-lactamase ancestral sequence was shown to correctly fold into a canonical β-lactamase structure with enhanced denaturation temperature and catalytic efficiencies (Risso et al. 2013). In addition, Sharma et al. reconstructed ancestral sequences of rpoB and showed that ancestral sequences were capable of detecting remote homologous sequences, and of reflecting their phylogenomic diversity (Sharma et al. 2014). Similarly, an HMM profile search was considered to be the most successful procedure for detecting remote homology (Madera and Gough 2002). Therefore, in the present study, we used both methods to detect remote novel putative β-lactamase-like sequences. The BLAST search analysis was executed with the extant β-lactamases, however, we did not obtain any extra sequences other than those retrieved by ancestral and HMM profile-based searches. Thus, in this study, we focused only on the HMM profile search and ancestral BLAST search analysis. In summary, ancestral sequence BLAST searches are better than extant sequences, while HMM profile searches are better than ancestral searches in terms of the number of homologue sequences.
In the BLAST search analysis of ancestral sequences, we identified hypothetical, unnamed, and partially annotated sequences, which shows the respective sequence identity (≥30%) and query coverage (≥50%) of queried ancestral sequences. We chose this identity threshold because a common rule of thumb is that two sequences are homologous if they are >30% identical (Stormo 2009). Moreover, it was previously suggested that protein–protein alignment with e-values < 0.001 can reliably be used to infer homology (Stormo 2009). The HMM profile search also produced numerous unnamed and hypothetical sequences, but the results were filtered on the basis of e-value (identity and coverage criteria were not applicable to the HMM profile search).
The PATRIC database contains genomes with AMR (antimicrobial resistance) data, which offers BLAST matches to antimicrobial resistance (AMR) related proteins (curated at ARDB, the Antibiotic Resistance Genes Database and CARD, the Comprehensive Antibiotic Resistance Database). Therefore, in order to verify the presence of homologous sequences of putative β-lactamases, in this study, we performed local BLAST matches (putative β-lactamases and PATRIC AMR-related protein data as a query and database, respectively). Based on selected threshold criteria (mentioned in Materials and Methods) we selected homologous sequences. When putative β-lactamases have homologous sequences in the PATRIC database, this indicates that these are antimicrobial resistance sequences. A total of 32,032 putative β-lactamase sequences (identified through BLAST matches of ancestral sequences and an HMM profile search) shows homologues in the PATRIC database. Supplementary table S1, Supplementary Material online, presents the list of putative β-lactamase sequences and their homologues which are present (abbreviated as “Y” in supplementary table S1, Supplementary Material online) or absent (abbreviated as “N” in supplementary table S1, Supplementary Material online) in the PATRIC database.
The occurrence of the putative β-lactamase sequence in the metagenomic database was considered as dark-matter. The term “dark-matter” indicates unannotated, unclassified metagenomic sequences where sufficient information is lacking about their sources and functions. The metagenomic databases help to discover novel protein families and are a valuable resource for identifying and annotating remote homologous sequences (Lobb et al. 2015). Previously, several novel antibiotic resistance genes, including genes responsible for β-lactam resistance were identified in remote Alaskan soil (Allen et al. 2009). To this end, our current study suggested a vast repository of unannotated β-lactamases in the human microbiome and environmental metagenomic databases, which illustrates the need for further experimentation with a view to their proper characterization. Apart from metagenomics, in this study, we found evidence that the human microbiome reference genome and NCBI’s nr databases, along with the respective organism, are also potential reservoirs of putative β-lactamases. In this study, we found a greater number of putative β-lactamases from the nr data sets than from other selected databases.
We predicted functional sites in the detected homologous sequences and our results provided evidence to support the hypothesis that the identified putative sequences would probably be functional β-lactamases. Our results revealed signature motif characteristics of each group of β-lactamases (S69XXK, S131DN, and K235XG in Class A, H131,133,222,291 in class B, S111XXK and Y209XN in class C, and S58XXK, K194XG in class D, where “X” represents any amino acid). This result is consistent with previous studies summarized by Bush (Bush 2013). These class-specific motifs indicate that these regions are either structurally or functionally important for β-lactamase activity. These regions can, therefore, be used to develop diagnostic probes. The functional domain that we recognized can be used as a fingerprint for recognizing new relatives and predicting their likely functional properties. The identified putative novel sequences are uncharacterized and unnamed, thus, in future, experiments should be conducted on these novel sequences to provide more detailed information.
Phylogenetic trees of different classes of β-lactamases (A–D) strongly support the existence of new clades. The topology of these trees suggests that the novel clades are distantly related to the extant β-lactamase enzymes. Overall, the findings from this study suggest that the human microbiome, environmental metagenomic, and NCBI’s nr databases offer a new and broadened reservoir of previously unseen β-lactamases, which are highly diverse from extant enzymes. These extra diverse novel sequences are likely to encode enzymes that provide resistance to diverse antibiotics.
Finally, our approach to identifying putative β-lactamases could obviously be applied to other protein families, and other medical applications can be envisaged, where resistance to chemical challenges has recently emerged during evolution.
Conclusions
The principal conclusions of this study can be summarized as follows:
The results of this study indicate that the putative ancestral sequences provide an intuitive way of inspecting homologous sequences in microbial communities, and the HMM-based profile search is far better in detecting remote homologs.
β-lactamases are more diverse than currently understood, and unannotated putative β-lactamase-like sequences are found in the metagenomic database.
Metagenomic putative β-lactamase protein sequences are evolutionarily distant from known β-lactamases. Based on sequence similarity, conserved amino acid residues and β-lactamase domains, these putative β-lactamases are likely to work like known β-lactamases.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Authors’ Contributions
V.K. analyzed data and wrote the manuscript, A.P. edited the manuscript, A.L and J.M.R provided helpful comments, P.P and D.R. were responsible for research concept, planning, and design. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
This work was funded by “Infectiopole Sud,” Méditerranée Infection, whose contribution is gratefully acknowledged. We are grateful to the reviewer for useful comments and suggestions.
Literature Cited
- Alderson RG, Barker D, Mitchell JBO.. 2014. One origin for metallo-β-lactamase activity, or two? An investigation assessing a diverse set of reconstructed ancestral sequences based on a sample of phylogenetic trees. J Mol Evol. 79(3–4):117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen HK, Moe LA, Rodbumrer J, Gaarder A, Handelsman J.. 2009. Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J. 3(2):243–251. [DOI] [PubMed] [Google Scholar]
- Ambler R. 1980. The structure of β-lactamases. Philos Trans R Soc Lond B Biol Sci. 289(1036):321–331. [DOI] [PubMed] [Google Scholar]
- Bateman A, et al. , 1999. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27(1):260–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. 1989. Classification of β-lactamases: groups 1, 2a, 2b, and 2b’. Antimicrob Agents Chemother. 33(3):264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. 2013. The ABCD’s of β-lactamase nomenclature. J Infect Chemother. 19(4):549–559. [DOI] [PubMed] [Google Scholar]
- Bush K, Jacoby GA, Medeiros AA.. 1995. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother. 39(6):1211–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella GS, Silla MJM, Gabaldón T.. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics (Oxford, England) 25(15):1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Costa VM, et al. , 2011. Antibiotic resistance is ancient. Nature 477:457–461. [DOI] [PubMed] [Google Scholar]
- Eddy SR, Crooks G, Green R, Brenner S, Altschul S.. 2011. Accelerated profile HMM searches. PLoS Comput Biol. 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, et al. , 2014. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 58(1):212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BG, Barlow M.. 2004. Evolution of the serine β-lactamases: past, present and future. Drug Resist Updates 7(2):111–123. [DOI] [PubMed] [Google Scholar]
- Heritage J, M'Zali FH, Gascoyne-Binzi D, Hawkey PM.. 1999. Evolution and spread of SHV extended-spectrum beta-lactamases in Gram-negative bacteria. J Antimicrob Chemother. 44(3):309–318. [DOI] [PubMed] [Google Scholar]
- Iqbal HA, Feng Z, Brady SF.. 2012. Biocatalysts and small molecule products from metagenomic studies. Curr Opin Chem Biol. 16(1–2):109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb B, Kurtz DA, Moreno-Hagelsieb G, Doxey AC.. 2015. Remote homology and the functions of metagenomic dark matter. Front Genet. 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madera M, Gough J.. 2002. A comparison of profile hidden Markov model procedures for remote homology detection. Nucleic Acids Res. 30(19):4321–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39:D225–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. , 2015. CDD: nCBI’s conserved domain database. Nucleic Acids Res. 43(Database issue):D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M, et al. , 2000. Crystal structure of the class D beta-lactamase OXA-10. Nat Struct Biol. 7(10):918–925. [DOI] [PubMed] [Google Scholar]
- Philippon A, Dusart J, Joris B, Frère J.. 1998. The diversity, structure and regulation of beta-lactamases. Cell Mol Life Sci. 54(4):341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP.. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabausch U, et al. , 2013. Functional screening of metagenome and genome libraries for detection of novel flavonoid-modifying enzymes. Appl Environ Microbiol. 79(15):4551–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16(6):276–277. [DOI] [PubMed] [Google Scholar]
- Risso VA, Gavira JA, Mejia-Carmona DF, Gaucher EA, Sanchez-Ruiz JM.. 2013. Hyperstability and substrate promiscuity in laboratory resurrections of precambrian β-lactamases. J Am Chem Soc. 135(8):2899–2902. [DOI] [PubMed] [Google Scholar]
- Sharma V, Colson P, Giorgi R, Pontarotti P, Raoult D.. 2014. DNA-dependent RNA polymerase detects hidden giant viruses in published databanks. Genome Biol Evol. 6(7):1603–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD. 2009. An introduction to sequence similarity (‘homology’) searching. Curr Protoc Bioinformatics 3(3.1):3.1.1–7. [DOI] [PubMed] [Google Scholar]
- Wattam AR, et al. , 2014. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42:D581–D591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke MS, Lovering AL, Strynadka NC.. 2005. Beta-lactam antibiotic resistance: a current structural perspective. Curr Opin Microbiol. 8(5):525–533. [DOI] [PubMed] [Google Scholar]
- Wright GD. 2007. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 5(3):175–186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.