

Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant disease caused by germline inactivating mutations of TSC1 or TSC2. In TSC-associated tumors of the brain, heart, skin, kidney and lung, inactivation of both alleles of TSC1 or TSC2 leads to hyperactivation of the mTORC1 pathway. The TSC/mTORC1 pathway is a key regulator of cellular processes related to growth, proliferation and autophagy. We and others have previously found that mTORC1 regulates microRNA biogenesis, but the mechanisms are not fully understood. Microprocessor, a multi-protein complex including the nuclease Drosha, processes the primary miR transcript. Using a dual-luciferase reporter, we found that inhibition of mTORC1 or downregulation of Raptor decreased Microprocessor activity, while loss of TSC2 led to a striking increase (∼5-fold) in Microprocessor activity. To determine the global impact of TSC2 on microRNAs we quantitatively analyzed 752 microRNAs in Tsc2-expressing and Tsc2-deficient cells. Out of 259 microRNAs expressed in both cell lines, 137 were significantly upregulated and 24 were significantly downregulated in Tsc2-deficient cells, consistent with the increased Microprocessor activity. Microprocessor activity is known to be regulated in part by GSK3β. We found that total GSK3β levels were higher in Tsc2-deficient cells, and the increase in Microprocessor activity associated with Tsc2 loss was reversed by three different GSK3β inhibitors. Furthermore, mTOR inhibition increased the levels of phospho-GSK3β (S9), which negatively affects Microprocessor activity. Taken together these data reveal that TSC2 regulates microRNA biogenesis and Microprocessor activity via GSK3β.
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder characterized by benign tumors of the brain, heart, kidney and skin, as well as neurologic manifestations (seizures, autism and intellectual disability) and pulmonary lymphangioleiomyomatosis (LAM), a destructive cystic lung disease (1). The TSC proteins, TSC1 (hamartin) and TSC2 (tuberin), form a complex with TBC1D7 to regulate the activity of the mammalian/mechanistic target of Rapamycin complex 1 (mTORC1) via Rheb, a small GTPase that is the target of TSC2’s GTPase activating domain (2). Activation of mTORC1 in TSC1- or TSC2-deficient cells leads to a decrease in autophagy and a cascade of catabolic processes, including increases in protein translation, lipid synthesis and nucleotide synthesis (3,4).
MicroRNAs (miRNA or miR) are small RNA molecules (around 22 nucleotides) with functions in most cellular pathways. In cancer, a global decrease in miR expression is often observed (5–7). Each miR can regulate multiple genes, providing a mechanism through which complex cellular functions can be coordinated (8). MicroRNA biogenesis is regulated at multiple steps. Microprocessor, a nuclear complex that includes the nuclease Drosha and its partner DGCR8, processes the primary miR transcript (pri-miR) to the precursor miR (pre-miR) by recognizing and cleaving at stem-loop structures in the pri-miR and cleaving at both the 5′ and the 3′ ends of the stem-loop (9). Microprocessor activity is known to be regulated by multiple mechanisms including Yap, which plays a role in cell density dependent regulation of Microprocessor activity and GSK3β, which binds directly to the Microprocessor complex and facilitates Microprocessor activity (10,11).
We previously found that mTOR inhibition with Rapamycin impacts the levels of multiple miRs in TSC2-deficient LAM-patient derived cells, which we termed ‘Rapa-miRs’, including increases in pro-survival ‘onco-miRs’ (miR-21 and miR-29b) (12,13). These findings suggested that induction of oncogenic miR could be a mechanism underlying the partial responses observed when TSC-associated tumors are treated with mTOR inhibitors. To elucidate the mechanisms through which the TSC proteins regulate miR levels, we examined the activity of Microprocessor using a dual-luciferase reporter assay. Here, we report that Tsc2 loss increases Microprocessor activity whereas Rapamycin and Torin 1 decrease Microprocessor activity. A global analysis of the impact of Tsc2 on microRNA biogenesis revealed that 259 microRNAs were expressed in both Tsc2-expressing and Tsc2-deficient mouse embryonic fibroblasts (MEFs). Of these microRNAs, 137 were upregulated and 24 downregulated in Tsc2-deficienct cells. This is consistent with increased Microprocessor activity in Tsc2 deficient-cells. GSK3β protein levels (including the nuclear fraction) were higher in Tsc2-deficient cells, and treatment with a GSK3β inhibitor blocked Microprocessor activity. Furthermore, mTOR inhibition increased the levels of phospho-GSK3β (S9), which negatively affects Microprocessor activity (11). Together these data point to a novel mechanism through which TSC2 and mTOR regulate miR biogenesis via GSK3β.
Results
Microprocessor activity is mTORC1 dependent
To determine whether mTORC1 regulates Microprocessor activity, we used HeLa cells stably expressing a Microprocessor reporter (10). This dual activity reporter contains a portion of pri-miR-125b-1 that forms a stem-loop within the 3′ UTR of the Renilla luciferase gene. Cleavage of this stem-loop destabilizes the Renilla luciferase mRNA resulting in decreased Renilla luminescence. The construct also contains the Firefly luciferase gene under a separate HSV-TK promoter. The Firefly luciferase mRNA is insensitive to Microprocessor activity, allowing for internal normalization of cell transfection. The level of Microprocessor activity is determined by the ratio of Firefly luminescence (the control) to Renilla luminescence (which is Microprocessor dependent), with the values positively correlating with the Microprocessor activity (10).
It has been previously shown that Microprocessor activity is cell density sensitive, via a Yap-dependent mechanism (10). Therefore, in a 96-well plate, we plated HeLa cells stably transfected with Microprocessor reporter construct at two densities: 5000 cells/well in a 96-well plate ‘lower density’ and 10 000 cells/well ‘higher density’. Crystal violet staining validated that the higher density had approximately 40% more cells/well (Fig. 1A). We confirmed the previous finding that Microprocessor activity is higher at higher cell densities (10), with a nearly 2-fold increase in the Firefly/Renilla luminescence ratio in the DMSO control cells at higher density (Fig. 1B). Treatment with mTORC1 inhibitor, Rapamycin, decreased Microprocessor activity by ∼30% in the lower density condition (P < 0.001, Fig. 1B) with no effect on the cells plated at higher density. Torin 1, an mTOR kinase inhibitor, which blocks both mTORC1 and mTORC2, decreased Microprocessor activity by ∼50% at lower cell density and by ∼40% at higher cell density (Fig. 1B). These data confirm that Microprocessor activity is cell density sensitive and demonstrate for the first time that it is regulated by mTORC1.
Figure 1.

Microprocessor activity is mTORC1 dependent. (A) Phase contrast images of HeLa cells with stable expression of the dual luciferase reporter (pri-miR-125b-1) plated in a six-well plate at 150 000 cells/well; ‘lower density’ and 300 000 cells/well; ‘higher density’ (left panel) and quantitation of cell number by crystal violet staining at both densities ‘lower’ (5000 cells/well of 96-well plate) and ‘higher’ (10 000 cells/well of 96-well plate) after 24 h treatment with Rapamycin (20 nm) or Torin 1 (250 nm) (right panel). (B) Microprocessor activity as determined by the ratio of Firefly luminescence to Renilla luminescence in HeLa cells stably transfected with the full-length construct of primary miRNA-125b-1 (pri-miR-125b-1 construct), at lower and higher density after treatment with DMSO (control), Rapamycin or Torin 1 for 24 h. (C) Microprocessor activity as determined by the ratio of Firefly luminescence to Renilla luminescence in HeLa cells transiently transfected with either the pri-miR-125b-1 construct or the control construct lacking the stem loop of primary miRNA-125b-1 (Δ-stem-loop construct), after treatment with DMSO, Rapamycin or Torin 1 for 24 h, at higher and lower densities, normalized to DMSO control. (D) Kinetics of Microprocessor activity, at 2, 8 and 24 h after Rapamycin and Torin 1 treatments, in HeLa cells with stable expression of the dual luciferase reporter (pri-miR-125b-1). Significance was determined by Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm that these results are specific to the nuclease activity of Microprocessor, we transiently expressed a construct that lacks the stem-loop (which is cleaved by Drosha) and compared it with the full-length construct including the stem-loop (pri-miR-125b-1 construct). Consistent with the data observed with the stably transfected cells, Rapamycin (an allosteric mTORC1 inhibitor) and Torin 1 (an mTOR catalytic inhibitor) decreased Microprocessor activity in the cells transiently expressing the pri-miR-125b-1 construct, with a greater effect at lower cell density settings. No effect of Rapamycin or Torin 1 was observed in cells expressing the control Δ-stem-loop construct, confirming that mTORC1 inhibition decreases miR processing by Microprocessor (Fig. 1C).
To further confirm that mTORC1 regulates the Microprocessor activity, we introduced siRNA for Raptor, an essential component of mTORC1, in HeLa cells stably expressing the Microprocessor reporter construct. Downregulation of Raptor led to significant decrease in Microprocessor activity (Supplementary Material, Fig. S1A and B). We confirmed these results using MEFs with 4-Hydroxytamoxifen-inducible knockout of Raptor, in which Raptor knockout also decreased Microprocessor activity (Supplementary Material, Fig. S1C and D). Interestingly, siRNA downregulation of Rictor and Rictor-inducible knockdown also decreased Microprocessor activity (Supplementary Material, Fig. S1A–D).
To define the kinetics of the Microprocessor activity regulation by mTORC1, we measured Microprocessor activity at 2, 8, and 24 h after Rapamycin and Torin 1 treatments, using the cells with stable expression of the dual luciferase reporter (pri-miR-125b-1). Both drugs decreased Microprocessor activity at 24 h, but not at earlier time points. Consistent with the prior data, Rapamycin decreased Microprocessor activity at lower cell density but not at higher cell density, while Torin 1 decreased Microprocessor activity at both lower and higher cell density, with a greater effect at lower cell density (Fig. 1D).
Microprocessor activity is TSC2 dependent
To determine whether Microprocessor activity is regulated by TSC2, upstream of mTORC1, we used MEFs derived from Tsc2flox/flox-Rosa26-CreERT2 mice. These MEFs were treated with either 4-Hydroxytamoxifen (4HT) to delete Tsc2 (Tsc2 KO MEFs) or with ethanol (EtOH) as control (Tsc2 WT MEFs). Western immunoblot confirmed the depletion of Tsc2 upon 4HT treatment (Fig. 2A). The dual-luciferase reporter was then transiently expressed in these cells. Microprocessor activity was almost 5-fold higher in the Tsc2-deficient cells (P < 0.001) at lower density and ∼2-fold higher at higher density (P < 0.01) (Fig. 2B). Expression of the control Δ-stem-loop construct revealed no Tsc2-dependent difference in activity, as expected. Treatment of these Tsc2-deficient cells with Rapamycin did not significantly decrease Microprocessor activity, although there was a trend toward lower activity at the higher cell density. Treatment with Torin 1 decreased Microprocessor activity at both densities in the Tsc2-deficient cells, but had no effect on Microprocessor activity in the cells with wild-type Tsc2 expression levels.
Figure 2.
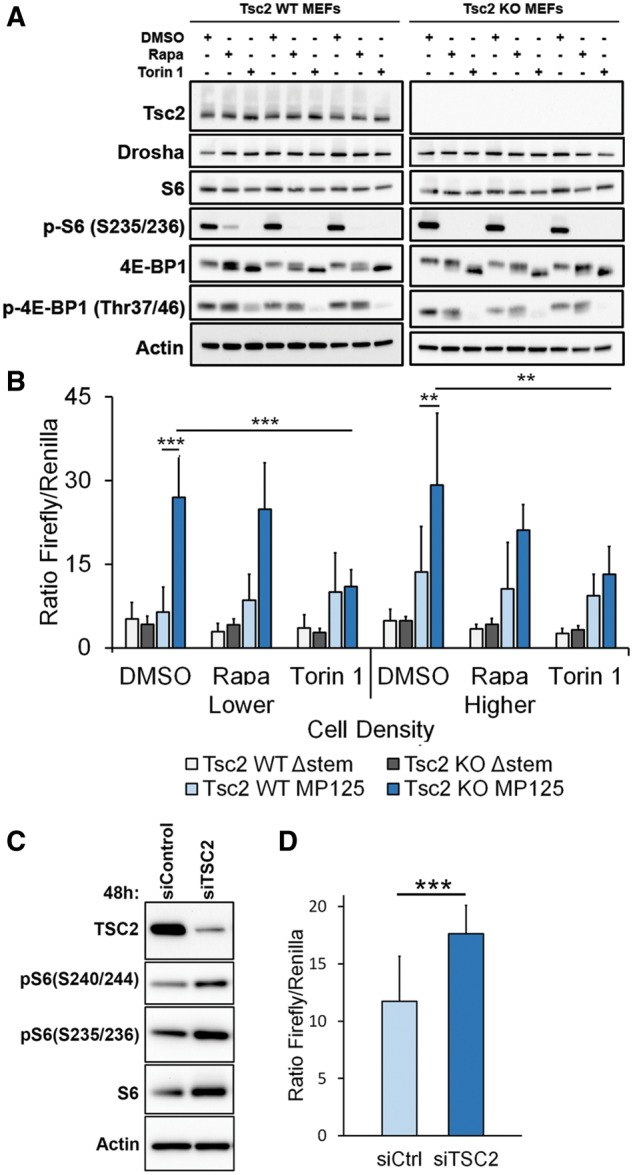
Microprocessor activity is TSC2 dependent. (A) MEFs derived from Tsc2flox/flox-Rosa26-CreERT2 mice were treated with 4-hydroxytamoxifen (4-HT) to delete Tsc2 (Tsc2 KO MEFs) or ethanol control (Tsc2 WT MEFs). Immunoblots show depletion of Tsc2, with markers of mTORC1 activation (phospho-S6, phospho 4EBP-1), and Drosha levels with three replicates for each cell line and condition. (B) The dual-luciferase reporters with either the full-length construct including the stem-loop (pri-miR-125b-1 construct) or the Δ-stem-loop construct (control), were transiently expressed in the MEFs with or without Tsc2 depletion. Microprocessor activity (as determined by the ratio of Firefly luminescence to Renilla luminescence) was measured at two cell densities: lower (5000 cells/well of a 96-well plate) and higher (10 000 cells/well of a 96-well plate), after treatment for 24 h with Rapamycin (20 nm), Torin 1 (250 nm) or DMSO control. (C) Immunoblot of HeLa cells with 48 h treatment of siRNA targeting TSC2. (D) Microprocessor activity in HeLa cells stably transfected with the full-length Microprocessor reporter construct after 48 h of siRNA for TSC2 or non-targeting control. Significance was determined by Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
In order to determine if the same phenomenon is observed in human cells, we used siRNA to downregulate TSC2 in HeLa cells stably expressing the Microprocessor activity construct (Fig. 2C). As expected, TSC2 knockdown led to a significant (P < 0.001) increase in Microprocessor activity (Fig. 2D).
mTORC1 inhibition decreases levels of small RNA
To determine the functional impact of mTORC1 inhibition on the expression of small RNAs, we used a Qubit assay from Invitrogen, which allows for selective quantification of small RNA over large mRNA or rRNA. The assay uses a proprietary dye that generates fluorescence upon binding to small RNA. Control standards, with known RNA concentrations, were used for calibration. Treatment of the Tsc2-deficient MEFs (Tsc2 KO MEFs) reduced levels of small RNA by approximately 30% after Rapamycin treatment (P < 0.05) and 50% after Torin 1 treatment (P < 0.01). Similar trends were observed in TSC2 expressing cells (Tsc2 WT MEFs) (Fig. 3).
Figure 3.

mTOR inhibition decreases levels of small RNA. Quantification of small RNA as measured by Qubit in Tsc2 WT and Tsc2 KO MEFs after treatment with DMSO control, Rapamycin (20 nm) or Torin 1 (250 nm) for 24 h. The cells were seeded at lower density as defined in Figure 1A. Significance was determined by Student’s t-test on three biological replicates, *P < 0.05, **P < 0.01.
Drosha protein levels and nuclear localization are not affected by loss of TSC2 or rapamycin treatment
To identify the mechanism of the decreased Microprocessor activity, we first examined Drosha protein levels in stably transfected HeLa cells treated with Rapamycin or Torin 1. No change in Drosha protein was observed after 24 h of treatment with either Rapamycin or Torin 1, the time point at which we observed the decrease in Microprocessor activity (Fig. 4A and B). At 3 days, Torin 1 decreased Drosha protein levels by ∼30% (P < 0.01), but Rapamycin had no effect (Fig. 4C). At 7 days, Torin 1 decreased Drosha levels by ∼20% and Rapamycin still had no effect, despite continued suppression of mTORC1 as evaluated by the phosphorylation of ribosomal protein S6 (Fig. 4D and E).
Figure 4.

Drosha protein levels and nuclear localization are not affected by loss of TSC2 or Rapamycin treatment. (A) Drosha protein levels as seen by western immunoblot analysis of HeLa cells treated with DMSO (control), Rapamycin (20 nm) or Torin 1 (250 nm) for 24 h. (B) Quantification of fold change in Drosha protein levels in HeLa cells shown in A. (C) Western immunoblot after 3 days of Rapamycin (20 nm) and Torin 1 (250 nm). (D) Western immunoblot after 7 days of Rapamycin (20 nm) and Torin 1 (250 nm). (E). Fold change of Drosha protein levels in HeLa cells treated with Rapamycin (20 nm) or Torin 1 (250 nm) for 3 and 7 days. (F) Immunoblot analysis of cytoplasmic and nuclear fractions of HeLa cells treated with Rapamycin (20 nm) or Torin 1 (250 nm) for 24 h. GAPDH and CREB serve as controls for the cytoplasmic and nuclear fractions, respectively. A–F. Experiments were performed on three biological replicates. (G) Western immunoblot of Tsc2 WT MEFs and Tsc2 KO MEFs. (H) Immunoblot analysis of cytoplasmic and nuclear fractions of Tsc2 WT MEFs and Tsc2 KO MEFs. GAPDH and CREB serve as controls for the cytoplasmic and nuclear fractions, respectively. Significance was determined by Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
Because Drosha can shuttle between the cytoplasm and the nucleus (14–16), we also used cellular fractionation to investigate the effect of mTOR inhibition on nuclear localization of Drosha. No change in nuclear Drosha levels was observed with 24 h of Rapamycin or Torin 1 treatment (Fig. 4F).
Drosha protein level and nuclear localization were also similar between Tsc2-expressing (Tsc2 WT MEFs) and Tsc2-deficient cells (Tsc2 KO MEFs) (Fig. 4G and H, respectively). Furthermore, treatment with Rapamycin or Torin 1 in these cells did not affect Drosha protein levels (Fig. 2A).
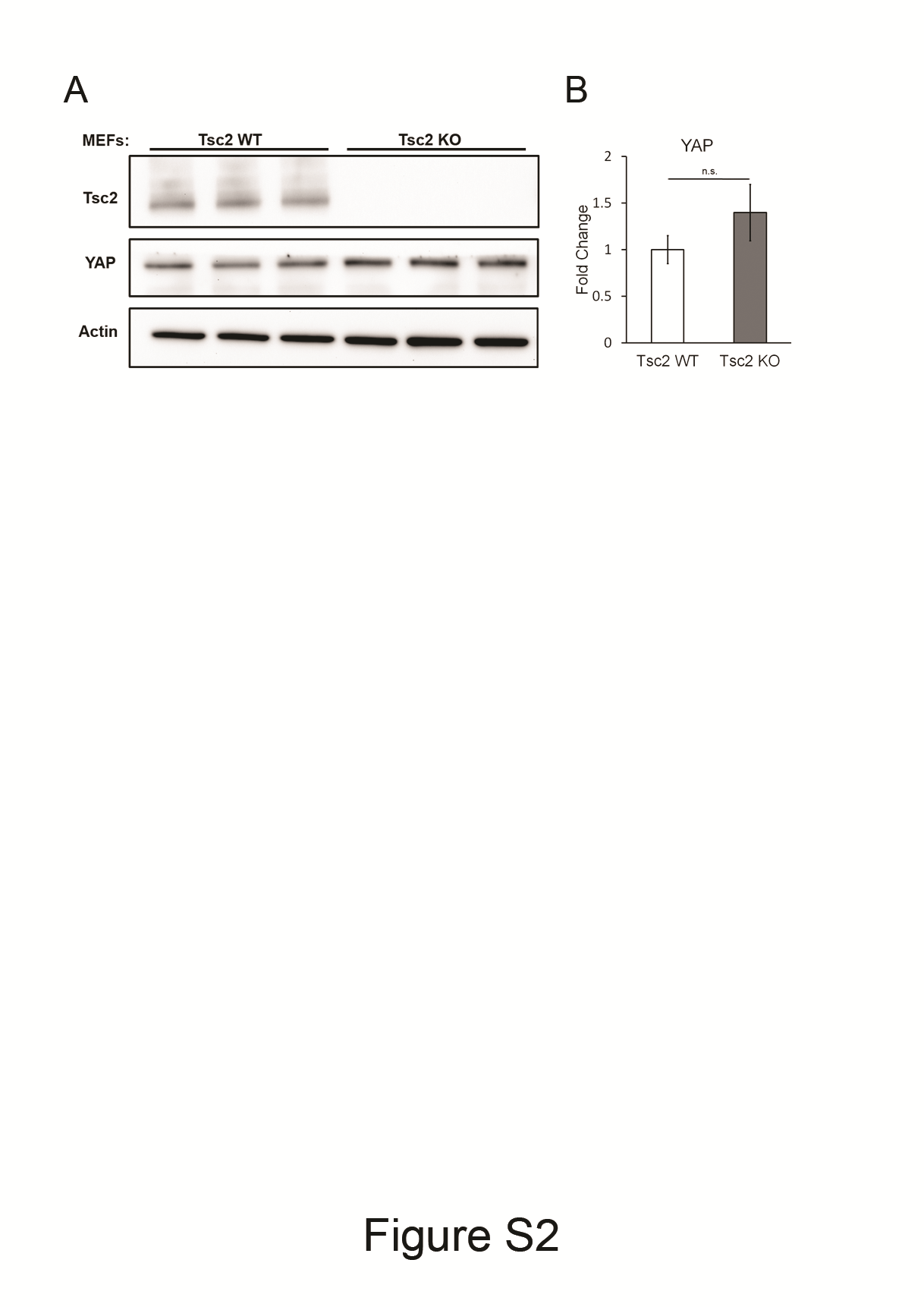
Because YAP regulates Microprocessor activity in a cell density-dependent manner (10), and has been previously shown to be upregulated in TSC2-deficient cells (17), we analyzed the protein level of YAP in Tsc2 WT and Tsc2-deficient MEFs. We observed a trend of higher level of YAP in Tsc2-deficient cells, but the results were not significant (Supplementary Material, Fig. S2). Furthermore, the nuclear level of YAP was not different in these conditions between Tsc2-expressing and Tsc2-deficient MEFs (Fig. 4H), leading us to conclude that YAP is unlikely to be mediating the effects of TSC2 on Microprocessor activity.
mTOR inhibition leads to a global decrease of microRNAs in Tsc2-deficient cells
To determine the global impact of TSC2 and mTOR on miR biogenesis, we quantitatively analyzed 752 miRs in Tsc2-deficient MEFs treated for 48 h with DMSO control or Torin 1 (250 nm) and Tsc2-expressing MEFs treated with DMSO control. We detected an average of 306 miRs per sample. Figure 5A shows a heat map of the top 50 differentially expressed microRNAs. Comparing Tsc2-expressing MEFs to Tsc2-deficient MEFs, out of 259 miRs that were expressed in both cell lines, 161 miRs were differentially expressed with an unadjusted P-value of 0.05 and 105 were differentially expressed using the Benjamini–Hochberg false discovery rate (FDR) correction. Of the 161 differentially expressed miRs using the P < 0.05, 137 were upregulated in Tsc2-deficient MEFs versus 24 downregulated in Tsc2-deficient MEFs (Fig. 5B left panel). Overall, more miRs were upregulated in Tsc2-deficient cells, consistent with the elevated activity of Microprocessor in Tsc2-deficient cells observed in Figure 2. In Tsc2-deficient cells treated with Torin 1 (250 nm, 48 h) versus DMSO control, 264 miRs were expressed. Of these, 105 were significantly different with a P-value <0.05 and 71 were differentially expressed after the FDR correction. Of the 105 differentially expressed miRs, 81 were upregulated by Torin 1 and 24 were downregulated (Fig. 5B right panel).
Figure 5.

Analysis of global changes in microRNA expression in Tsc2-deficient cells. (A) Heat map and unsupervised hierarchical clustering of Tsc2 KO MEFs treated with DMSO control or Torin 1 (250 nm) for 48 h, compared with Tsc2 WT MEFs treated with DMSO control, showing the top 50 differentially expressed microRNAs. (B) Volcano plot of Tsc2 KO MEFs versus Tsc2 WT MEFs, both treated with DMSO control (left panel) and of Tsc2 KO MEFs treated with DMSO control versus Tsc2 KO MEFs treated with Torin 1 (right panel) show the relation between the log 10 P-values and the log 2 fold change of detected microRNAs.
GSK3β inhibition decreases Microprocessor activity in mTOR hyperactive cells
It was recently reported that GSK3β increases Microprocessor activity by direct interaction with DGCR8 and p72 in the presence of primary microRNA (11). Phosphorylation of GSK3β by Akt or S6K1 at S9 negatively regulates the activity of GSK3β (18). To determine if TSC2 regulates GSK3β in our cells, we analyzed protein levels of total GSK3β and phospho-GSK3β (S9) by immunoblot. Total GSK3β levels were higher in the Tsc2-deficient cells (Tsc2 KO MEFs) (Fig. 6A). Rapamycin and Torin 1 increased phospho-GSK3β (S9) levels to a greater extent in the Tsc2-deficient MEFs than the Tsc2-expressing control MEFs (Tsc2 WT MEFs) (Fig. 6A). Interestingly, GSK3β (S9) levels were higher in the Torin 1-treated cells compared with the Rapamycin treated cells, paralleling the greater impact of Torin 1 on Microprocessor activity, as shown in Figure 2B. Quantitative protein levels of phospho-GSK3β (S9) in Tsc2-deficient cells treated with DMSO control or Torin 1 are shown in Figure 6B. We also performed cytoplasmic and nuclear fractionation on the cells and found increased nuclear and cytoplasmic GSK3β in the Tsc2-deficient cells (Tsc2 KO MEFs) (Fig. 6C).
Figure 6.

GSK3β inhibition decreases Microprocessor activity in Tsc2-deficient cells. (A) Western immunoblot of Tsc2 WT MEFs and Tsc2 KO MEFs treated with DMSO control, Rapamycin (20 nm) or Torin 1 (250 nm) for 24 h. (B) Quantification of fold change of pGSK3β protein levels in Tsc2 KO MEFs treated with DMSO control, or Torin 1 (250 nm) for 24 h. Experiment was performed on three biological replicates for DMSO control and four biological replicates for Torin 1 treatment. Significance was determined by Student’s t-test, *P < 0.05. (C) Western immunoblot of cytoplasmic and nuclear fractions of Tsc2 WT MEFs and Tsc2 KO MEFs. (D) Microprocessor activity as determined by the ratio of Firefly luminescence to Renilla luminescence in Tsc2 WT MEFs and Tsc2 KO MEFs under different drug treatment conditions: CHIR99021 (2 um), AR-A014418 (5 um), lithium chloride (10 um) or MK2206 (10 um) for 48 h. Experiment was performed on six biological replicates and significance was determined by Student’s t-test, *P < 0.05, **P < 0.01. (E) Working model illustrating that the increased Microprocessor activity associated with TSC2 deficiency is mTORC1 and GSK3β dependent.
To determine whether GSK3β regulates Microprocessor activity in Tsc2-deficient cells, we transiently expressed the dual luciferase reporter and treated the cells for 48 h with three GSK3β inhibitors: AR-A014418 (5 um), CHIR99021 (2 um) or lithium chloride (10 um). Given that Microprocessor activity is sensitive to cell density (10) and the Tsc2-deficient cells are larger than the Tsc2-expressing cells, we plated twice as many cells per well of the Tsc2-expressing cells to achieve similar cell density. Importantly, the reporter construct contains the Firefly luciferase gene as an internal control of transfection efficiency. Again, as in Figure 2B, we observed increased Microprocessor activity in the Tsc2-deficient cells (approximately a 3-fold increase, P < 0.01, Fig. 6D). All three inhibitors decreased Microprocessor activity in the Tsc2-deficient cells (Tsc2 KO MEFs) at 48 h by at least 2-fold, close to the level of Microprocessor activity in the Tsc2-expressing cells (Fig. 6D). Interestingly, the GSK3β inhibitors had no significant effect on Microprocessor activity in the Tsc2-expressing MEFs (Fig. 6D), and treatment of HeLa cells with CHIR99021 did not affect Microprocessor activity (Supplementary Material, Fig. S3). Since Akt regulates GSK3β activity (19), we also examined the impact of MK2206 (10 um), an allosteric Akt inhibitor, on Microprocessor activity. MK2206 similarly inhibited Microprocessor activity in the Tsc2-deficient MEFs, but not in the Tsc2-expressing MEFs (Fig. 6D).
Taken together these data suggest that the increased Microprocessor activity associated with TSC2 deficiency is mTOR and GSK3β dependent (Fig. 6E).
Discussion
We have previously found that levels of some microRNAs are increased by treatment with Rapamycin for 24 h, including certain oncomiRs (miR-21 and miR-29), while levels of other microRNAs are decreased (12,13). To elucidate the mechanisms of this mTOR-dependent regulation of microRNA biosynthesis, we assayed the activity of Microprocessor, the multiprotein complex that includes Drosha (a type III RNAse) and DGCR8. Microprocessor cleaves the stem loop of pri-miRNA to form pre-miRNA via the nuclease activity of Drosha. We report here that Microprocessor activity is TSC2 and mTOR dependent. mTORC1 inhibition decreased Microprocessor activity by 30–50% in both human and mouse cells, and Tsc2-deficient cells were found to have 3–5 fold higher Microprocessor activity compared with Tsc2-expressing cells. mTORC1 is known to regulate a balance between multiple anabolic processes, including macromolecular synthesis, nucleotide synthesis and nutrient storage, and catabolic processes such as autophagy (3,4). These data indicate that mTORC1 also plays a critical role in regulating the processing of microRNA, with the potential for widespread impact on gene expression.
An important strength of the dual-luciferase reporter that we used to assay Microprocessor activity is the ability to control for transfection efficiency and other variables that may differ with cell density and/or mTORC1 activation. Our data (Fig. 1B) confirm the prior discovery that Microprocessor activity is cell density dependent. Because of this, our experiments were carefully controlled for cell density, with the strongest effects of Rapamycin on Microprocessor seen at lower cell density, while Torin 1 impacted Microprocessor activity at both lower and higher cell density. Furthermore, downregulation of Raptor, an essential component of mTORC1, decreased Microprocessor activity confirming the involvement of mTORC1 in the regulation of microRNA biogenesis (Supplementary Material, Fig. S1).
Our data support a model in which increased GSK3β levels in Tsc2-deficient cells promotes Microprocessor activity (model, Fig. 6D). GSK3β was recently found to bind DGCR8 and to phosphorylate Drosha (11,20). GSK3β levels were higher in our Tsc2-deficient MEFs, similarly to recent data from Cao et al., who found higher total levels of GSK3β in Tsc2-deficient melanocytes, with lower nuclear beta-catenin (21). Previously, Mak et al. found that the TSC1/TSC2 complex physically interacts with GSK3β, and Flavin et al. and Mak et al. have shown that human TSC2-deficient tumors (including angiomyolipomas and LAM) have high levels of nuclear beta-catenin (22,23). We found that three different GSK3β-inhibitors decreased Microprocessor activity in Tsc2-deficient cells to approximately the level of activity in Tsc2-expressing cells.
Surprisingly, despite the striking effect of mTOR kinase activity inhibition with Torin 1 on Microprocessor activity (Fig. 1), global analysis of microRNA showed that more miRs were upregulated than downregulated by Torin 1 treatment. The reasons for this apparently contrasting data are currently unknown. The timing of the microRNA analysis is one potential factor, since miRs are long lived (24). Another possibility is that Torin 1 impacts multiple steps in microRNA biogenesis, in addition to Microprocessor activity, leading to a global increase in specific miRs. Finally, it is important to note that the global analysis of miRs assays relative levels of individual miRs, while our assay of small RNA did reveal a decrease in small RNA with Torin 1 treatment (Fig. 3B); therefore, it is possible that specific miRs are upregulated by Torin 1 while the total quantity of miRs is decreased. It is interesting that Rapamycin treatment of Tsc2 KO MEFs led to a decrease in small RNA, but does not decrease the activity of the Microprocessor reporter in Tsc2 KO MEFs (Fig. 2B). Potential explanations for this include the possibility that other small RNAs may be impacted by Rapamycin, since small RNA includes miRNA as well as small nucleolarRNA, piwi-interacting RNA and others, and/or that Rapamycin regulates microRNA biogenesis via additional Microprocessor independent mechanisms. Further studies will be needed to address these hypotheses.
It was previously shown by Ye et al. that 4 days of Rapamycin treatment increases Drosha levels via the E3 ubiquitin ligase MDM2 in Tsc1-deficient MEFs (25). In contrast, we found no change in Drosha levels upon Rapamycin treatment of Tsc2-deficient MEFs at 24 h, 3 days or 7 days. We also found no changes in MDM2 levels in our cells with Rapamycin treatment (data not shown), again in contrast to the prior work. The reasons for these differences are currently unclear, but could involve differences between Tsc1 and Tsc2-deficient cells, or differences in growth conditions since Microprocessor activity is clearly regulated by cell density (10). We also note that Ye et al. did not measure Microprocessor activity.
Regulation of Microprocessor activity by cell density was previously shown to be YAP dependent. In low cell density YAP sequesters p72, a regulatory component, from the Microprocessor complex, and transfers it from the nucleus to the cytoplasm. At higher cell density Yap is retained in the cytoplasm, allowing p72 to bind to the Microprocessor and increase its activity (10). Since Yap has also been shown to be upregulated in TSC1/TSC2-deficient cells due to impairment of autophagy (17), we considered the possibility that TSC2 regulates Microprocessor activity via YAP. However, in our cells we did not observe a significant TSC2-dependent or mTOR-dependent change in YAP total protein levels or nuclear localization. It remains possible that under different conditions, YAP may participate in the regulation of Microprocessor activity in TSC2-deficient cells.
Inhibition of mTOR kinase activity with Torin 1, which inhibits both mTORC1 and mTORC2, had a greater impact on Microprocessor activity than inhibition of mTORC1 with Rapamycin. This is consistent with our findings that both mTORC1 and mTORC2 inhibit Microprocessor activity, and with the fact that mTORC1 is only partially inhibited by Rapamycin (26,27).
GSK3β is known to be regulated by mTORC2 via Akt, where mTORC2 phosphorylates Akt at S473, which results in increased inhibitory phosphorylation of GSK3β at S9. Furthermore, mTORC1 has been implicated in regulation of GSK3β via Akt and S6K (18,28). We found that downregulation of Raptor increased phosphorylation of Akt (S473) and increased phosphorylation of GSK3β at S9, which is associated with GSK3β inhibition, suggesting that mTORC1 inhibition may be impacting Microprocessor activity via mTORC2.
In summary, our data position mTORC1 and mTORC2 as key regulators of Microprocessor activity and microRNA biogenesis, with broad implications for cell growth, tumorigenesis and cellular homeostasis.
Materials and Methods
Cell lines and culture
Tsc2flox/flox-Rosa26-CreERT2 mice were generated by crossing Tsc2flox/flox mice (gift from Michael Gambello) with Rosa26-CreERT2 mice (Jackson Laboratory). MEFs were then isolated from Tsc2flox/flox-Rosa26-CreERT2 embryos at E13-E14 and were treated with 4-hydroxytamoxifen (4HT) (Sigma) (2 um for 1 week). Clones with knockout of Tsc2 (Tsc2 KO MEFs) were selected and cultured at 37°C in 5% CO2 in DMEM containing 4.5 g/l glucose, pyruvate and glutamine supplemented with 10% FBS. Cells treated with ethanol were used as controls (Tsc2 WT MEFs). Cell lines were validated by immunoblot analysis for Tsc2 expression and phosphorylation of the ribosomal subunit S6 (235/236) in serum free conditions. HeLa cells stably expressing a Microprocessor reporter have been reported previously (10). A construct that lacks the stem-loop (which is cleaved by Drosha) was transiently expressed using FuGENE 6 (Promega) or Lipofectamine 3000 (Invitrogen) and compared with the full-length construct including the stem-loop (pri-miR-125b-1 construct). MEFs with inducible knockdown of Raptor and Rictor were a gift from Michael Hall (29). These MEFs were treated with 4HT (1 um, 4 days of treatment) to knockdown Raptor or Rictor. Cells treated with ethanol were used as controls. Cell lines were validated by immunoblot analysis for Raptor, Rictor expression and phosphorylation of Akt and GSK3β. For all cell lines, MycoAlert (Lonza, Walkersville, MD) was used to detect mycoplasma contamination. Cells were re-tested every other month and were consistently negative for mycoplasma.
Antibodies and drugs
The following antibodies were used: S6, P-S6 (S235/236), 4E-BP1, p-4E-BP1 (Thr47/46), Drosha, Akt, p-Akt (S-473), TSC2, GSK3β, p-GSK3β (S9), YAP, GAPDH and CREB (all from Cell Signaling Technology) and β-actin (Sigma). Rapamycin was purchased from LC Laboratories and Torin 1 from Tocris. GSK3β inhibitors: AR-A014418, CHIR99021 and MK2206 were from Selleckchem, and lithium chloride was from Sigma.
Cell proliferation by crystal violet assay
Cells were seeded at 5000 or 10 000 cells/well in 96-well plates and processed with crystal violet (Sigma). Cells were fixed for 5 min with 10% formalin and then stained with 0.5% crystal violet diluted in distilled water for 20 min. Crystal violet was then removed and cells were washed with H2O. Plates were dried completely and crystal violet stain was solubilized using 100 ul of methanol and absorbance was read at 540 nm.
Microprocessor activity assay
Microprocessor activity using the dual-luciferase reporter was normalized to the control Firefly luciferase activity (10). Luciferase assays were performed using the dual-luciferase reporter system (Promega).
Immunoblotting
For western blot analyses, cells were lysed in 1 × RIPA (Cell Signaling Technology) containing protease and phosphatase inhibitors (Sigma). The lysates were normalized to protein concentration, resolved by electrophoresis on a 4–12% Bis–Tris gel (Invitrogen), and transferred to a PVDF membrane. Blots were blocked with 5% milk or bovine serum albumin (BSA), incubated with primary then secondary antibodies and chemiluminescence was visualized using Super Signal ECL reagent (Pierce) on SynGene G-BOX gel documenting system. Nuclear and cytoplasmic fractionation was performed using Cell LyticTM NuCLEARTM Extraction kit from Sigma.
Small-interfering RNA transfections
siRNA Smartpools targeting TSC2, Raptor, Rictor, GSK3β and nontargeting controls were obtained from Dharmacon (Lafayette, CO, USA) and used at 60 μm final concentration. Cells were transfected with Lipofectamine-RNAiMAX Reagent Invitrogen (Carlsbad, CA, USA) according to the manufacturer’s protocol.
Small RNA assay
Tsc2 WT and KO MEFs were seeded on 6-well plates at lower density, as defined in Figure 1A. The cells were trypsinized and counted following Rapamycin (20 nm), Torin 1 (250 nm) or DMSO control treatments. A total of 300 000 cells were used for miRNA isolation using miRCURYTM RNA Isolation Kit (Exiqon), followed by Qubit measurement. Qubit microRNA assay kit (Molecular Probes) was used for quantification of small RNA.
microRNA expression profiling
microRNAs were isolated using the miRCURY RNA Cell and Plant Isolation kit (Exiqon) from equal number of cells under various treatment conditions, then sent to Exiqon for real-time PCR panel analysis of microRNA. Each RNA sample was successfully reverse transcribed (RT) into cDNA and run on the miRCURY LNATM Universal RT microRNA PCR Mouse/Rat panel I + II of 752 microRNAs using ExiLENT SYBR® Green master mix. All assays were inspected for distinct melting curves. Measurements of 5 Cq less than the negative control and with Cq < 37 were included in the data analysis. All data were normalized to the RNU5G (RNU5G Cq–Assay Cq).
Statistical analyses
Data are presented as the mean ± standard deviation (SD). Normally distributed data were analyzed for statistical significance with Student’s unpaired t-test. Statistical significance was defined as P < 0.05. For microRNA expression profiling t-test was performed and both raw P-values and q-values adjusted for multiple testing by the Benjamini–Hochberg correction were assessed. The normal distribution of the data was assessed by a Shapiro–Wilk normality test.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Michael Gambello for providing the Tsc2flox/flox mice and Michael Hall for providing Raptor and Rictor inducible knockout MEFs. We also thank Andras Boeszoermenyi for helpful discussions. Finally, we acknowledge the National Institutes of Health and The LAM Foundation for their financial support of this work.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (DK102146 to E.P.H.); Postdoctoral Fellowship from The LAM Foundation (LAM00102F01–14 to H.C.L.).
References
- 1. Crino P.B., Nathanson K.L., Henske E.P. (2006) The tuberous sclerosis complex. N. Engl. J. Med., 355, 1345–1356. [DOI] [PubMed] [Google Scholar]
- 2. Duvel K., Yecies J.L., Menon S., Raman P., Lipovsky A.I., Souza A.L., Triantafellow E., Ma Q., Gorski R., Cleaver S. (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell, 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Henske E.P., Jóźwiak S., Kingswood J.C., Sampson J.R., Thiele E.A. (2016) Tuberous sclerosis complex. Nat. Rev. Dis. Primers, 2, 16035.. [DOI] [PubMed] [Google Scholar]
- 4. Laplante M., Sabatini D.M. (2012) mTOR signaling in growth control and disease. Cell, 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen C.Z. (2005) MicroRNAs as oncogenes and tumor suppressors. N. Engl. J. Med., 353, 1768–1771. [DOI] [PubMed] [Google Scholar]
- 6. Garzon R., Calin G.A., Croce C.M. (2009) MicroRNAs in Cancer. Annu. Rev. Med., 60, 167–179. [DOI] [PubMed] [Google Scholar]
- 7. Croce C.M. (2009) Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet., 10, 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bartel D.P. (2009) MicroRNAs: target recognition and regulatory functions. Cell, 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winter J., Jung S., Keller S., Gregory R.I., Diederichs S. (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol., 11, 228–234. [DOI] [PubMed] [Google Scholar]
- 10. Mori M., Triboulet R., Mohseni M., Schlegelmilch K., Shrestha K., Camargo F.D., Gregory R.I. (2014) Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell, 156, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fletcher C.E., Godfrey J.D., Shibakawa A., Bushell M., Bevan C.L. (2016) A novel role for GSK3beta as a modulator of Drosha microprocessor activity and microRNA biogenesis. Nucleic Acids Res., 45, 2809–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trindade A.J., Medvetz D.A., Neuman N.A., Myachina F., Yu J., Priolo C., Henske E.P. (2013) MicroRNA-21 is induced by rapamycin in a model of tuberous sclerosis (TSC) and lymphangioleiomyomatosis (LAM). PLoS One, 8, e60014.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lam H.C., Liu H., Baglini C.V. (2017) Rapamycin-induced miR-21 promotes mitochondrial homeostasis and adaptation in mTORC1 activated cells. Oncotarget, 8, 64714–64727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tang X., Zhang Y., Tucker L., Ramratnam B. (2010) Phosphorylation of the RNase III enzyme Drosha at Serine300 or Serine302 is required for its nuclear localization. Nucleic Acids Res., 38, 6610–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dai L., Chen K., Youngren B., Kulina J., Yang A., Guo Z., Li J., Yu P., Gu S. (2016) Cytoplasmic Drosha activity generated by alternative splicing. Nucleic Acids Res., 44, 10454–10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang Q., Li W., She H., Dou J., Duong D.M., Du Y., Yang S.H., Seyfried N.T., Fu H., Gao G.. et al. (2015) Stress induces p38 MAPK-mediated phosphorylation and inhibition of Drosha-dependent cell survival. Mol. Cell, 57, 721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liang N., Zhang C., Dill P., Panasyuk G., Pion D., Koka V., Gallazzini M., Olson E.N., Lam H., Henske E.P.. et al. (2014) Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med., 211, 2249–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H.H., Lipovsky A.I., Dibble C.C., Sahin M., Manning B.D. (2006) S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell, 24, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manning B.D., Toker A. (2017) AKT/PKB Signaling: navigating the Network. Cell, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang X., Li M., Tucker L., Ramratnam B. (2011) Glycogen synthase kinase 3 beta (GSK3beta) phosphorylates the RNAase III enzyme Drosha at S300 and S302. PLoS One, 6, e20391.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao J., Tyburczy M.E., Moss J., Darling T.N., Widlund H.R., Kwiatkowski D.J. (2016) Tuberous sclerosis complex inactivation disrupts melanogenesis via mTORC1 activation. J. Clin. Invest., 127, 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flavin R.J., Cook J., Fiorentino M., Bailey D., Brown M., Loda M.F. (2011) beta-Catenin is a useful adjunct immunohistochemical marker for the diagnosis of pulmonary lymphangioleiomyomatosis. Am. J. Clin. Pathol., 135, 776–782. [DOI] [PubMed] [Google Scholar]
- 23. Mak B.C., Kenerson H.L., Aicher L.D., Barnes E.A., Yeung R.S. (2005) Aberrant beta-catenin signaling in tuberous sclerosis. Am. J. Pathol., 167, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bail S., Swerdel M., Liu H., Jiao X., Goff L.A., Hart R.P., Kiledjian M. (2010) Differential regulation of microRNA stability. RNA, 16, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ye P., Liu Y., Chen C., Tang F., Wu Q., Wang X., Liu C.G., Liu X., Liu R., Liu Y.. et al. (2015) An mTORC1-Mdm2-Drosha axis for miRNA biogenesis in response to glucose- and amino acid-deprivation. Mol. Cell, 57, 708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thoreen C.C., Sabatini D.M. (2009) Rapamycin inhibits mTORC1, but not completely. Autophagy, 5, 725–726. [DOI] [PubMed] [Google Scholar]
- 27. Choo A.Y., Yoon S.O., Kim S.G., Roux P.P., Blenis J. (2008) Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. U. S. A., 105, 17414–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Breuleux M., Klopfenstein M., Stephan C., Doughty C.A., Barys L., Maira S.M., Kwiatkowski D., Lane H.A. (2009) Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther., 8, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cybulski N., Zinzalla V., Hall M.N. (2012) Inducible raptor and rictor knockout mouse embryonic fibroblasts. Methods Mol. Biol., 821, 267–278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.