

Abstract
Research in two fronts has enabled the development of therapies that provide significant benefit to cancer patients. One area stems from a detailed knowledge of mutations that activate or inactivate signaling pathways that drive cancer development. This work triggered the development of targeted therapies that lead to clinical responses in the majority of patients bearing the targeted mutation, although responses are often of limited duration. In the second front are the advances in molecular immunology that unveiled the complexity of the mechanisms regulating cellular immune responses. These developments led to the successful targeting of immune checkpoints to unleash anti-tumor T cell responses, resulting in durable long-lasting responses but only in a fraction of patients. In this review we discuss the evolution of research in these two areas and propose that intercrossing them and increasing funding to guide research of combination of agents represent a path forward for the development of curative therapies for the majority of cancer patients.
Introduction
The scientific community united against a common enemy in 1971 when President Nixon signed a bill initiating the “War on Cancer”, which provided funding for scientific research focused on improving our understanding and treatment of cancer. Without doubt, the intervening years were followed by great advances in the elucidation of the molecular mechanisms that regulate growth and death of normal cells, including a deep understanding of how these pathways progressively go awry during the development of cancer. This understanding led to the era of genomically-targeted therapies and “precision medicine” in the treatment of cancer. Genomically-targeted therapies can result in remarkable clinical responses. The ability of cancer cells to adapt to these agents by virtue of their genomic instability and other resistance mechanisms eventually leads to disease progression in the majority of patients nonetheless. Unraveling the mechanisms by which cancer cells become resistant to drugs and developing new agents to target the relevant pathways have become logical next steps, in this approach for cancer treatment. However, given the genetic and epigenetic instability of cancer cells, it is likely that each new drug or combination of drugs targeting the tumor cells will meet with more complex mechanisms of acquired resistance. Recent findings suggest that T cells, bearing antigen receptors that are randomly generated by random rearrangement of gene segments followed by selective process that generate a vast repertoire of T cell clones, provide sufficient diversity and adaptability to match the complexity of tumors. Discoveries regarding regulation of T cell responses have provided key principles regarding immune checkpoints that are being translated into clinical success, with durable responses and long-term survival greater than 10 years in a subset of patients with metastatic melanoma as well as yielding promising results in several other tumor types. These advances and the perspective of combining genomically-targeted agents and immune checkpoint therapies, we are finally poised to deliver curative therapies to cancer patients. To support this goal and accelerate these efforts, changes in directions of research support and funding may be required.
Precision Medicine: Targeting the Drivers
In the past three decades enormous strides have been made in elucidating the molecular mechanisms involved in the development of cancer (Hanahan and Weinberg, 2011). It is now clear that the oncogenic process involves somatic mutations that result in activation of genes that are normally involved in regulation of cell division and programmed cell death, as well as inactivation of genes involved in protection against DNA damage or driving apoptosis (Bishop, 1991; Solomon et al., 1991; Weinberg, 1991; Knudson, 2001). These genetic links led to the decision early in the war on cancer to undertake sequencing of cancer genomes to provide a comprehensive view of somatic mutational landscapes in cancer and identify possible therapeutic targets. Infrastructure and funding were provided to coordinate the sequencing efforts. It has become apparent that the level of somatic mutations differs widely between and within different tumor types ranging from very low rates in childhood leukemias to very high rates in tumors associated with carcinogens (Alexandrov et al., 2013).
Mutations can be divided into two broad classes: those whose products “drive” tumorigenesis in a dominant fashion, and “passengers” with no obvious role in the tumor causation. The Cancer Genome Atlas (TCGA) projects have enabled identification of many of these mutations (Chen et al., 2014; Cancer Genome Atlas Research Network, 2014). This has allowed for the rational design of drugs that target and selectively interfere with oncogenic signaling pathways. This approach has revolutionized cancer medicine by moving away from the “one size fits all” approach – for instance traditional chemotherapy, which attacks all dividing cells including both cancer, differentiating or regenerating normal cells – to a more personalized strategy of treating patients with a specific drug only if their cancer bears particular molecular mutations that are target of that drug.
As an example of genomically-targeted therapies, an inhibitor against BRAF was developed when it was discovered that approximately 40–60% of cutaneous melanomas carry mutations in BRAF, which induces constitutive activation of the MAPK pathway (Curtin et al., 2005; Davies et al., 2002). In a randomized phase III trial comparing a BRAF-inhibitor (vemurafenib) versus dacarbazine, the vemurafenib treatment group had a response rate of approximately 48% versus 5% in the dacarbazine arm (Chapman et al., 2011). However, the median duration of response was short, only 6.7 months (Sosman et al., 2012). Another oncogenic pathway that has been targeted is the tyrosine kinase chromosomal rearrangement, which results in the fusion oncogene EML4-ALK that is found in approximately 5% of NSCLC patients (Soda et al., 2007). The EML4 fusion partner mediates ligand-independent oligomerization and/or dimerization of ALK, resulting in constitutive kinase activity. Standard chemotherapies in this subgroup of patients have been associated with response rates of up to 10% (Hanna et al., 2004). Crizotinib, a tyrosine kinase inhibitor targeting ALK (Kwak et al., 2010), was shown to elicit a response rate of approximately 65% with a median duration of response of less than 8 months in a phase III trial (Shaw et al., 2013). Although there was a significant increase in progression-free survival for patients treated with crizotinib, regrettably there was no overall survival benefit in the interim analysis. Therefore, while the concept of targeting “driver mutations” has great merit and has demonstrated clinical responses, the reality remains that the majority of patients treated with these agents will derive short-term clinical responses with eventual development of resistance mechanisms that lead to disease progression and death.
Mechanisms operative in acquired resistance fall into three main categories: alterations in the targeted gene (as a result of mutation, amplification, or alternative splicing); other changes that do not affect the original target but re-activate the signaling pathway involved (i.e. NRAS and MEK mutations in BRAF-mutant melanoma); and changes that activate alternate pathways (such as activation of growth factor receptors). Considerable effort has gone into finding ways to enhance efficacy of genomically-targeted therapies. One effort involves multiple agents that target different molecules in the same pathway, such as the combination of a BRAF-inhibitor plus a MEK-inhibitor (Larkin et al., 2014; Robert et al., 2015). This approach helps to reduce compensatory feedback loops as well as to block the development of resistance due to mutations downstream that pathway. A different strategy consist of blocking parallel pathways to prevent emerging resistance (Martz et al., 2014). Still, the chief challenge of these combinatorial approaches is the multiplicity of resistance mechanisms and the fact that different mechanisms may be in operation in different cells due to intratumor heterogeneity. Given these observations, it is difficult to envision realistic approaches to effectively overcome the myriad of resistance mechanisms that may arise in the course of cancer treatment. The continued evolvability of the tumor cells and their mechanisms of escape from targeted therapies raise the question as to whether combinations of genomically-targeted agents will ever be curative.
Advantages of Mobilizing T cells for Cancer Therapy
As the knowledge of the intricate biology of cancer has progressed, so has the understanding of the fundamental cellular and molecular mechanisms that orchestrate the interplay of the innate and adaptive arms of the immune system. In a simplistic way, the innate system is comprised primarily of cytokines, the complement system and phagocytes such as macrophages, neutrophils, dendritic cells and natural killer (NK) cells. Cells of the innate immune system have hard-wired receptors to detect products of infectious microorganisms and dying cells. Macrophages and neutrophils provide an early defense against microorganisms, while dendritic cells provide a key interface to the adaptive immune system, comprised of B and T cells with their somatically generated, clonally expressed repertoire of antigen receptors.
The understanding of the basic principles governing the controlling immunity provided the rational for the development of powerful strategies to actively engage the immune system for cancer therapy. Strategies to unleash T cells against tumors are particularly compelling, as the activity of these cells present important features that are advantageous over other cancer therapies. The first is their specificity. T cells express antigen receptors that recognize cell surface complexes of MHC molecules and peptides sampled from virtually all the proteins in the cell, and are not limited to peptide antigens derived from cell surface molecules. The second feature is memory. Primary T cell responses are generally followed by the production of long-lived memory T cells with accelerated kinetics of secondary response if the antigen recurs. Finally, the T cell response is adaptable and can accommodate not only tumor heterogeneity, but also responses to novel antigens expressed by recurring tumors. It has been calculated that the somatic recombination process that generates the antigen receptors of T cells can generate as many as 1015 different receptors (Davis and Bjorkman, 1988). Of this theoretical number, each individual human has perhaps 109 different receptors. The immense size of the repertoire suggests that the immune system is indeed well-equipped to deal with mutability and adaptability of cancer.
Harnessing T Cell Responses to Tumor Antigens
With the advent of genomic and cDNA expression cloning methods, and sequencing of peptides eluted from tumor cell MHC molecules, an avalanche of tumor antigens defined by tumor specific T cells have been identified in both mice and in humans. Most of these are shared between cancer cells of different individuals, and fall into four groups: products of oncogenic viruses (Epstein-Barr virus in certain leukemias and human papilloma virus in cervical and some head and neck cancers); antigens related to tissue specific differentiation molecules (tyrosinase and related proteins in melanoma and prostate specific antigen and prostatic acid phosphatase in prostate cancer); molecules normally expressed only during fetal development (carcino-embryonic antigen in colon cancer, α-fetoprotein in liver cancer); and cancer-testes (CT) antigens, which are normally expressed during gametogenesis but are found in many cancer cells as a result of changes in epigenetic regulation (MAGE, NY-ESO-1).
Additionally, somatic mutations also can result in the generation of tumor specific peptides with the potential to bind MHC molecules and therefore be recognized by the immune system as neoantigens (Sjoblom et al., 2006; Segal et al., 2008). The analysis of the epitope landscape of breast and colon carcinoma cells revealed that the products of 7–10 mutant genes in colorectal and breast cancer, respectively, have the potential for binding to HLA-A*0201 alone. Since each heterozygote individual carries as many as 6 different HLA class I genes, this means an average of 42–60 potential neoantigens that can be presented to T cells. In support to these estimates, recent studies have demonstrated that neoantigens generated by somatic mutation are recognized by T cells in both mouse and human cancers (Linnemann et al., 2015; Gros et al., 2014; Tran et al., 2014; Gubin et al., 2014).
At first, as a result of earlier studies identifying shared antigens, the field of cancer immunotherapy became focused on developing therapeutic vaccines to expand T cells against these shared antigens expressed on tumors. Many studies focused on stimulating T cell responses with peptides, proteins, whole tumor cells including those modified to express cytokines, DNA, recombinant viral-based vaccines or antigen-pulsed dendritic cells, given alone or in combination with various adjuvants or cytokines. Although these trials were conducted with the best available science at the time and provided promising anecdotal evidence that induction of immune responses could elicit clinical benefit, they remained largely negative and generally failed to show objective clinical responses (see Rosenberg et al., 2004 for review). Enthusiasm waned somewhat as the number of failed clinical trials mounted.
Many reasons might have contributed to the failure of these vaccination strategies, including choice of antigen, failure to provide adequate costimulation, or functional inactivation of tumor-reactive T cells (Melero et al., 2014). A number of T cell extrinsic suppressive mechanisms such as TGFβ, FoxP3+ regulatory T cells (Treg), and tryptophan metabolites (IDO) that can hamper anti-tumor responses have also been identified, and there have been efforts to minimize the suppressive effects of these in pre-clinical and clinical studies.
Unraveling the Complexity of T Cell Activation
Another contributing factor to the failure of earlier cancer vaccine trials was perhaps the lack of understanding and appreciation of the full complexity of cell intrinsic pathways that regulate T cell activation. By the late 1980s it was known that simple engagement of peptide/MHC complexes by the antigen receptor is insufficient for activation of T cells and may render them anergic (Jenkins and Schwartz, 1987; Mueller et al., 1989). In order to become fully activated, T cells must encounter antigen in the context of antigen presenting cells (APC) such as dendritic cells, which provide costimulatory signals mediated by B7 molecules (B7–1 and B7–2) that will engage their ligand, CD28, in the T cell (Greenwald et al., 2005). Thus, T cells specific for a tumor antigen will not be activated by an initial encounter with tumor cells or may even be rendered anergic since, with the exception of a few lymphomas, tumors do not express costimulatory B7 molecules (Townsend and Allison, 1993). Thus, tumors are essentially invisible to T cells until the T cells are activated as a result of cross-priming by dendritic cells that present tumor antigens acquired from dying tumor cells. Simultaneous recognition of antigen/MHC complexes and costimulatory ligands by T cells initiates a complex set of genetic programs that result in cytokine production, cell cycle progression and production of anti-apoptotic factors that result in proliferation and functional differentiation of T cells. Consistent with the importance of both antigen receptor and costimulatory signals in initiating anti-tumor responses, many therapeutic vaccines now incorporate both antigen and dendritic cells or agents that enhance costimulatory signaling.
By the mid-90s it became clear that T cell priming elicits not only programs leading to induction of T cell responses, but also a parallel program that will eventually stop the response. The critical inhibitory program is mediated by CTLA-4, a homolog of CD28 that also binds B7–1 and B7–2, although with much greater avidity than that CD28. Expression of the ctla-4 gene is initiated upon T cell activation, and it traffics to and accumulates in the immunological synapse, eventually attenuating or preventing CD28 costimulation by competition for B7 binding and negative signaling (Walunas et al.,1994; Krummel and Allison, 1995). The fact that ctla-4 knockout mice suffer from a rapid and lethal lymphadenopathy (Waterhouse et al., 1995; Tivol et al., 1995; Chambers et al., 1997) speaks for a negative role for CTLA-4 in limiting T cell responses to prevent damage to normal tissues.
Thus, activation of T cells as a result of antigen receptor signaling and CD28 costimulation is followed not only by induction of genetic programs leading to proliferation and functional differentiation, but also induction of an inhibitory program mediated by CTLA-4, which will ultimately stop proliferation. Extrapolating this paradigm to anti-tumor T cell responses, if eradication of the tumor has not been completed by the time that the inhibitory signal of CTLA-4 is triggered, the T cells will be turned off and will be unable to complete the task. Importantly, this also suggests that after this program is initiated, vaccines used to stimulate antigen receptor signaling may actually serve to strengthen the “off” signal as a result of additional induction of ctla-4 expression by antigen receptor signaling. In any event, this suggests the importance of shifting strategies for cancer immunotherapy from activating T cells to unleashing them.
Inactivating the Brakes to Increase Anti-Tumor Immunity
Consistent with the observations that CD28 and CTLA-4 had opposing effects on T cell responses in vitro, in the late 90s it was found that while blocking antibodies to CD28 impaired anti-tumor responses in mice, blocking antibodies to CTLA-4 enhanced anti-tumor responses in mouse tumor models (Leach et al., 1996). In fact, the treatment of mice with anti-CTLA-4 antibodies as monotherapy results in complete tumor rejection and long lived immunity. Later on, mechanistic studies revealed that anti-tumor activity was associated with increased ratio of both CD4 and CD8 effector cells to FoxP3+ regulatory T cells (Quezada et al., 2006). The success of CTLA-4 blockade in these initial studies raised two compelling points. First, since the target molecule was on the T cell and not the tumor cell, it was feasible to imagine that the same strategy would work on many different histologic tumors as well as on tumors caused by different genetic lesions. Second, taking into consideration that CTLA-4 inhibited CD28-mediated costimulation by a cell intrinsic mechanism (Peggs et al., 2009), its blockade could allow for enhanced T cell costimulation, which in turn would increase the efficacy of tumor vaccines as well as agents that kill tumor cells under conditions that promote inflammatory responses. These possibilities were further supported by the results of a series of studies, in different mouse models, including the demonstration that blockade of CTLA-4 was not limited to any particular tumor type, but was rather broadly effective. CTLA-4 also was able to synergize with a vaccine consisting of tumor cells engineered to express the cytokine GM-CSF to eradicate tumors (Hurwitz et al., 1998; van Elsas et al., 1999). Finally, CTLA-4 could be combined with local delivery of irradiation, cryoablation, or an oncolytic virus to induce systemic tumor immunity and eradication of distant metastases (Zamarin et al., 2014; Waitz et al., 2012; Tang et al., 2014). These preclinical studies supported the development of clinical anti-CTLA-4 therapy.
Immune Checkpoint Therapy: The Clinical Success
CTLA-4 blockade was translated to the clinic with a fully human antibody to human CTLA-4 (ipilimumab, Medarex,Bristol-Myers Squibb). Tumor regression was observed in Phase I/II trials in patients with a variety of tumor types, including melanoma, renal cell carcinoma, prostate cancer, urothelial carcinoma, and ovarian cancer (Yang et al., 2007; Hodi et al., 2008; Carthon et al., 2010; van den Eertwegh et al., 2012). Two Phase III clinical trials with ipilimumab were recently completed in prostate cancer, the first in patients with castrate-resistant prostate cancer who had not received prior chemotherapy treatment and the second in a more advanced disease setting, in which patients with castrate-resistant prostate cancer presented disease that had progressed on chemotherapy treatment. The former trial is yet to be reported. The latter trial reports the lack of statistical significance (p-value of 0.053) to indicate a survival benefit for patients who received ipilimumab treatment. However, subset analyses indicate that patients who have favorable clinical characteristics such as lack of liver metastases do benefit from ipilimumab therapy (Kwon et al., 2014). Two Phase III clinical trials with anti-CTLA-4 (ipilimumab) were also conducted in patients with advanced melanoma and demonstrated improved overall survival for patients treated with ipilimumab (Hodi et al., 2010; Robert et al., 2011). Importantly, these trials indicate long-term durable responses with greater than 20% of treated patients living for more than 4 years, including a recent analysis indicating survival of 10 years or more for a subset of patients (Schadendorf et al., 2013). The FDA approved ipilimumab as treatment for patients with melanoma in 2011.
The clinical success of anti-CTLA-4 opened a new field termed “immune checkpoint therapy” as additional T cell intrinsic pathways were identified and targeted for clinical development (Sharma et al., 2011, Pardoll et al., 2012). Another T cell intrinsic inhibitory pathway identified after CTLA-4 was that mediated by PD-1 (programmed death 1) and its ligand PD-L1. PD-1 was initially cloned in 1992 in a study of molecules involved in negative selection of T cells by programed cell death in the thymus (Ishida et al., 1992). Its function as an immune checkpoint was not established until 2000 upon identification of its ligands (Freeman et al., 2000). PD-L1 was then shown to protect tumor cells by inducing T cell apoptosis (Dong et al., 2002). Later, preclinical studies in animal models evaluated anti-PD-1 and anti-PD-L1 antibodies as immune checkpoint therapies to treat tumors (Keir et al., 2008).
Much like CTLA-4, PD-1 is expressed only in activated T cells. However, unlike CTLA-4, PD-1 inhibits T cell responses by interfering with T cell receptor signaling as opposed to outcompeting CD28 for binding to B7. PD-1 also has two ligands, PD-L1 and PD-L2. PD-L2 is predominantly expressed on antigen presenting cells while PD-L1 can be expressed on many cell types including cells comprising the immune system, epithelial cells and endothelial cells. Antibodies targeting PD-L1 have shown clinical responses in multiple tumor types including melanoma, renal cell carcinoma, non-small cell lung cancer (Brahmer et al., 2012) and bladder cancer (Powles et al., 2014). Similarly, Phase I clinical trials with a monoclonal antibody against PD-1 demonstrated clinical responses in multiple tumor types including melanoma, renal cell carcinoma, non-small cell carcinoma (Topalian et al., 2012), Hodgkin’s lymphoma (Ansell et al., 2015) and head and neck cancers (Seiwert et al., 2014). Recently, a large Phase I clinical trial with an anti-PD-1 antibody known as MK-3475 showed response rates of ~37–38% in patients with advanced melanoma, including patients who had progressive disease after prior ipilimumab treatment (Hamid et al., 2013), triggering the approval of MK-3475 (pembroluzimab, Merck) by the FDA in September 2014. A Phase III clinical trial that treated patients with metastatic melanoma with a different anti-PD-1 antibody (nivolumab, Bristol-Myers Squibb, BMS) also demonstrated improved responses and overall survival benefit as compared to chemotherapy treatment (Caroline et al., 2015). Nivolumab was FDA-approved for patients with metastatic melanoma in December 2014. In addition, nivolumab was FDA-approved in March 2015 for patients with previously treated advanced or metastatic non-small cell lung cancer based on a Phase III clinical trial, which reported an improvement in overall survival for patients treated with nivolumab as compared to patients treated with docetaxel chemotherapy.
Since CTLA-4 and PD-1 regulate different inhibitory pathways on T cells, combination therapy with antibodies targeting both molecules was tested and found to improve anti-tumor responses in a pre-clinical murine model (Curran et al., 2010). A recently reported Phase I clinical trial with anti-CTLA-4 in combination with anti-PD-1 also demonstrated tumor regression in ~50% of treated patients with advanced melanoma, in most cases with tumor regression of 80% or higher (Wolchok et al., 2013). There are ongoing clinical trials with anti-CTLA-4 (ipilimumab, BMS or tremelimumab, MedImmune/Astrazeneca) plus anti-PD-1 or anti-PD-L1 in other tumor types, with preliminary data indicating promising results (Hammers et al., 2014; Callahan et al., 2014), which highlight this combination as an effective immunotherapy strategy for cancer patients.
As with other cancer therapies, immune checkpoint therapies may lead to side-effects and toxicities (see Postow et al., 2015; Gao et al., 2015 for recent reviews). Briefly these side-effects consist of immune-related adverse events that are defined by inflammatory conditions, including dermatitis, colitis, hepatitis, pancreatitis, pneumonitis and hypophysitis. These side-effects can be managed and usually involves administration of immunosuppressive agents such as corticosteroids, which do not appear to interfere with clinical benefit that is derived from the immune checkpoint agents. The profile of side-effects that occur with both anti-CTLA-4 and anti-PD-1/PD-L1 antibodies are similar; however, the side-effects appear to occur more frequently in the setting of anti-CTLA-4 therapy as compared to anti-PD-1 and anti-PD-L1 therapies. The continued success of immune checkpoint therapies in the clinic will require education of the oncology community regarding recognition and treatment of the side-effects elicited by these agents.
Novel Immunologic Targets for Cancer Immunotherapy
Although blockade of the CTLA-4 and PD-1/PD-L1 pathways are furthest along in clinical development, they only represent the tip of the iceberg in the realm of potential targets that can serve to improve anti-tumor responses. Ongoing studies on regulation of immune responses have led to the identification of multiple other immunologic pathways that may be targeted for the development of therapies, either as monotherapy or in combination strategies, for the successful treatment of cancer patients. These include immune checkpoints or inhibitory pathways, as well as co-stimulatory molecules, which act to enhance immune responses. A partial list of new immune checkpoints that are being evaluated in pre-clinical tumor models and/or in the clinic with cancer patients include LAG-3 (Treibel et al., 1990), TIM-3 (Sakuishi et al., 2010) and VISTA (Wang et al., 2011), while co-stimulatory molecules include ICOS (Fan et al., 2014), OX40 (Curti et al., 2013) and 4–1BB (Melero et al., 1997).
Of these emerging immune checkpoints, LAG-3 is the furthest along in clinical development with a fusion protein (IMP321, Immuntep) and an antibody (BMS-986016, BMS) in clinical trials. The fusion protein was tested as monotherapy in patients with renal cell carcinoma, which was well-tolerated and led to stabilization of disease in some patients (Brignone et al., 2009). IMP321 was also tested in combination with paclitaxel chemotherapy in patienst with metatstic breast cancer, which led to an objective response rate of 50% (Brignone et al., 2010). Based on these promising results, a Phase III clinical trial is expected to begin accrual in 2015. Other clinical trials are ongoing with an antibody against LAG-3 (BMS-986016), which is also being tested in combination with anti-PD-1 (nivolumab) (NCT01968109, clinicaltrials.gov). TIM-3 is another immune checkpoint for which agents are being developed for clinical testing. Pre-clinical studies indicate that TIM-3 is co-expressed with PD-1 on tumor-infiltrating lymphocytes and combination therapy targeting these two pathways improves anti-tumor immune responses (Sakuishi et al., 2010). Finally, an antibody targeting VISTA was recently shown to improve anti-tumor immune responses in mice (Le et al., 2014), with clinical development soon to follow. Again, these agents represent only a partial list of the immune checkpoints agents that are currently under development for clinical testing, with expectations that they will be tested in combination strategies based on in-depth analyses of human tumors to provide an understanding of co-expression of these, and other immunologic targets, to guide rational combinations
As regards the co-stimulatory molecules, OX40 and 41BB, which are members of the TNF-receptor superfamily, are furthest along in clinical development. A murine anti-OX40 antibody, given as a single dose, was tested in a Phase I clinical trial and found to have an acceptable safety profile as well as evidence of anti-tumor responses in a subset of patients (Curti et al., 2013). Humanized antibodies against OX40 are expected to enter clinical trial in 2015. Anti-41BB (BMS-663513) is a fully humanized monoclonal antibody that has been tested in a Phase I/II study in patients with melanoma, renal cell carcinoma and ovarian cancer, with promising clinical responses as well as toxicities, especially at higher doses, which led to re-evaluation of the dose and schedule of treatment (Sznol et al., 2008). Currently, there are 5 clinical trials with anti-41BB (urelumab, BMS-663513) that are recruiting patients with various tumor types (clinicaltrials.gov), including combination with anti-PD-1 (nivolumab), with data expected to be presented from these trials during the next 1–2 years. The third co-stimulatory molecule is inducible costimulator (ICOS), a member of the CD28/B7 family, whose expression increases on T cells upon T cell activation. ICOS+ effector T cells (Teff), as opposed to ICOS+ regulatory T cells (Treg), increase after patients receive treatment with anti-CTLA-4 (Liakou et al., 2008), correlating with clinical benefit in a small retrospective study (Carthon et al., 2010). ICOS thus may serves as a pharmacodynamic biomarker to indicate that anti-CTLA-4 has “hit its target” enhancing T cell activation (Ng Tang et al., 2013). Also, the association of agonistic targeting of ICOS and blockade of CTLA-4 can lead to improved anti-tumor immune responses and tumor rejection in mice (Fan et al., 2014). Anti-ICOS antibodies are expected to enter into clinical trials in 2015. It is likely that combination therapy to simultaneously engage co-stimulatory pathways and limit inhibitory pathways will be a successful path forward to provide clinical benefit. Importantly, based on the profile of toxicities observed to date, it will be critical to closely monitor these combination strategies for potential adjusts of dosage and management of toxicities that may arise.
Reconciliation: Curative Therapeutic Combinations
The last few decades have witnessed the emergence of two effective, but fundamentally different strategies for cancer therapy, each with its own strengths and weaknesses. Genomic-guided identification of mutations that drive cancer has led to the development of drugs that result in remarkable responses in the majority of patients whose tumors have the targeted lesion, but the responses are relatively short-lived. As was the case with chemotherapies, it is not unreasonable that combinations of genomically-targeted agents will be more powerful against cancer than single agents. It is possible that the use of multiple agents may enhance their effectiveness in terms of increasing overall survival. However, the myriad of mechanisms of acquired resistance and the complexity of the target landscape due to inherent genomic instability may prove extremely hard to overcome through the sole use genomically-targeted strategies, attaining to achieve cure. In contrast, immune checkpoint therapy is inherently multivalent, since targeting a single checkpoint can potentially release T cells with specificity for peptides derived from many different antigens present in a tumor, including differentiation, cancer testis, and even neoantigens generated by mutational events inherent in the genomic instability that drives cancer (Snyder et al., 2014; Linnemann et al., 2015). As a result of the generation of improved anti-tumor T cell responses, immune checkpoint therapy results in durable responses, but only in a fraction of patients. As discussed in the previous sections, it is certainly possible to target multiple immune checkpoints with different mechanisms for improved anti-tumor responses in greater numbers of patients. Will patients benefit from combination of these two strategies?
Efforts to combine molecularly-targeted agents and immunotherapy have already begun. A Phase I clinical trial with agents that inhibit receptor tyrosine kinases, sunitinib or pazopbnib, in combination with anti-PD-1 was recently reported and showed promising overall response rates of 40–50% in patients with metastatic RCC (Amin et al., 2014). These types of combinations will require further follow-up to evaluate for survival and durability of responses. An area that has not yet received enough attention is the immunological impact of genetically targeted agents. Vemurafenib, an FDA-approved BRAF-inhibitor used for the treatment of melanoma has been shown to increase expression of tumor antigens and MHC molecules (Frederick et al., 2013), increasing the sensitivity of the tumor cells to immune attack. Vemurafenib also has potent effects on T cells, enhancing the effects of antigen-mediated activation, perhaps as a result of enhanced activation of the MAP kinase pathway after T cell antigen receptor signaling (Atefi et al., 2014). These data suggest that certain agents may be well-suited for combination with immunotherapy. However, a clinical trial testing a BRAF-inhibitor (vemurafenib) in combination with anti-CTLA-4 (ipilimumab) was terminated due to hepatotoxicity (Ribas et al., 2013). A second clinical trial with a BRAF-inhibitor (dabrafenib) in combination with anti-CTLA-4 (ipilimumab) is currently ongoing and preliminary data indicate that this combination appears to be well-tolerated (Puzanov et al., 2014), which highlight the need to consider differences in drugs, dose and/or schedule when evaluating agents for combination strategies. Understanding how different genetically targeted agents affect the responsiveness to immunotherapy may help guide choices of combinations of drugs.
From a mechanistic perspective, it is possible that combination strategies with immune checkpoint therapies and genomically-targeted agents will result in induction of immune memory, leading to more durable control of tumor growth than what is achievable with either modality alone. Genomically-targeted therapies with high objective response rates actually could serve as ‘cancer vaccines’, inducing the killing of tumor cells and resulting in the release of tumor antigens and neoantigens, which can then be presented by APCs to tumor-specific T cells (Figure 1). These T cells would become activated but also upregulate inhibitory checkpoints such as CTLA-4 and PD-1, which can be blocked with antibodies to permit enhanced anti-tumor T cell responses, including memory T cell responses to enable long-term control of disease and possible cure. In addition, the use of targeted agents to directly kill tumor cells, with release of tumor antigens, may focus the activated immune response generated by immunotherapy agents on tumor antigens rather than self-antigens expressed on normal tissues, resulting in fewer adverse events. Furthermore, identification of neoantigens may result in the development of personalized vaccines comprised of these neoantigens for novel vaccine strategies plus immune checkpoint agents (Gubin et al., 2014; Tran et al., 2014; Linnemann et al., 2015).
Figure 1.
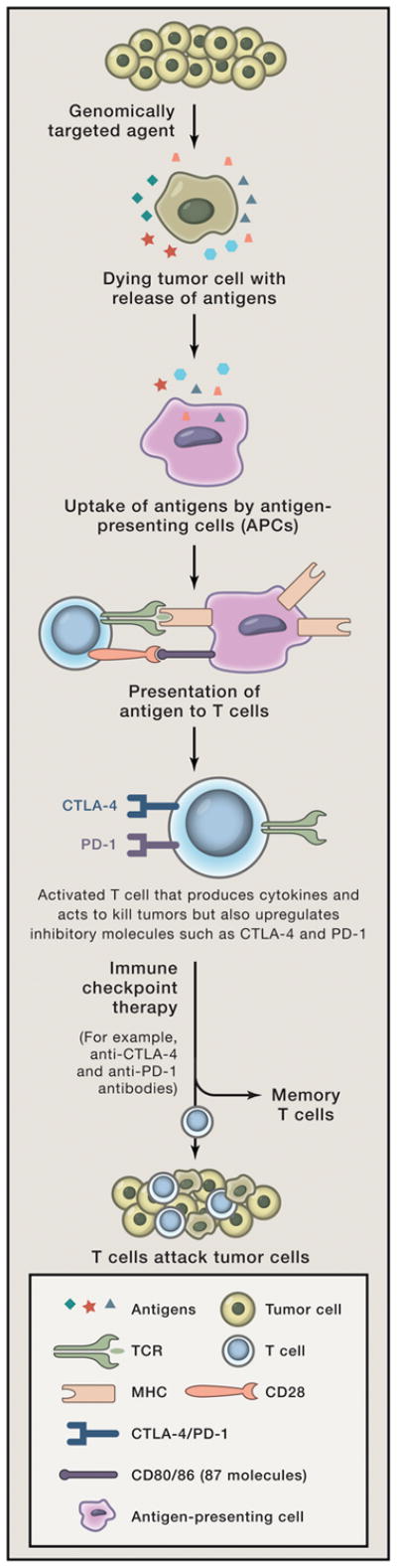
Depiction of tumor cells dying as a result of genomically-targeted therapies with release of tumor antigens. Tumor antigens are taken up by antigen-presenting cells (APCs) and presented in the context of B7 costimulatory molecules to T cells. T cells recognize antigens on APCs to become activated. Activated T cells also upregulate inhibitory checkpoints such as CTLA-4 and PD-1. Immune checkpoint therapy prevents attenuation of T cell responses thereby allowing T cells to kill tumor cells. Development of memory T cells that can reactivate in the presence of recurrent tumor.
Although it is clear that clinical responses can be elicited with immune checkpoint therapies or genomically-targeted agents, it appears that genomically-targeted agents alone tend to improve median survival without providing long-term durable responses (Figure 2, blue line). Targeting immune checkpoints improves median survival, but remarkably also provide long-term durable responses, raising the tail of the survival curve (Figure 2, green line). When combined, these therapies are likely to have additive or even synergistic therapeutic effect that not only would potentially further improve median survival, but also raise the tail of the survival curve, increasing the number of patients that appreciate long-term clinical benefit (Figure 2, red line).
Figure 2.

Depiction of Kaplan-Meier survival curve with genomically-targeted agents (blue line) as compared to standard therapies (purple line) indicating an improvement in median overall survival but lack of durable responses; improved median overall survival and durable responses in a fraction of patients treated with immune checkpoint therapy (green line); possibility for improved median overall survival with durable responses for the majority of patients in the setting of combination treatment with genomically-targeted agents and immune checkpoint therapy (red line)
A Future of Curative Cancer Therapies
Federal funding for research has been overwhelmingly directed towards genomically-targeted therapies as compared to immune checkpoint therapies. The fundamental research that led to the identification of CTLA-4 as an immune checkpoint, as well as the pre-clinical studies showing the potential of its blockade in cancer therapy, were funded by the National Cancer Institute, but since then there have been no major initiatives to accelerate progress in this area. Given the durability of the responses that have been obtained with immune checkpoint therapies, it seems reasonable also to allocate enough funds and resources to research focused on immune checkpoint therapies and combination therapy of genomically-targeted agents and immunotherapy with promising curative potential. Efforts to determine the impact of genomically-targeted therapies on the immune system should also be prioritized as they will help to identify which agents can enhance anti-tumor T cell responses and guide the choice of combinations from the two classes of agents. At this stage, it does not seem a stretch to say that increasing funding to combination therapies will be key to development of new safe treatments that may prove to be curative for many patients with many types of cancer.
Acknowledgments
Drs. Sharma and Allison are founders and advisors for Jounce Therapeutics. Dr. Sharma also serves as a consultant for Bristol-Myers Squibb, Amgen and Glaxo SmithKline. Dr. Allison is an inventor of intellectual property owned by the University of California, Berkeley, and licensed to Bristol-Myers Squibb and has received royalties from Bristol-Myers Squibb. Our work has been supported by the SU2C-CRI Dream Team Cancer Immunotherapy Grant (JPA and PS), PCF Challenge Grant in Immunology (JPA and PS), NCI/NIH 1-R01 CA1633793-01 (PS) and Cancer Prevention Research in Texas grants (JPA and PS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin A, Plimack ER, Infante JR, Ernstoff MS, Rini BI, McDermott DF, Knox JJ, Pal SK, Voss MH, Sharma P, et al. Nivolumab (anti-PD-1; BMS-93+669, ONO-438) in combination with sunitinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (MRCC) J Clin Oncol. 2014;32:5s. (suppl: abstr 4010) [Google Scholar]
- Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372(4):311–19. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–57. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JM. Molecular themes in oncogenesis. Cell. 1991;64:235–248. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignone C, Escudier B, Grygar C, marcu M, Treibel F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin Cancer Res. 2009;15:6225–31. doi: 10.1158/1078-0432.CCR-09-0068. [DOI] [PubMed] [Google Scholar]
- Brignone C, Gutierrez M, Mefti F, Brian E, Jarcau R, Cvitkovic F, Bousetta N, Medioni J, Gligorov J, Grygar C, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med. 2010;8:71. doi: 10.1186/1479-5876-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Bendell JC, Chan E, Morse M, Pillai RN, Bono P, Jaeger D, Evans TRJ, Chau I, Calvo E, et al. Phase I/II, open – label study of nivolumab (anti-PD-1; BMS-936558, ONO-4538) as monotherapy or combined with ipilimumab in advances or metastatic solid tumors. J Clin Oncol. 2014;32:5s. (suppl: abstr TPS3114) [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, Ku G, Troncoso P, Logothetis CJ, Allison JP, et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res. 2010;16(10):2861–71. doi: 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7(6):885–95. doi: 10.1016/s1074-7613(00)80406-9. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Higgs RE, Bhagwat SV, Buchanan S, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014;9(5):e989293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–80. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuswen T, Fox BA, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73(24):7189–98. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334(6181):395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Fan X, Quezada S, Sharma P*, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211(4):715–25. doi: 10.1084/jem.20130590. * Equal contribution corresponding authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, He Q, Subudhi S, Aparicio A, Zurita-Saavedra A, Lee DH, Jimenez C, Suarez-Almazor M, Sharma P. Review of immune-related adverse events in prostate canecr patients treated with ipilimumab: MD Anderson experience. Oncogene. 2015 doi: 10.1038/onc.2015.5. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–59. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammers JH, Plimack ER, Infante JR, Ernstoff MS, Rini BI, McDermott DF, Razak ARA, Pal SK, Voss MH, Sharma P, et al. Phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC) J Clin Oncol. 2014;32:5s. doi: 10.1200/JCO.2016.72.1985. (suppl: abstr 4504) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. Review. [DOI] [PubMed] [Google Scholar]
- Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol. 2004;22(9):1589–97. doi: 10.1200/JCO.2004.08.163. [DOI] [PubMed] [Google Scholar]
- Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, Macrae S, Nelson M, Canning C, Lowy I, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci USA. 2008;105(8):3005–10. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998;95(17):10067–71. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165(2):302–19. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995 Aug 1;182(2):459–65. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–12. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, et al. Combined vemurafenib and combimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Le MI, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, Wang L. VISTA regulates the devlopmentof protective antitumor immunity. Cancer Res. 2014;74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C, Sharma P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi T cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U S A. 2008;105(39):14987–92. doi: 10.1073/pnas.0806075105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015 Jan;21(1):81–5. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winter PS, Wang T, Alley HM, et al. Systematic identification of signaling pathways with potential to confer anticancer drug resistance. Sci Signal. 2014;7(357):ra121. doi: 10.1126/scisignal.aaa1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen Y, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, Mittler RS, Chen L. Monoclonal antibodies against the 4-1BB T cell activation molecule eradicate established tumors. Nat Med. 1997;3(6):682–5. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–24. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- Mueller DL, Jenkins MK, Schwartz RH. An accessory cell-derived costimulatory signal acts independently of protein kinase C activation to allow T cell proliferation and prevent the induction of unresponsiveness. J Immunol. 1989;142(8):2617–28. [PubMed] [Google Scholar]
- Ng Tang D, Shen Y, Sun J, Wen S, Wolchok JD, Yuan J, Allison JP, Sharma P. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti-CTLA-4 therapy. Cancer Immunol Res. 2013;1:229–234. doi: 10.1158/2326-6066.CIR-13-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012 Mar 22;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–25. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015 Jan 20; doi: 10.1200/JCO.2014.59.4358. (published online ahead of print). Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–62. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- Puzanov I, Callahan MK, Linette GP, Patel SP, Luke JJ, Sosman JA, Wolchok JD, Hamid O, Minor DR, Orford KW, et al. Phase I study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation-positive unresectable or metastatic melanoma (MM) J Clin Oncol. 2014;32:5s. (suppl; abstr 2511) [Google Scholar]
- Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116(7):1935–45. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hodi FS, Callahan MK, Konto C, Wolchok J. Hepatotoxicity with combination fo vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Robert C, Thomas L, Bondarenko I, O'Day S, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Lebbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 patwhays to reverse T cell exhaustion and restore anti-tumro immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Chen TT, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. J Clin Oncol. 2015 Feb 9; doi: 10.1200/JCO.2014.56.2736. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008 Feb 1;68(3):889–92. doi: 10.1158/0008-5472.CAN-07-3095. [DOI] [PubMed] [Google Scholar]
- Seiwert TY, Burtness B, Weiss J, Gluck I, Eder JP, Pai SI, Dolled-Filhart M, Emancipator K, Pathiraja K, Gause C, et al. A phase Ib study of MK-3475 in patients with human papillomavirus (HPV)-associated and non-HPV-associated head and neck (H/N) cancer. J Clin Oncol. 2014;32(5s) (suppl; abstr 6011) [Google Scholar]
- Solomon E, Borrow J, Goddard AD. Chromosome aberrations and cancer. Science. 1991;254(5035):1153–60. doi: 10.1126/science.1957167. [DOI] [PubMed] [Google Scholar]
- Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat Rev Cancer. 2011;11(11):805–12. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for linical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznol M, Hodi FS, Margolin K, McDermott DF, Ernstoff MS, Kirkwood JM, Wojtaszek C, Feltquate D, Logan T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA) J Clin Oncol. 2008;26(suppl 15) abstract 3007. [Google Scholar]
- Tang C, Wang X, Soh H, Seyedin S, Cortez MA, Krishnan S, Massarelli E, Hong D, Naing A, Diab A, et al. Combining radiation and immunotherapy: a new systemic therapy for solid tumors? Cancer Immunol Res. 2014;2(9):831–8. doi: 10.1158/2326-6066.CIR-14-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259(5093):368–70. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- Tran E, Torcutte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. Cancer Immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treibel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. LAG-3: a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171:1393–1405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Eertwegh AJ, Versluis J, van den Berg HP, Santegoets SJ, van Moorselaar RJ, van der Sluis TM, Gall HE, Harding TC, Jooss K, Lowy I, et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: a phase I dose-escalation study. Lancet Oncol. 2012;13(5):509–17. doi: 10.1016/S1470-2045(12)70007-4. [DOI] [PubMed] [Google Scholar]
- van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–66. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitz R, Fassò M, Allison JP. CTLA-4 blockade synergizes with cryoablation to mediate tumor rejection. Oncoimmunology. 2012;1(4):544–546. doi: 10.4161/onci.19442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–13. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu LF, Gondek D, Wang Y, Fava RA, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208:577–92. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. Tumor suppressor genes. Science. 1991;254(3035):1138–46. doi: 10.1126/science.1659741. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, Suri KB, Levy C, Allen T, Mavroukakis S, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30(8):825–30. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Sci Transl Med. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. 2014;6(226):226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]