

Abstract
Concomitant activation of the Wnt pathway and suppression of Mapk signaling by two small molecules in the presence of LIF (2i/L) induces a naïve state in mouse embryonic stem cells (ESCs) that resembles the inner cell mass (ICM) of the pre-implantation embryo1. Since the ICM exists only transiently in vivo, it remains unclear how sustained propagation of naïve ESCs in vitro affects their stability and functionality. Here we show that prolonged culture of male ESCs in 2i/L results in irreversible epigenetic and genomic changes that impair their developmental potential. Additionally, we find that female ESCs cultured in conventional serum/LIF (S/L) media phenocopy male ESCs cultured in 2i/L. Mechanistically, we demonstrate that Mek1/2 inhibition is predominantly responsible for these effects, in part through downregulation of DNA methyltransferases and their associated cofactors. Finally, we show that replacement of the Mek1/2 inhibitor with a Src inhibitor preserves the epigenetic and genomic integrity as well as developmental potential of ESCs. Taken together, our data suggest that, while short-term suppression of Mek1/2 in ESCs helps maintain an ICM-like epigenetic state, prolonged suppression results in irreversible changes that compromise their developmental potential.
Dysregulation of Wnt/Mapk signaling as well as DNA methylation have been linked to cellular transformation and chromosomal instability in ESCs2–5. Therefore, we sought to determine whether sustained perturbation of the Mapk/Wnt pathway and associated DNA hypomethylation during 2i/L culture impacts the stability and functionality of ESCs. Specifically, we utilized three isogenic male ESC lines (129S6 × C57B6 F1) that were derived in 2i/L and then cultured in S/L for 4 additional passages (p) (Fig. 1a). Each ESC line was subsequently passaged onto a feeder layer of irradiated MEFs in either 2i/L or S/L and propagated for an additional 6 or 16 passages (final p10 or p20, respectively). To assess the reversibility of any observed changes, we also switched the p20 2i/L-cultured ESC lines back into S/L for an additional 3 or 10 passages.
Figure 1. Erosion of genomic imprints in 2i/L-cultured male ESCs and S/L-cultured female ESCs.
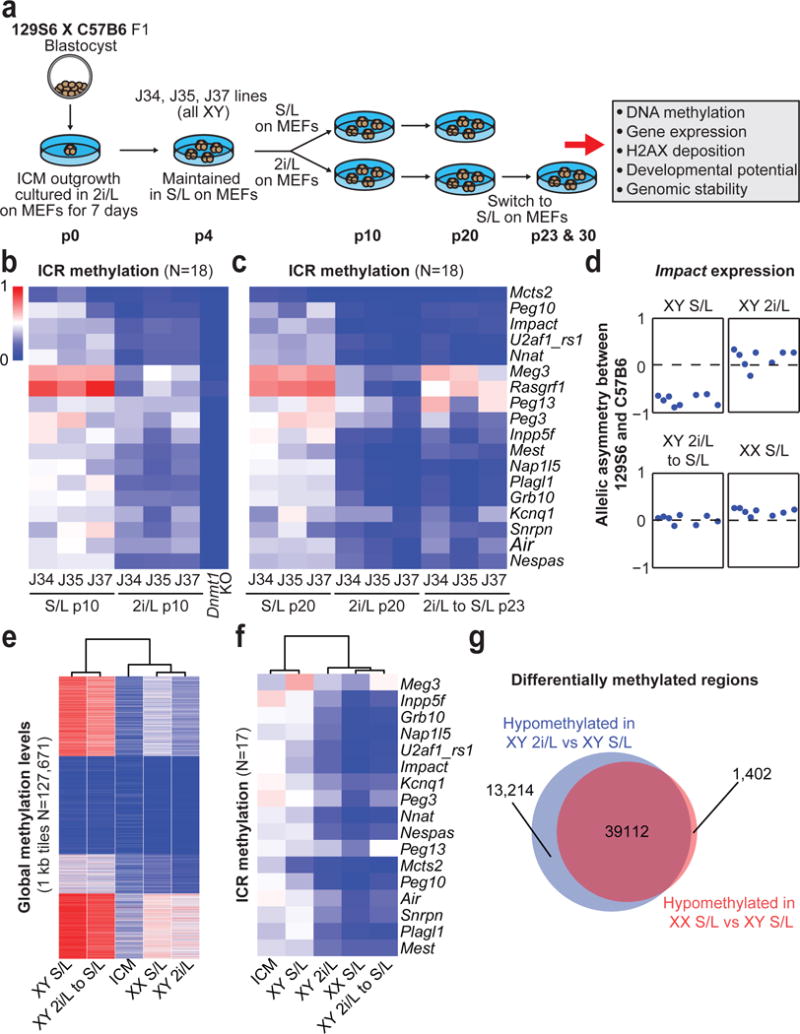
(A) Schematic of experimental design. p, passage.
(B,C) ICR methylation levels in ESCs at p10 (B) and p20, 23 (C).
(D) Allelic expression of the imprinted gene Impact.
(E,F) Global (D) and ICR (E) methylation levels in male ESCs at p20 and 23 (n=3 biological replicates) and female ESCs at p6 (n=3 biological replicates). ICM, Inner cell mass.
(G) Overlap of differentially methylated regions.
We assessed global DNA methylation patterns in our cultured ESCs using reduced representation bisulfite sequencing (RRBS). Similar to previous reports6–8, global methylation levels of 2i/L-cultured ESCs were lower than S/L-cultured ESCs and became remethylated when switched back to S/L (Extended Data Fig. 1a–c). This demethylation and subsequent remethylation occurs across most methylated features including CpG islands, shores, SINEs, LINEs and LTRs (Extended Data Fig. 1d,e). In contrast, 2i/L culture caused a progressive and irreversible erosion of DNA methylation at the majority of imprinted control regions (ICRs), resulting in the biallelic expression of the imprinted gene, Impact (Fig. 1b–d and Extended Data Fig. 1f,g). Additionally, we confirmed biallelic expression of additional imprinted genes using a Mus musculus × Mus spretus F1 stem cell line cultured in 2i/L for 6 passages (Extended data Fig. 1h). Considering that genomic imprinting is essential for development, these results suggest that 2i/L-cultured ESCs may.
Female ESCs cultured in S/L exhibit attenuated Mapk signaling, increased Wnt signaling, and upregulation of transcription factors associated with a naïve-like state when compared to male ESCs cultured in S/L9. Furthermore, female ESCs cultured in S/L were reported to be hypomethylated at imprinted and non-imprinted loci6,10,11, although the extent of hypomethylation appears to be variable12. We therefore compared the methylation status of our male ESCs with three isogenic (129S6 × C57BL6 F1) female ESC lines that were cultured in S/L for 6 passages. At the time of analysis, our female ESC lines retained both X chromosomes, were hypomethylated globally and at ICRs, and expressed Impact biallelically (Fig. 1d–f and Extended Data Fig. 1i). Unsupervised clustering based on global methylation levels revealed that S/L-cultured female ESCs clustered with ICM cells and 2i/L-cultured male ESCs (Fig. 1e). However, when the same samples were clustered based on ICR methylation levels, female ESCs clustered with 2i/L-cultured male ESCs but apart from ICM cells and S/L-cultured male ESCs (Fig. 1f). Moreover, we noticed a substantial overlap between differentially methylated regions that distinguish male versus female ESCs grown in S/L, and those that distinguish male ESCs grown in S/L versus 2i/L (Fig. 1g).
Concomitant suppression of Mapk signaling and activation of Wnt signaling may drive additional epigenetic aberrations such as aberrant histone deposition. Accumulation or loss of the histone variant H2A.X has been shown to impact the developmental potential of pluripotent stem cells13. To examine whether S/L-cultured female ESCs or 2i/L-cultured male ESCs accumulate aberrant H2A.X binding patterns, we performed ChIP-Seq for H2A.X. Relative to S/L-cultured male ESCs, we found that female ESCs and 2i/L-cultured male ESCs lack H2A.X binding at 38,925 and 51,442 regions, respectively (Extended Data Fig. 2a–d). Of regions where H2A.X was lost in both male 2i/L-cultured ESCs and female ESCs (12,179 regions), a significant number of genes associated with gastrulation, organ development and germ layer formation were found (Extended Data Fig. 2c–e). Additionally, switching 2i/L-cultured male ESCs to S/L did not restore the majority of H2A.X binding, suggesting that H2A.X depletion is irreversible at these sites, similar to ICR methylation (Extended Data Fig. 2a,b).
To determine whether the developmental potential of ESCs is impacted by the erosion of ICR methylation and/or aberrant H2A.X deposition, we injected the different ESC lines into 2n or 4n host blastocysts (Fig. 2a and Supplementary Table 1,2). We found that 2i/L-cultured male ESCs and S/L-cultured female ESCs gave rise to fewer pups compared to S/L-cultured male ESCs upon 2n blastocyst injections (Fig. 2b), and several of the pups we obtained displayed phenotypes associated with aberrant imprinting14 (inset of Fig. 2b and Extended Data Fig. 3a). Moreover, adult chimeras derived from 2i/L-cultured male ESCs or S/L-cultured female ESCs had significantly less agouti coat color, implying a poor contribution to adult tissues (Fig. 2c). Nevertheless, some low-grade chimeras produced germline offspring, demonstrating their ability to produce functional germ cells (Extended Data Fig. 3b). Similarly, both 2i/L-cultured male ESCs and S/L-cultured female ESCs were 5-6 times less efficient at producing entirely ESC-derived (all-ESC) pups compared to S/L-cultured male ESCs when 4n blastocyst injections were performed (Fig. 2d and Extended Data Fig. 3c–e). These results are consistent with a previous report, which showed that early-passage female ESCs are slightly less effective at producing all-ESC pups relative to male ESCs15. Strikingly however, none of the all-ESC pups generated from 2i/L-cultured male or S/L-cultured female ESCs survived beyond birth (Fig. 2d and Extended Data Fig. 3d), and our p20 J37 2i/L-cultured ESC line even failed to generate full term all-ESC pups. J37 ESCs cultured in 2i/L for 16 passages also failed to produce well-differentiated teratomas, suggesting that mutations acquired in this line compromised their differentiation potential (Extended Data Fig. 4a,b).
Figure 2. Prolonged 2i/L culture impairs the developmental potential of ESCs.
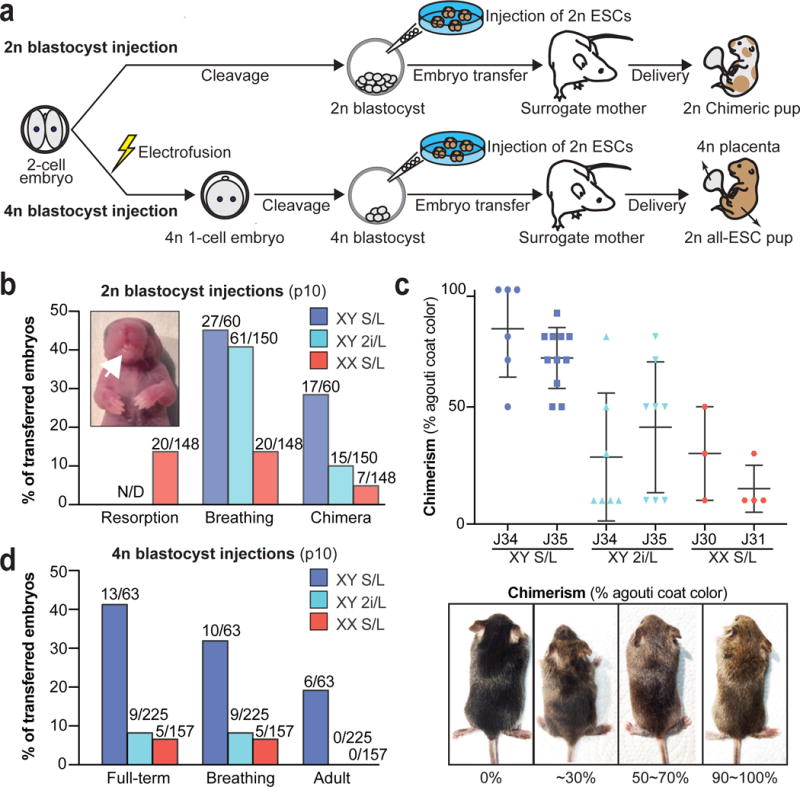
(A) Schematic of blastocyst injection protocol.
(B) 2n blastocyst injections. Numbers of animals obtained per total number of transferred embryos are shown. White arrow indicates the protruding tongue of a female pup. N/D, not determined. Details in Supplementary Table 1.
(C) Quantification of coat color chimerism of the chimeras from (B) (top). Representative images of the varying degrees of chimerism (below).
(D) 4n blastocyst injections. Numbers of animals obtained per total number of transferred embryos are shown. Details in Supplementary Table 2.
Modulation of Wnt/Mapk signaling2,3, DNA hypomethylation4,5, and H2A.X loss16 have been shown to impact genomic stability. We therefore performed karyotype analysis (Supplementary Table 3) and array Comparative Genomic Hybridization (aCGH) on our ESC lines cultured in 2i/L. ESCs propagated in 2i/L for 10 passages had mostly diploid chromosome counts (40,XY) (Fig. 3a, top row). However, 2i/L-cultured ESC lines at p20 exhibited recurrent chromosomal aberrations including trisomy 6, 8, 19, and loss of the Y chromosome in many, if not all cells analyzed, while S/L-cultured ESCs at p20 remained euploid (Fig. 3a–c and Extended data Fig. 5a). To exclude the possibility that these abnormalities are specific to our cell lines or culturing techniques, we analyzed a 129/Ola ESC line that had been independently maintained in 2i/L conditions17 and found several chromosomal aberrations including trisomy 6 and 8 (Extended Data Fig. 5b). Additionally, despite previous reports demonstrating that irradiated MEFs do not affect ESCs in a co-culture system18, we passaged one of our ESC lines in 2i/L for 16 passages without feeders. As expected, every cell karyotyped acquired trisomy 6 and 8, confirming that the 2i/L-derived chromosomal aberrations are not due to co-culture with MEFs (Extended data Fig. 5c). Considering these unexpected chromosomal abnormalities, we performed aCGH on DNA isolated from a rare full term all-ESC pup that was generated using p20 2i/L-cultured ESCs. We did not find evidence for gross copy number abnormalities in this pup (Extended Data Fig. 5d), even though the parental bulk cultures had chromosomal aberrations in every cell counted (Fig. 3a middle row, 3b). We surmise that rare euploid ESCs present in the culture experienced a strong positive selection during development, yet succumbed postnatally due to the epigenetic aberrations present. To confirm this, we performed RRBS on cultured explanted keratinocytes derived from chimeric mice produced with 2i/L-cultured ESCs and found they were hypomethylated at several imprinted loci (Extended data Fig. 5e).
Figure 3. Impact of prolonged 2i/L culture on chromosomal stability.
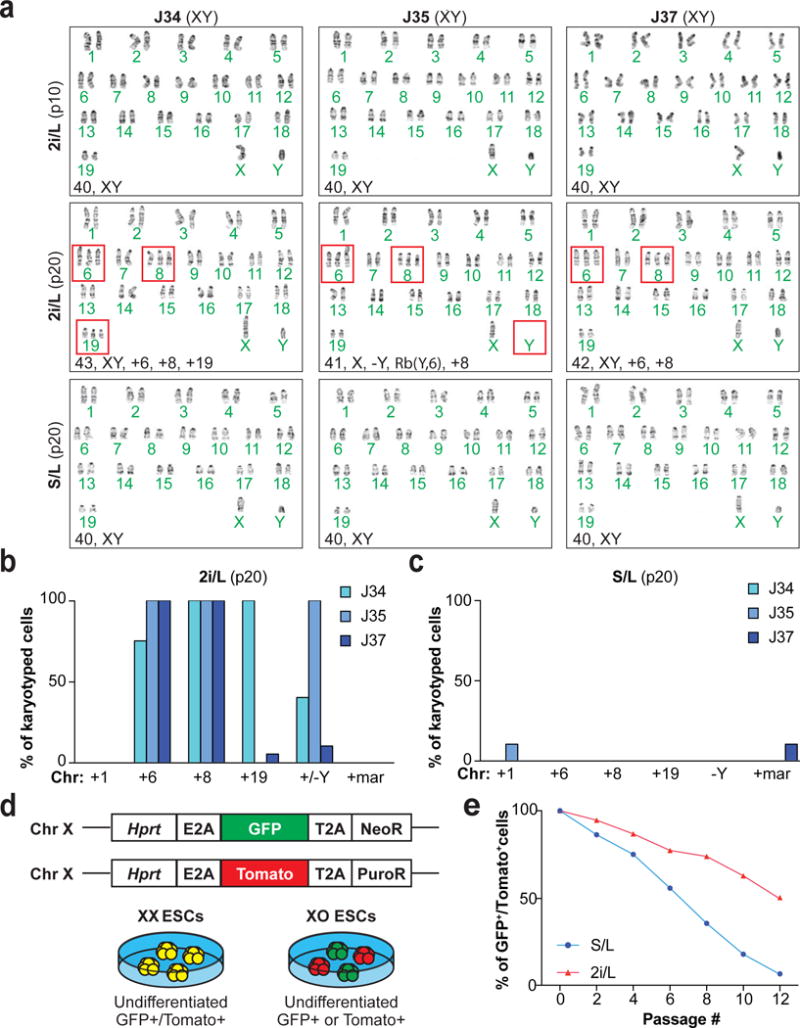
(A) Karyotyping analysis of male ESC lines. Red boxes indicate abnormal chromosomes detected.
(B–C) Quantification of the chromosomal abnormalities found in 2i/L-cultured (B) and S/L-cultured (C) male ESCs at p20.
(D) Schematic of the XGXT ESC line and expected differences between XX and XO ESCs.
(E) Quantification of X chromosome loss in XGXT ESCs cultured in S/L or 2i/L.
Female ESCs tend to lose one of their two X chromosomes with culture, thus becoming XO10. The loss of an X chromosome in XX ESCs has been associated with downregulation of naïve-like genes and elevated DNA methylation, similar to male ESCs cultured in S/L9,11. We hypothesized that the naïve-like state of XX female ESCs—particularly as it relates to decreased Mapk signaling and increased Wnt signaling—is responsible for the selective pressure to lose an X chromosome. Thus, if the chemical inhibition of Mek1/2 and Gsk3α/β via 2i/L culture attenuates X chromosome loss in female ESCs, we would conclude that suppressed Mapk signaling and/or elevated Wnt signaling in XX ESCs drives female ESCs to become XO. To test this hypothesis, we developed a dual reporter female ESC line (XGXT) and measured X chromosome loss during S/L and 2i/L culture (Fig. 3d and Extended data 6a–c). Indeed, when we exposed XGXT ESCs to 2i/L, a substantial fraction of GFP/Tomato double-positive cells persisted relative to XGXT ESCs cultured in S/L (Fig. 3e).
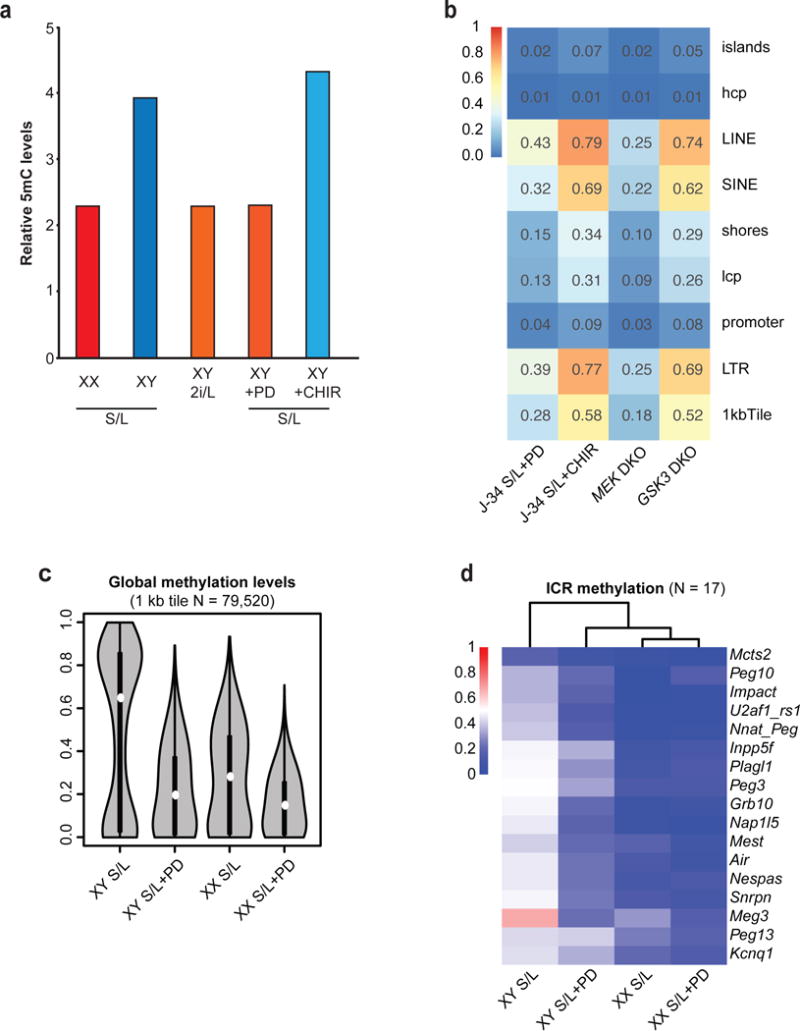
It remains unclear whether Mapk suppression, Wnt activation, or a combination of these activities is responsible for the epigenetic abnormalities we observed. Therefore, we performed RRBS on male ESCs cultured in S/L with one of the two inhibitors used in 2i/L culture: the Mek1/2 inhibitor (S/L+PD) or Gsk3α/β inhibitor (S/L+CHIR). We found that only ESCs exposed to S/L+PD acquired methylation levels similar to ESCs cultured in 2i/L (Fig. 4a and Extended Data Fig. 7a). To confirm these results, we analyzed DNA methylation levels in male pluripotent stem cells deficient for Mek1 and Mek2 (Mek DKO) or Gsk3α and Gsk3β (Gsk3 DKO). We found that Mek DKO stem cells are hypomethylated globally and at ICRs while Gsk3 DKO stem cells remain largely unchanged (Fig. 4b,c and Extended Data Fig. 7b). Notably, global methylation, including at ICR loci, was further reduced in female ESCs cultured in S/L+PD when compared to female ESCs cultured in S/L (Extended Data Fig. 7c,d).
Figure 4. Mek1/2 suppression underlies the epigenetic and chromosomal changes observed in ESCs.
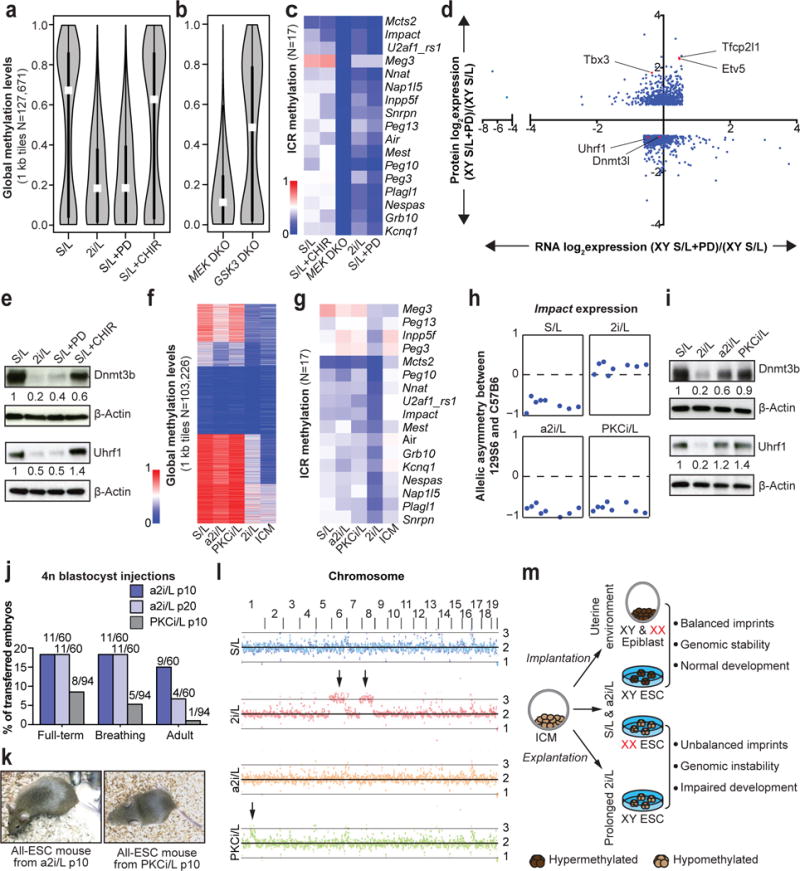
(A) Global methylation levels of male ESC cultured in indicated conditions (n=3 biological replicates). White dots, median value.
(B) Global methylation levels of Mek1/2 double knock-out (DKO) iPSCs and GSk3α/β DKO ESCs cultured in S/L. White dots, median value.
(C) ICR methylation levels in male pluripotent stem cells with the indicated pharmacological or genetic perturbations.
(D) Differential protein levels between S/L and S/L+PD (>1.5-fold) are plotted along the y-axis and the corresponding differentially expressed RNAs are plotted along the x-axis.
(E) Western blot analysis of Dnmt3b and Uhrf1 in the labeled consitions.
(F–G) Global (F) and ICR (G) methylation levels in male ESCs cultured in indicated conditions. ICM, inner cell mass.
(H) Allelic expression of the imprinted gene Impact.
(I) Western blot analysis of Dnmt3b and Uhrf1 in the labeled conditions.
(J) 4n blastocyst injections. Numbers of animals obtained per total number of transferred embryos are shown. Details in Supplementary Table 2.
(K) Adult all-ESC mice generated using a2i/L-cultured (left) or PKCi/L-cultured (right) male ESCs.
(L) Chromosomal copy number analysis by whole genome sequencing. Black arrows indicate trisomies 6 and 8 in 2i/L-cultured ESC lines and the partial amplification of chromosome 1 in PKCi/L-cultured ESC lines, respectively.
(M) Graphical summary of results.
In order to understand how Mek1/2 inhibition induces global hypomethylation, we performed RNA-sequencing and quantitative large-scale proteomics of male ESCs cultured in either S/L or S/L+PD for 3 passages (Fig. 4d). PD treatment led to the upregulation of transcription factors that have been associated with naïve pluripotency (Extended Data Fig. 8a). Additionally, all major DNA methyltransferases (Dnmt1, Dnmt3a, Dnmt3b) and associated cofactors (Dnmt3l, Uhrf1) were downregulated at the RNA and protein level in PD-treated ESCs (Fig. 4e and Extended Data Fig. 8b). These observations are consistent with the previous notion that reduced levels of Uhrf119 and Dnmt3a/Dnmt3b7 underlie 2i/L-induced hypomethylation.
We next tested whether Mek1/2 suppression is sufficient to drive the chromosomal abnormalities we observed in male ESCs cultured in 2i/L. Karyotypes of our male ESCs cultured in S/L+PD for 16 passages revealed that every counted cell had some karyotypic abnormality, contrary to ESCs exposed to S/L+CHIR (Extended Data Fig. 8c,d). To determine whether the genomic hypomethylation driven by Mek1/2 inhibition in ESCs is sufficient to select for the recurring karyotypic aberrations, we derived ESCs harboring conditional alleles for Dnmt1, Dnmt3a and Dnmt3b (Dnmt cTKO). When we deleted the Dnmts and cultured the ESCs in serum for 15 passages, we found that they remained euploid, suggesting that Mek1/2 suppression drives the selection for certain chromosomal abnormalities independently of DNA methylation. (Extended Data Fig. 8e,f).
Considering the impact of continual Mek1/2 inhibition on ESCs, we determined whether previously reported alternative culture conditions lacking Mek1/2 inhibition20,21 resemble naïve ESCs while maintaining their developmental potential. We tested two defined media conditions using our ESCs supplemented with the PKC inhibitor Gö6976 (PKCi/L), or with the Src inhibitor CGP77675 in combination with CHIR (a2i/L) (Extended data Fig. 9a). While both PKCi/L and a2i/L conditions were transcriptionally more similar to S/L than 2i/L, a2i/L-cultured ESCs clustered with 2i/L-cultured ESCs when the ESCs were grouped using a previously published set of pluripotency and differentiation genes22 (Extended Data Fig. 9b–e). RRBS analysis revealed that ESCs cultured in PKCi/L and a2i/L were globally hypermethylated and retained proper methylation of ICRs, thus preserving monoallelic expression of Impact (Fig. 4f–h and Extended data Fig. 9f,g). The hypermethylation of a2i/L- and PKCi/L-cultured ESCs coincided with increased Dnmt3b and Uhrf1 protein levels relative to 2i/L-cultured ESCs (Fig. 4i). Additionally, when we cultured our female XGXT ESC line in a2i/L, we observed an intermediate level of X chromosome loss when compared to S/L+PD and S/L (Extended data Fig. 9h).
We next assessed the developmental potential of PKCi/L- and a2i/L-cultured ESCs by 4n blastocyst injections. Consistent with the preservation of monoallelic ICR methylation and euploid karyotype under these conditions, we were able to generate several germline-competent, adult all-ESC mice using ESCs cultured in a2i/L for 6 or 16 passages (Fig. 4j,k and Extended data Fig. 9i,j). Unexpectedly, we were able to generate only one adult all-ESC mouse using ECSs cultured in PKCi/L for 6 passages (Fig. 4j,k), even though these ESCs maintained monoallelic ICR methylation and remained euploid (Extended data Fig 9k). It is unclear as to why these PKCi/L generated so few all-ESC mice, however by p20 in PKCi/L, these ESCs did select for a large duplication on chromosome 1 (Fig 9i and Extended data Fig 9k).
Different conditions have recently been discovered that allow for the propagation of naïve human ESCs23,24. Similar to 2i/L-cultured mouse ESCs, naïve human ESCs are globally hypomethylated, lose ICR methylation, and acquire karyotypic abnormalities25. Since inhibition of MEK1/2 via PD is common among current naïve human culture conditions, we modified the naïve media “t2iLGöY”26 by replacing PD with the Src inhibitor used in a2i/L culture (termed a2iLGöY). We then monitored the maintenance of the naïve state in human ESCs carrying a naïve-specific ΔPE-OCT4-EGFP reporter23 upon exposure to t2iLGöY or a2iLGöY. As expected, t2iLGöY maintained GFP expression in the majority of ΔPE-OCT4-EGFP ESCs. However, exposure of ΔPE-OCT4-EGFP ESCs to a2iLGöY led to a rapid loss OCT4-GFP expression and differentiation, indicating that SRC inhibition cannot replace MEK1/2 inhibition in this culture system (Extended Data Fig. 10a,b).
Our results demonstrate that attenuation of Mapk signaling via Mek1/2 suppression is sufficient to induce global hypomethylation of DNA in ESCs, thus recapitulating the naïve epigenetic state within the ICM. Based on our data, we suggest that the levels of Mapk signaling capture distinct epigenetic states ranging from the naïve state that resembles the ICM to the primed state that resembles the post-implantation epiblast. While inhibition of Mek1/2 maintains ESCs in a naive epigenetic state, inhibition of Src appears to maintain ESCs in an epigenetic state that lies in between naïve and primed (Fig 4m). Considering that Mek1/2 inhibition suppresses Mapk signaling to a greater extent than Src inhibition21, our results imply that the degree of Mapk signaling dictates the epigenetic state and chromosomal stability of ESCs. Comparing targets of Mek1/2 and Src will be informative to understand how Mapk signaling facilitates the capture of different epigenetic states as well as drive the selection of particular chromosomal aberrations in pluripotent stem cells. These comparative studies should also provide valuable insights into the epigenetic regulation of early embryonic development and may lead to strategies to generate stable, human naïve ESCs.
METHODS
Animal care and use
All mice used in the study were housed and bred in Specific Pathogen Free (SPF) rooms located in the AAALAC-accredited Center for Comparative Medicine vivarium at Massachusetts General Hospital. All mice were housed in ventilated cages on a standard 12:12 light cycle. All procedures involving mice adhered to the guidelines of the approved Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC) protocol no. 2006N000104.
ESC derivation and cell culture
Blastocysts were isolated from the uterine horns of pregnant females at 3.5 dpc using a dissection microscope. Individual blastocysts were plated onto feeder cells in 2i/L medium and cultured for 7 days. Emerging ICM outgrowths were directly picked into S/L medium on feeders to establish stable ESC lines as previously reported11. ESC lines were maintained by dissociating cells with 0.25% trypsin every 4 days and re-plating them onto irradiated MEFs. S/L medium contains KO DMEM (Invitrogen) supplemented with 15% FBS (Hyclone), 1mM L-glutamine, 100 μM non-essential amino acids (Invitrogen), 1X PenStrep (Invitrogen), 0.1 mM beta-mercaptoethanol (Invitrogen), and 103 IU LIF (in house). 2i/L medium contains a 1:1 mixture of DMEM/F12 supplemented with N2 (Invitrogen) and Neurobasal media with glutamine (Invitrogen) supplemented with B27 (Invitrogen), 1X PenStrep (Invitrogen), 103 IU LIF (in house), 1 μM PD0325901 (Tocris) and 3 μM CHIR99021 (Tocris). a2i/L medium contains a 1:1 mixture of DMEM/F12 supplemented with N2 (Invitrogen) and Neurobasal media with glutamine (Invitrogen) supplemented with B27 (Invitrogen), 1X PenStrep (Invitrogen), 103 IU LIF (in house), 1.5 μM CGP77675 (Tocris) and 3 μM CHIR99021 (Tocris). PKCi/L medium contains a 1:1 mixture of DMEM/F12 supplemented with N2 (Invitrogen) and Neurobasal media with glutamine (Invitrogen) supplemented with B27 (Invitrogen), 1X PenStrep (Invitrogen), 103 IU LIF (in house) and 5 μM Gö6976 (Tocris). Irradiated MEFs were used for all culture media to ensure consistent conditions, unless noted. All lines were periodically tested for Mycoplasma using the Lonza mycoalert kit and found to be negative.
iPSC generation
Mek1fl/;Mek2−/− or F1 Mus musculus × Mus spretus MEFs were transduced with lentiviruses that facilitate the constitutive expression of M2rtTA and inducible expression of Oct4, Klf4, Sox2 and c-Myc27. Infected MEFs were plated onto irradiated MEF feeders in S/L media supplemented with 2 μg/ml doxycycline until iPSC colonies emerged. Doxycycline-independent, clonal iPSCs were picked and expanded. To remove the floxed Mek1 allele to generate Mek1/2 DKO iPSCs, we transiently transfected via electroporation a plasmid driving Cre recombinase and subcloned. Mek1/2 double knock-out iPSCs were verified by Western blot and maintained on irradiated MEF feeders in S/L.
Generation of Dnmt1/3a/3b triple knock out (TKO) ESCs
Dnmt cTKO ESCs were generated as described using blastocysts obtained from a Dnmt1fl/fl4, Dnmt3afl/fl28 and Dnmt3bfl/fl29 intercross. To remove the floxed alleles, we transiently transfected via electroporation a plasmid driving Cre recombinase and subcloned. Dnmt TKO ESCs were verified by Western blot and maintained on irradiated MEF feeders in S/L.
Reduced representation bisulfite sequencing (RRBS)
Genome-wide basepair-level methylation was assayed using Reduced Representation Bisulfite Sequencing (RRBS)30. Briefly, genomic DNA was digested using the MspI restriction enzyme and subjected to bisulfite treatment, which converts unmethylated cytosine residues to uracil. Genomic fragments were sequenced using next-generation sequencing, and aligned to the mm9 reference genome. Methylation levels of individual CpGs were measured by observing bisulfite conversion in the aligned read as compared to the reference genome. On average, 1.596M CpGs were covered per sample, at a mean depth of 14.31 reads/CpG.
Region-level methylation values were computed by pooling coverage-weighted methylation levels in CpGs that were measured by at least 3 reads in 80% of samples. A consensus set of regions was defined using regions that had at least 5 reads in every sample. Genomic methylation plots used methylation levels of 127,671 non-overlapping 1kb tiles. Heatmaps show methylation levels of 1kb tiles clustered using k-means clustering (k=5), and samples clustered by hierarchical clustering. Promoters were defined as 1kb up- and downstream from RefSeq transcription start sites. LINEs, SINEs, LTRs and IAPs were defined using RepBase. ICRs were defined as a set of 17 canonical ICRs covered by RRBS as used in31.
Violin plots were created using the ‘vioplot’ R package. The white dot represents the median. The box extends from the 25th percentile to the 75th percentile of the data (the interquartile range). Whiskers extend from the box to the most extreme data point, which is no more than 1.5 times the interquartile range. Heatmaps were created using the ‘pheatmap’ R package and clustering was performed using euclidean distance and complete linkage.
RNA-sequencing and analysis
RNA-Seq libraries were constructed from RiboZero-selected RNA using NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs) and sequenced on an Illumina HiSeq2500 instrument. Transcriptome mapping was performed with STAR version 2.3.032 using the Ensembl 67 release exon/splice-junction annotations. Approximately 55-90% of reads mapped uniquely. Read counts for individual genes were produced using the unstranded count feature in HTSeq v.0.6.033. Differential expression analysis was performed using the exactTest routine of the edgeR package34 after normalizing read counts and including only those genes with cpm > 1 for one or more samples35. Differentially expressed genes were defined based on the criteria of >2-fold change in expression value and false discovery rate (FDR) < 0.05. Allelic expression asymmetry was quantified via samtools mpileup at exonic SNP sites that differentiated C57B6 from 129S6 reference genomes (indels excluded)36. To measure allelic asymmetry in expression, we used the asymmetry metric calculated as the ratio (129S6 − C57B6)/(129S6 + C57B6), where 129S6 and C57B6 are the numbers of reads assigned at a given SNP to 129S6 and C57B6 allele, respectively. This measure of allelic asymmetry ranges from −1 (C57B6 only) to 1 (129S6 only), with 0 corresponding to fully balanced biallelic expression.
RNA-seq and analysis of Interspecific iPSCs
Total RNA specimens were isolated from the interspecific iPSCs using (please specify kits or methods), and their integrity [RNA Integrity Numbers (RIN) ≥ 9.4] was confirmed using Agilent Tapestation with RNA High Sensitivity tapes. Illumina sequencing libraries with 6-bp single index were constructed from 1 ug total RNA using the TruSeq Standard Total RNA LT kit with the Ribo-Zero Gold ribosomal RNA depletion module (Illumina). Libraries were subjected to size distribution analysis (200 – 500 bp, peaking at 280-290 bp) using Tapestation with the High Sensitivity D1000 tapes and quantified using the Illumina library qPCR quantification kit (KAPA Biosystems). Equal concentrations (4 nM) of libraries were pooled and subjected to Illumina NextSeq deep sequencing using High Output Sequencing Kit version 2 (paired-end, 75 nt + 75 nt). The fastq format raw reads data files were generated by the Illumina BaseSpace Sequencing Hub and subjected to pre-alignment quality control using the fastQC software (Babraham Institute). The fastq reads were aligned to the GRCm38/mm10 Mus musculus musculus (C57BL/6) reference genome sequence using the STAR aligner (version 2.4.2.a) with the UCSC annotation GTF file and the sjdbOverhang parameter 74 for genome indexing and alignment. The aligned reads were sorted and subjected to duplicates removal using the sambamba37 implementation of SAMtools38, resulting in 82-95 uniquely-mapped reads per RNA specimen. These reads were assigned to mm10-annotated genes and counted using the Bioconductor package Rsubread33 (69.1-71.2% successfully assigned reads), and the gene counts were normalized to generate the cpm (counts per million reads) values using the negative binominal distribution model implemented by edgeR35. Tracks of the uniquely mapped reads were directly inspected using the Integrative Genomics Viewer39, which reports paternal (M. spretus) and maternal (mm10 – Mus musculus musculus) allelic counts for each SNP. Statistical significance of Allelic counts at multiple SNPs in each gene were examined by t-test (unpaired, two-tail, homoscedastic assumption).
Whole-genome sequencing
Genomic DNA (1 ug) was sonicated using a Covaris S2 sonicator (Covaris) to ~350 bp peak fragments. To avoid PCR-derived bias of read counts, gDNA fragments were subjected to PCR-free synthesis of Illumina NextSeq deep sequencing libraries with the i7 index using the TruSeq DNA PCR-free LT Sample Preparation Kit - Set A (Illumina). Libraries were quantified using KAPA Illumina library quantification qPCR kit (KAPA Biosystems), and equal concentrations (4 nM) of libraries were pooled. Size distributions of gDNA fragments and deep sequencing libraries were examined using Agilent Tapestation with Genomic DNA screening tapes (Agilent Technologies). Single-read deep sequencing (76 nt) was performed using Illumina NextSeq 500 sequencer with the high output v2 kit. Fastq files were subjected to a quality control process using Trim Galore! and fastQC (Babraham Institute), which excluded raw quality reads (Phred score < 20) and deleted the Illumina adaptor sequences and the last one nucleotide at the 3′ end to avoid nucleotide base biases. The trimmed/QCed fastq reads were aligned to the mouse GRCm38/mm10 reference genome sequence using the STAR aligner (ver. 2.4.2.a) with the genomic DNA alignment configuration, generating the bam format aligned reads files. The reads were extracted using the sambamba toolkit37. Numbers of the uniquely mapped reads were 46.2 – 59.6 million per sample, which corresponded to about 1.2X ~ 1.6X average depth of genome coverage. Reads assigned to each chromosome are counted to generate cnt files. The chromosome count data were subjected to ploidy analysis, as previously described40.
Native Chromatin immunoprecipitation (N-ChIP)
N-ChIP assay was performed as previously described41. Briefly, approximately 10 million ESCs were used for each ChIP and massive parallel sequencing (ChIP-Seq) experiment. Chromatin pellets were briefly digested with MNase (New England BioLabs) and the mononucleosomes were monitored by electrophoresis 0.5ug anti-H2A.X antibody (active motif 39689) was used per ChIP experiment. Co-purified DNA and whole cell extraction (WCE) input genomic DNA were subject to library construction, cluster generation and next-generation sequencing (Illumina Genome Analyzer II and HiSeq 2000). The output sequencing reads were filtered and pre-analyzed with Illumina standard workflow. After filtration, the qualified tags (in fastq format) were aligned to the mouse genome (mm9) with bowtie2 (2.0.6)42. Reads mapped to sex chromosomes were discarded. Aligned reads were used for peak calling using the RSEG algorithm (0.4.8) in mode 3 to identify differential binding regions relative to J34 S/L samples43. For cells cultured in 2i/L and then switched back to serum, the combination of all male S/L samples were used as control to reach a comparable throughput. The relative decreased regions were assigned to whole genome-wide 20kb windows to calculate a weighted average score and used for enumerating overlapping regions. Heatmap and unsupervised hierarchical clustering analysis were drawn in 500kb domains. GREAT was used to predict the putative target genes statistical relevance of associations between genomic regions and annotations44.
Blastocyst injections
Blastocyst (2n and 4n) injections were performed as previously described45. Briefly, female BDF1 mice were superovulated by intraperitoneal injection with PMS and hCG and mated to BDF1 stud males. Zygotes were isolated from females with a vaginal plug 24 h after hCG injection. For 2n injections, zygotes were cultured in vitro for 3 d in KSOM medium, and resultant blastocysts were injected with 10-15 ESCs before transfer into pseudopregnant recipient females. When newborn pups were not delivered until day 18 after the transfer, resorbed embryos were counted from the uteri of the recipient females and counted as “Resorption”. Surviving neonates with regular breathing was termed “Breathing” and two weeks later their chimerism was assessed based on their agouti coat color contributions.
For 4n injections, zygotes were cultured overnight until they reached the 2-cell stage, at which point they were electrofused. After 1 h, fused 1-cell embryos were carefully identified and separated from embryos that had failed to fuse, cultured in KSOM for another 2 d, and then injected with 15-20 ESCs before embryo transfer. C-section was performed at day 17 after the embryo transfer and embryos were termed “Full-term”, “Breathing” and “Adult” when they reached the full-term, established regular breathing after birth and survived past 5 weeks, respectively.
Teratoma assay
ESCs were grown to confluency in a single well of a confluent six well plate. Cells were then trypsinized and counted. 2×106 ESCs were resuspended in 300 μL of ESC media and injected subcutaneously into the flanks of a nu/nu immunocompromised mouse. Teratomas were monitored and removed once tumor size reached 1 cm or tumors became ulcerated. Tumors were then weighed and processed for hemaytoxylin and eosin staining Analysis of the tumor sections were blindly scored for the presence of each germ layer.
Array Comparative genomic hybridization (aCGH)
Genomic DNA was isolated from frozen cell pellets using DNeasy tissue kit (Qiagen) following the manufacturer’s protocol. Concentration and quality of isolated genomic DNA was assessed using a spectrophotometer (NanoDrop Technologies, Wilmington, DE) to assure absence of contaminating protein (260/A280 ratio is greater than or equal to 1.8) and absence of carbohydrate, lipid and residual phenol (260/A230 ratio greater than or equal to 2.0). Integrity of genomic DNA was confirmed by low voltage 0.6% agarose gel electrophoresis with mean band size of about 50Kb. Sample labeling was performed following Agilent’s recommendation for array CGH. Briefly, 1.5 μg of genomic DNA was digested with 5 units of Alu I and Rsa I (Promega) for 2 hours at 37°C. Labeling reactions were carried out with total amount of fragmented DNA for 3 hours at 37C using a BioPrime Array CGH Genomic Labeling Module (Invitrogen) with 3 umol Cy5-dUTP or Cy3-dUTP (Perkin Elmer). Labeled samples were purified, concentrated on a Centricon YM-30 column (Millipore), and then mixed with 10× blocking agent and 2× hybridization buffer (Agilent Technologies). Samples were then hybridized on a SurePrint G3 Mouse 4× 180K CGH Microarray chip that contains 170,305 distinct biological probes taken from the mouse genome. Hybridized arrays were imaged with Agilent G2565BA microarray scanner using default settings. Data were extracted and analyzed using Agilent’s Genome Workbench software. Aberrant regions (gains or losses) were identified using a build-in ADM-2 algorithm with aberration filter setting as minimum number of probes for gains or losses >= 20 and absolute log2 ratio for gains or losses >= 0.35.
Karyotyping
Mouse ESC lines were prepared for cytogenetic analysis by treatment with colcemid (Irvine Scientific, Santa Ana, CA) at a final concentration of 0.1 μg/ml for 3 hours to accumulate cells in metaphase. Cells were then exposed to 0.075 M KCl for 25 minutes at 37°C and fixed with 3:1 methanol:acetic acid. Air-dried slides were generated and G-banded following standard GTG banding protocols. Two levels of analysis were pursued: for all lines, a minimum of 10 metaphase cells were counted, and 2-4 karyotypes prepared. For lines with a normal or consistent straightforward result, no further analysis was done. For lines with a complex karyotype, additional metaphases were counted (total 20), and 10 karyotypes prepared to better characterize the variability.
Generation of XGXT ESC line
XGXT lines were generated by CRISPR/Cas mediated gene targeting in a newly derived 129SvJ × C57B6 F1 ESC line. Two homology constructs were designed to integrate either E2A-GFP-T2A-NeoR-STOP or E2A-Tomato-T2A-PuroR-STOP in frame with the last exon of Hprt. The constructs were generated via PCR amplification and Giboson assembly (New England Biolabs) of the respective fluorescent protein and resistance marker flanked on either end by 500bp of homology to the Hprt locus. An sgRNA was designed targeting the last exon of Hprt. Both homology constructs, the sgRNA and a construct containing Cas9 were cotransfected into early passage female ESCs with Lipofectamine 2000 (Thermo Fisher). Following transfection, dual selection with G418 and puromycin was applied for 10-14 days. Resistant clones were picked and targeting was confirmed by Southern blot.
Dot blot analysis for 5mC
Genomic DNA samples were prepared with 2-fold serial dilutions in TE buffer and then denatured in 0.4 M NaOH/10 mM EDTA at 95°C for 10 min and followed by adding an equal volume of cold 2 M ammonium acetate (pH 7.0). Denatured DNA samples were spotted on a nitrocellulose membrane in an assembled Bio-Dot apparatus (Bio-Rad) according to the manufacturer’s protocol. The membrane was washed with 2X SSC buffer and UV-crosslinked for 10 min. The membrane was then blocked with 5% non-fat milk for 1 hr and incubated with an anti-5mC antibody (Active Motif; 39649), washed, and then incubated with an HRP-conjugated secondary antibody. Chemiluminescence was measured, and subsequently stained with methlyene blue to detect the quantity of DNA loaded. Quantification was performed by image-J software analysis.
Mass spectrometry analysis
Mass spectrometry analyses were performed on an Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific) coupled to an Easy-nLC 1200 ultra-high pressure liquid chromatography (LC) pump (ThermoFisher Scientific). Peptides were separated at 300 nL/min using a self-packed analytical column (75 μm inner diameter) that was packed with 0.5 cm of Magic C18 resin (5 μm, 100 Å, Michrom Bioresources) followed by 35 cm of Sepax Technologies GP-C18 resin (1.8 μm, 120 Å). LC buffers consisted of 0.1% formic acid (buffer A) and 80% ACN with 0.1% formic acid and LC gradients were optimized to ensure equal elution of peptides throughout the analysis. Intact mass analysis of peptides (MS1) was performed in the Orbitrap (AGC target 1e6, 120,000 resolution, 100 ms maximum injection time) and used to select the 10 most abundant features for MS/MS (MS2) analysis. Candidate peaks were filtered based on charge sate ≥ 2 and monoisotopic peak assignment, and dynamic exclusion (60 second ± 7.5 ppm) was enabled. Additionally, only one charge state was selected for each precursor for analysis of non-phosphorylated peptides. Each precursor was isolated with using a quadrupole mass filter (0.5 Th width) and then fragmented with collision-induced dissociation (CID, 35 NCE) ion the ion trap with distinct settings for non-phosphorylated (AGC target = 2.5 × 104, maximum injection time = 200 ms) peptides. To alleviate the effects of precursor ion interference multiple fragment ions were isolated using synchronous precursor selection (SPS) prior to HCD (55 NCE, SPS notches = 8, AGC target = 2.2 × 105, maximum injection time of 150 ms for non-phosphorylated peptides) MS3 fragmentation and Orbitrap analysis (50,000 resolution).
Mass spectrometry data analysis
A compilation of in-house software was used to convert mass spectrometric data (Thermo “.raw” files) to mzXML format, as well as to correct monoisotopic m/z measurements and erroneous peptide charge state assignments46. Assignment of MS/MS spectra was performed using the SEQUEST algorithm47. Static modifications included TMT (229.16293 Da) on both the n-terminus of peptides and lysine residues and carbamidomethylation of cysteine residues (57.02146 Da). Peptide spectral matches were filtered to 1% false discovery rate (FDR) using the target-decoy strategy48, before being grouped into proteins which were then filtered to 1% FDR at the protein level as previously described46. All data analysis was performed using R.
Western blot analysis
Cells were lysed in modified RIPA buffer [50 mM Tris·HCl, pH 8, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 1 mM EDTA, 1× protease inhibitor mixture (Roche Diagnostics), 1× PhosSTOP phosphatase inhibitors (Roche Diagnostics), 0.01 U/μL benzonase (Novagen)]. Cell lysate was then sonicated for five 30-second pulses (with 30 second pauses) in a bath sonicator at 4°C (Diagenode). Lysates were cleared of cell debris by centrifugation at 20,000 × g for 5 minutes. Westerns were performed with anti-Dnmt1 (1:500 dilution; Cell Signaling, 5119S), anti-Uhrf1 (1:200 dilution; Santa Cruz, sc-98817), anti-Dnmt3a (1:500 dilution; Santa Cruz Biotechnology, 20703), anti-Dnmt3b (1:1000 dilution; Abcam, 13604), or anti-β-Actin (1:10,000 dilution; Cell Signaling Technology, 5125S).
Data Availability Statement
The accession number for the raw data files for the RRBS analysis reported in this paper is NCBI GEO: GSE97995.
The accession number for the raw data files for the RRBS analysis on female ESC lines is NCBI GEO: GSM1679872, GSM1679873, GSM1679874.
The accession number for the raw data files for the RNA-seq analysis reported in this paper is NCBI GEO: GSE97954, GSE93103.
The accession number for the raw data files for the Genome-sequencing analysis reported in this paper is NCBI SRA: SRP102419.
The accession number for the raw data files for the ChIP-seq analysis reported in this paper is NCBI GEO: GSE98771.
Extended Data
Extended Data Figure 1. Effect of maintaining the naïve state on DNA methylation in ESCs.
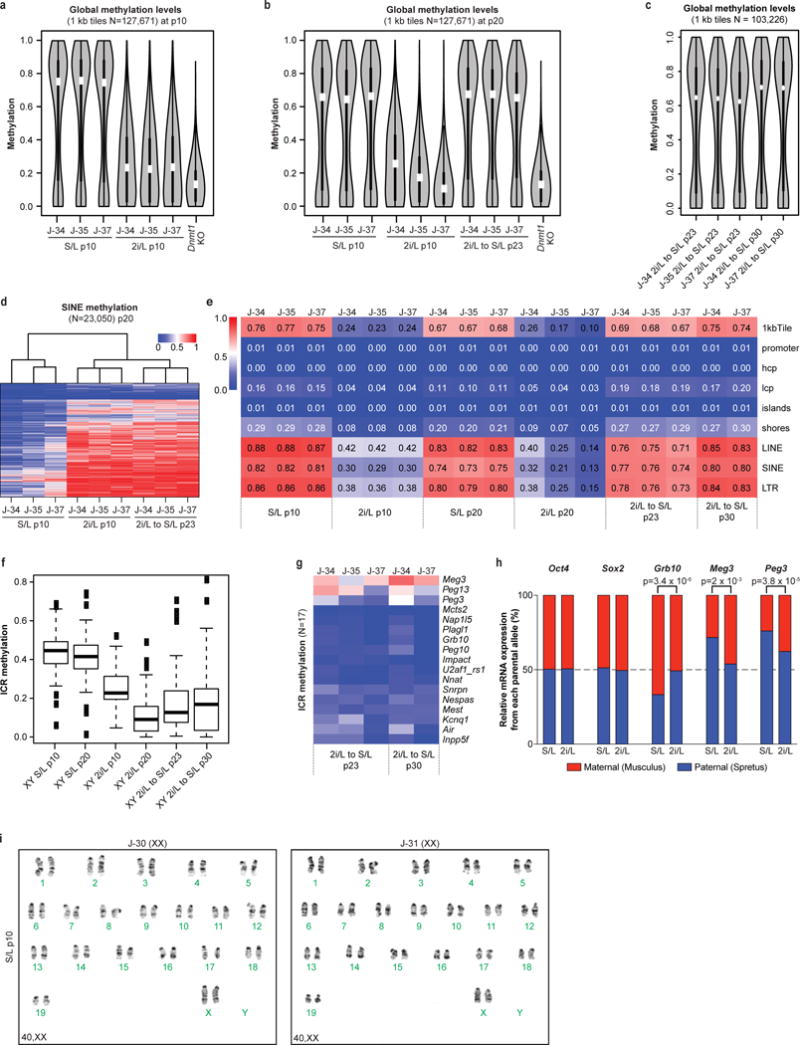
(a–c) Violin plots showing global methylation levels of ESCs at p10 (a), p20 (b) and after being placed back into S/L at p23 and 30 (c) by reduced representation bisulfite sequencing (RRBS). Hypomethylated Dnmt1 knock-out (KO) ESCs were included as control. White dots, median value.
(d) Heatmap of DNA methylation levels at SINE elements in ESCs.
(e) Median methylation levels of different genomic elements in the indicated conditions.
(f) Box plot showing methylation levels of ICRs cultured in each condition.
(g) ICR methylation levels after being re-exposed to S/L for the indicated passages.
(h) RNA sequencing was performed on a F1 Mus musculus × Mus spretus stem cell line cultured in 2i/L for 6 passages. Normalized read counts of mRNA transcripts expressed from the paternal or maternal alleles are shown based on multiple allele-discriminating SNPs for each gene (6 or more SNPs per gene). Statistical analysis (two-tail t-tests) for allelic biases of the SNPs is shown.
(i) Karyotyping results from two female ESC lines cultured in S/L for 10 passages.
Extended Data Figure 2. Effects of prolonged 2i/L culture on H2A.X deposition in ESCs.
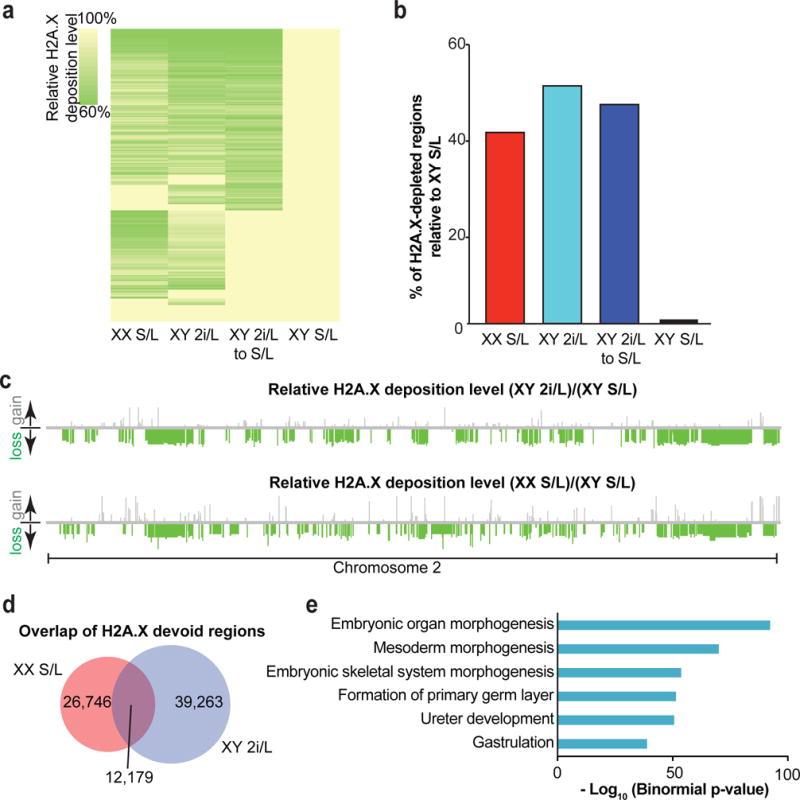
(a) Heatmap showing global H2A.X deposition loss relative to male ESCs cultured in S/L. Three ESC lines per condition (male S/L, male 2i/L, female S/L) at passage 10 and two ESC lines (male 2i/L to S/L) at passage 30 were analyzed (see Fig. 1a).
(b) Bar graph quantifying the percentage of H2A.X depleted regions relative to male ESCs cultured in S/L. Each bar represents median values of biological replicates of samples in (a).
(c) Relative H2A.X deposition on a representative chromosome. Relative losses (green bars) or gains (gray bars) of H2A.X are mapped to their location on chromosome 2.
(d) Overlap of devoid H2A.X regions common to both female ESCs cultured in S/L and male cells cultured in 2i/L.
(e) H2A.X devoid regions present in the overlap identified in (d) were significantly enriched for genes involved in the listed developmental pathways. The GREAT bioinformatic database was used to bin the genes identified into transcriptional networks.
Extended Data Figure 3. Developmental potential of ESCs cultured in S/L and 2i/L.
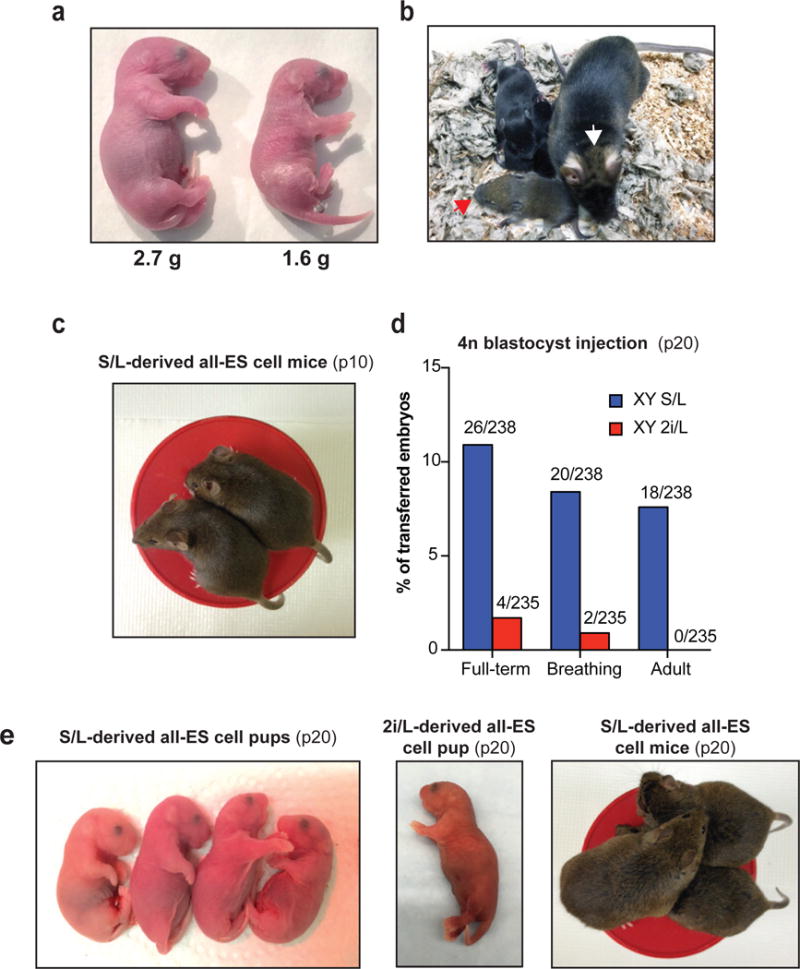
(a) Representative image of a chimeric pup derived from female ESCs cultured in S/L (left) compared to a pup obtained through natural mating (right). Body weight of each pup was shown below.
(b) Representative image of a low-grade adult chimera with patches of agouti hairs generated using female ESCs in S/L (white arrow). Her agouti germline offspring (red arrowhead) was generated by crossing her to a C57B6 wild-type mouse.
(c) Representative image of all-ESC adult mice generated from male ESCs cultured in S/L for 10 passages.
(d) Bar graphs showing percentages of transferred 4n blastocysts that survived to birth (“Full-term”), established regular breathing (“Breathing”) or survived past 5 weeks (“Adult”) using male ESCs grown in either S/L (blue bars) or 2i/L (red bars) until passage 20. The number of animals obtained per total number of transferred embryos is shown.
(e) Representative images of all-ESC neonates produced from male ESCs cultured in S/L (left) and 2i/L (middle) at passage 20. Representative image of all-ESC adult mice generated from ESCs cultured in S/L at passage 20 (right).
Extended Data Figure 4. Differentiation potential of male ESCs cultured in 2i/L.
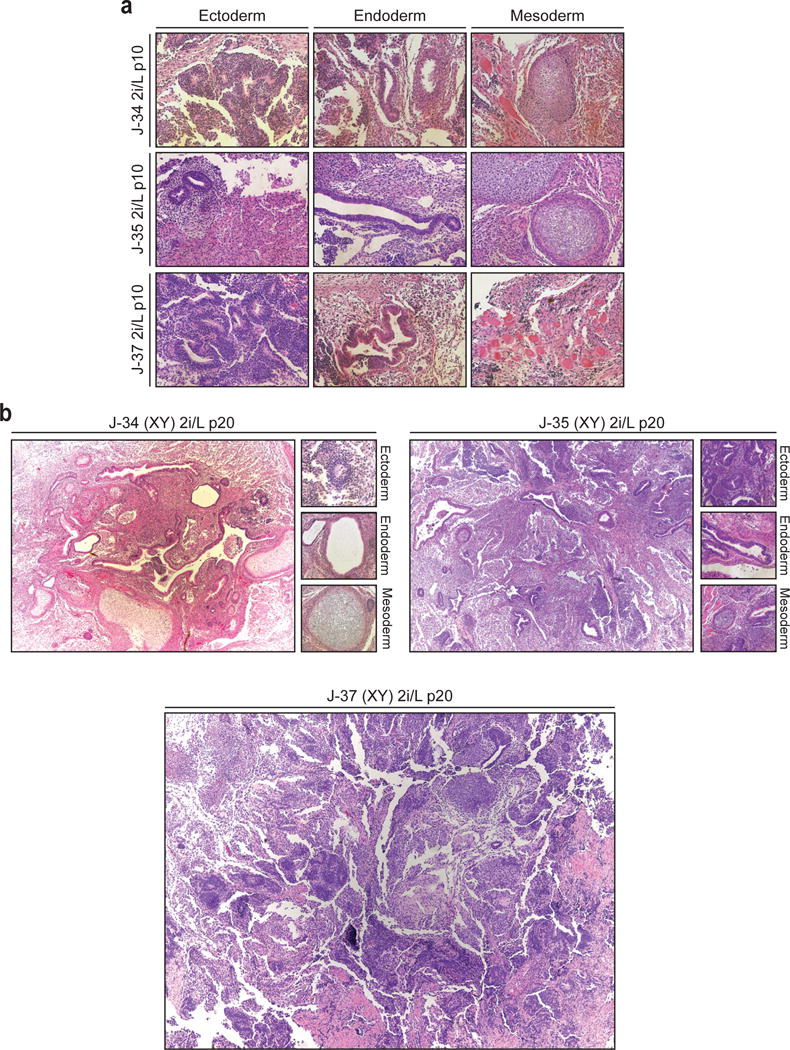
(a) Karyotype analysis of male ESC lines grown in 2i/L at p10.
(b) Representative images of teratomas produced with three male ESC lines grown in 2i/L at p20 with depiction of germ layer differentiation where detectable.
Extended Data Figure 5. Effects of prolonged 2i/L culture on chromosomal stability in ESCs.

(a) Array comparative genomic hybridization (aCGH) analysis of the J-35 ESC line cultured in S/L or 2i/L condition for the indicated passage numbers.
(b) Karyotype analysis of Rex1-dGFP ESC line grown in 2i/L for 8 passages after receiving the line. Red boxes indicate abnormal chromosomes detected.
(c) Karyotype analysis of J35 ESC line grown in 2i/L in the absence of a layer of MEF feeder cells for 16 passages (p20). Red boxes indicate abnormal chromosomes detected.
(d) aCGH analysis on DNA isolated from a newborn pup generated by 4n blastocyst injection using ESCs cultured in S/L or 2i/L at 20 passages.
(e) Combined dendrogram and heatmap depicting ICR methylation levels in ESCs (n=3 biological replicates) and keratinocytes using RRBS analysis. “2i/L Keratinocytes” were explantated from chimeric adult mice derived from male 2i/L ESC p10 and purified by drug selection (G418).
Extended Data Figure 6. Characterization of the XGXT ESC line.
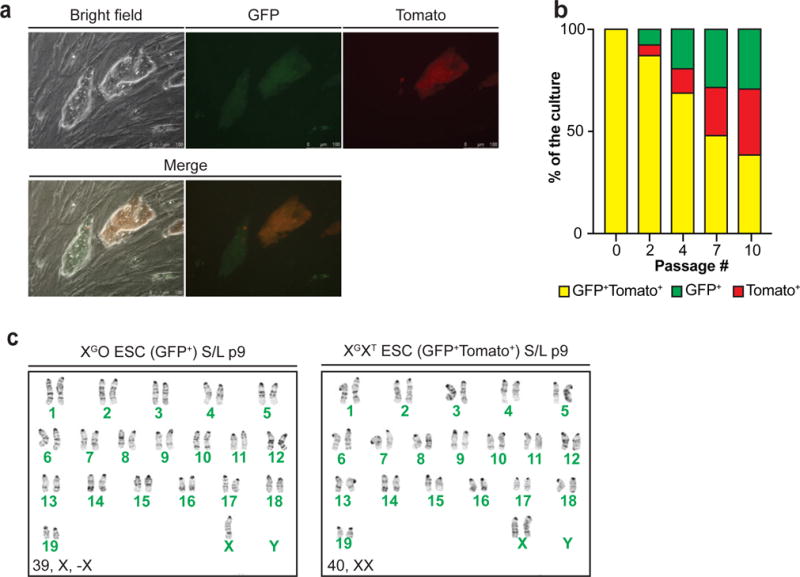
(a) Fluorescence microscopic images of XGXT ESCs. GFP/tdTomato double-positive cells (yellow cells in Merge 1 and 2) indicate two active X chromosomes while GFP+Tomato− colony depicts cells that lost one of the X chromosomes.
(b) GFP/tdTomato double-positive XGXT ESCs were sorted at passage 5 and plated in S/L (p6) before measuring the percentage of double-positive and single-positive cells at p8, 10, 13 and 16 using flow cytometry.
(c) Karyotype analysis of undifferentiated, XGO (GFP-positive) or XTO (tdTomato-positive) ESCs (left) and GFP/tdTomato double-positive XGXT ESCs (right). GFP/tdTomato double-positive XGXT ESCs were sorted at passage 5 and maintained in S/L for 9 passages before analyzed. This result confirms that the progressive loss of GFP or TdTomato signal was due to the loss of an X and not X-inactivation due to differentiation.
Extended Data Figure 7. Consequences of PD and CHIR treatment on DNA methylation.
(a) Global methylation levels of were analyzed by 5mC dot blot analysis for male and female ESC lines cultured in the indicated conditions for 6 passages.
(b) Median methylation levels at the indicated genetic elements following inhibition or loss of Mek1/2 or Gsk3α/β, respectively.
(c) Violin plots showing global methylation levels of male and female ESCs cultured in S/L for 6 passages and then cultured for an additional 3 passages in S/L supplemented with PD. White boxes indicate median methylation levels.
(d) Heatmap and dendrogram of ICR methylation levels in male and female ESCs shown in (c).
Extended Data Figure 8. Chromosomal aberrations that occur in 2i/L cultured ESCs are in part due to Mek1/2 inhibition.
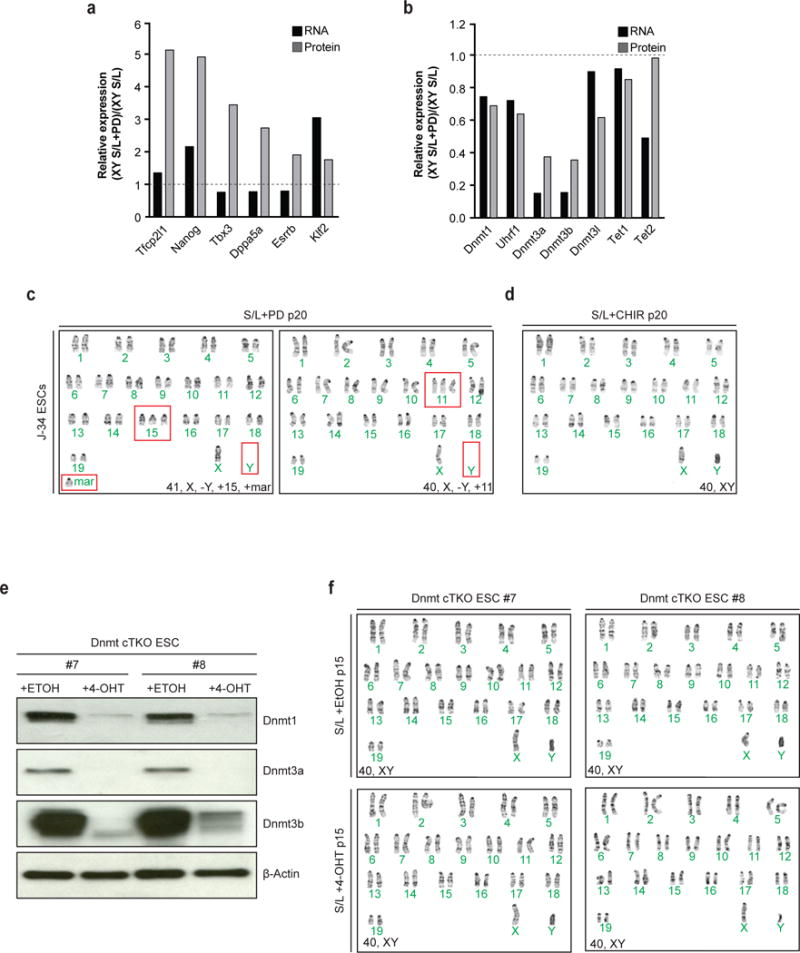
(a) Quantification of relative RNA and protein levels of pluripotency-related genes between male ESCs in S/L (n=2 technical replicates) and male ESCs in S/L+PD (n=2 technical replicates).
(b) Quantification of relative RNA and protein levels of genes that facilitate DNA methylation between male ESCs in S/L (n=2 technical replicatges) and male ESCs in S/L+PD (n=2 technical replicates) (see Fig. 4d).
(c–d) Karyotype analysis of male ESCs cultured in S/L for 4 passages and subsequently cultured in either S/L+PD (c) or S/L+CHIR (d) for additional 16 passages. +Mar stands for chromosomal fragment of unknown origin. Red boxes indicate abnormal chromosomes detected.
(e) Western blot analysis of a Dnmt cTKO ESC line for Dnmt1, Dnmt3a and Dnmt3b after treatment with either 4-OHT or EtOH for one passage.
(f) Karyotype analysis of a Dnmt cTKO ESC line that was treated with either 4-OHT or EtOH for one passage and subsequently cultured for 15 passages in S/L.
Extended Data Figure 9. Characterization of alternative culture conditions for ESCs.
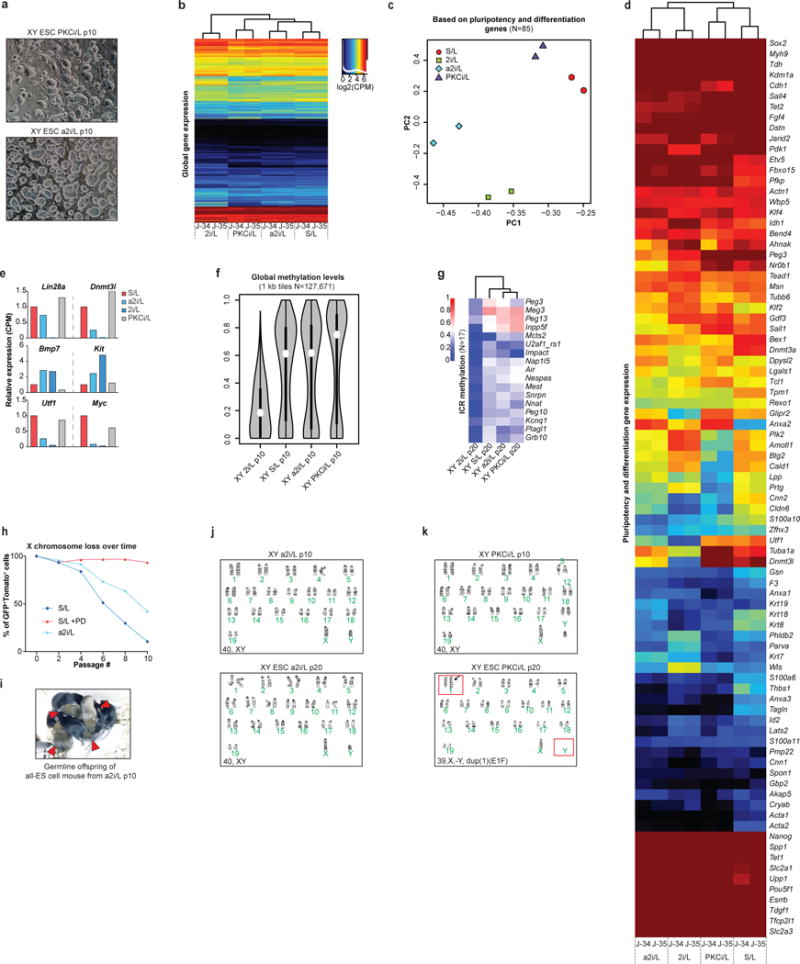
(a) Representative images of XY ESCs cultured in the indicated conditions.
(b) Heatmap and dendrogram of global gene expression levels in male ESCs cultured in the indicated conditions.
(c) Principal component analysis of ESCs cultured in S/L, 2i/L, a2i/L or PKCi/L using a previously published set of known pluripotency and differentiation genes22. Two male ESC lines maintained in S/L for 4 passages were switched to each condition and cultured for additional 6 passages (final passage 10).
(d) Heatmap and dendrogram showing expression levels of pluripotency and developmental genes22 in male ESC lines cultured in the indicated conditions.
(e) Relative expression levels of transcripts associated with naïve pluripotency in (d).
(f) Violin plots of global methylation levels in the indicated ESC lines. White dots, median value.
(g) Heatmap and dendrogram of ICR methylation levels in the indicated ESCs lines.
(h) Flow-cytometric analysis of GFP and TdTomato after culturing XGXT ESCs in S/L, S/L+PD and a2i/L for the indicated passages.
(i) Agouti germline offspring (red arrows), obtained from across between an all-ESC male generated from p10 a2i/L-cultured ESCs and a C57B6 female.
(j–k) Karyotype analysis of male ESCs cultured in a2i/L for 6 or 16 passages (j), or PKCi/L for 6 or 16 passages (k). Red boxes indicate abnormal chromosomes detected.
Extended Data Figure 10. MEK1/2-independent culture of naïve hESCs.
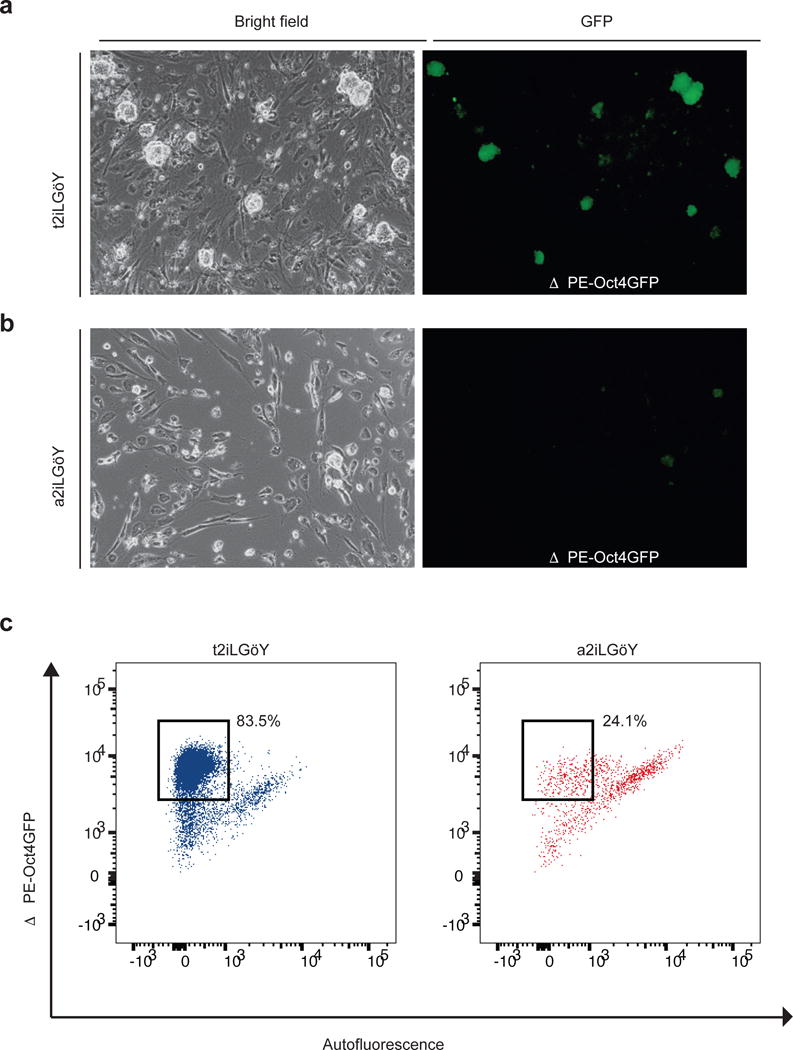
(a) Representative image of human ESCs (hESCs) carrying a naïve-specific OCT4-EGFP reporter (ΔPE-Oct4GFP)23 cultured in the presence of small molecule inhibitors of the ROCK, MEK1/2, GSK3 and PKC kinases (t2iLGöY)24. Bright field image (left) and GFP expression (right).
(b) Representative image of ΔPE-Oct4GFP hESCs cultured in the presence of small molecule inhibitors of the ROCK, SRC, GSK3 and PKC kinases (a2iLGöY). Bright field image (left) and GFP expression (right).
(c) Flow-cytometric analysis of EGFP expression in ΔPE-Oct4GFP hESCs cultured in t2iLGöY or a2iLGöY.
Supplementary Material
Acknowledgments
We thank members of the Hochedlinger lab for helpful suggestions as well as L. Barrett for critical reading of the manuscript. Additionally, we thank A. Galvin, M. Gesner and M. Handley at the Massachusetts General Hospital flow cytometry core. We also thank A. L. Hawkins, S. Wang, A. Aggarwal and C.C. Morton at Brigham and Woman’s Hospital CytoGenomics Core Laboratory for karyotyping analysis. We are grateful to K. Shioda from the Shioda lab for technical support for genome-sequencing. We are also grateful to W. Yu at The Sidney Kimmel Cancer Center Microarray Core Facility at Johns Hopkins University for aCGH analysis, and was supported by NIH grant P30 CA006973 entitled Regional Oncology Research Center. We thank T. Theunissen and R. Jaenisch for sharing human OCT4-GFP ESC lines and T. Graf for providing the Rex1-dGFP ESCs. A.J.H. is supported by an American Cancer Society – New England Division – Ellison Foundation Postdoctoral Fellowship (PF-15-130-01-DDC). B.D.S. is supported by an EMBO long-term Fellowship (#ALTF 1143-2015). T. Shioda is supported by NIH grant (R21ES024861). A.M. is a New York Stem Cell Foundation Robertson Investigator and supported by NIH grant (1P50HG006193) and the New York Stem Cell Foundation. K.H. was supported by funds from the MGH, HHMI, NIH (R01 HD058013-06) and the Gerald and Darlene Jordan Chair in Regenerative Medicine.
Footnotes
AUTHOR CONTRIBUTIONS
J.C. and A.J.H. performed bulk of the cell culture, teratomas assays, dot blot and flow-cytometry analysis; J.C., A.J.H. and S.K. performed the blastocyst injections; J.C., A.J.H. and J.C. generated the Mek1/2 DKO iPSC lines; R.M.W. generated the XGXT ESC line; J.S. and H.K. generated the Dnmt TKO ESC line; J.C., A.J.H. and J.B. conducted the Western blot analysis; J.B., C.M.R. and S.G. performed the proteomics analysis; B.D.S. performed experiments related to human ESC culture; J.O. and T.S. performed the genome sequencing analyses and analyzed the RNA-seq data of interspecies iPSC lines; K.C, H.G. A.M., A.G. and A.M. performed the RRBS analyses; A.S. and R.I.S. performed the RNA-seq analyses; K.L. and A.X. conducted the H2A.X ChIP-seq analysis.; J.C., A.J.H. and K.H. designed the experiments, interpreted the results and wrote the manuscript.
The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Ying QL, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raggioli A, Junghans D, Rudloff S, Kemler R. Beta-catenin is vital for the integrity of mouse embryonic stem cells. PLoS ONE. 2014;9:e86691. doi: 10.1371/journal.pone.0086691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, et al. Erk signaling is indispensable for genomic stability and self-renewal of mouse embryonic stem cells. Proc Natl Acad Sci USA. 2015;112:E5936–43. doi: 10.1073/pnas.1516319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 5.Gaudet F, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 6.Habibi E, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Ficz G, et al. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell. 2013;13:351–359. doi: 10.1016/j.stem.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leitch HG, et al. Naive pluripotency is associated with global DNA hypomethylation. Nat Struct Mol Biol. 2013;20:311–316. doi: 10.1038/nsmb.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulz EG, et al. The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell. 2014;14:203–216. doi: 10.1016/j.stem.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 10.Zvetkova I, et al. Global hypomethylation of the genome in XX embryonic stem cells. Nat Genet. 2005;37:1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]
- 11.Choi J, et al. DUSP9 Modulates DNA Hypomethylation in Female Mouse Pluripotent Stem Cells. Cell Stem Cell. 2017;20:706–719.e7. doi: 10.1016/j.stem.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hackett JA, et al. Synergistic mechanisms of DNA demethylation during transition to ground-state pluripotency. Stem Cell Reports. 2013;1:518–531. doi: 10.1016/j.stemcr.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu T, et al. Histone variant H2A.X deposition pattern serves as a functional epigenetic mark for distinguishing the developmental potentials of iPSCs. Cell Stem Cell. 2014;15:281–294. doi: 10.1016/j.stem.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141:1805–1813. doi: 10.1242/dev.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Celeste A, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wray J, et al. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol. 2011;13:838–845. doi: 10.1038/ncb2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamm C, Pijuan Galitó S, Annerén C. A comparative study of protocols for mouse embryonic stem cell culturing. PLoS ONE. 2013;8:e81156. doi: 10.1371/journal.pone.0081156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Meyenn F, et al. Impairment of DNA Methylation Maintenance Is the Main Cause of Global Demethylation in Naive Embryonic Stem Cells. Mol Cell. 2016;62:983. doi: 10.1016/j.molcel.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutta D, et al. Self-renewal versus lineage commitment of embryonic stem cells: protein kinase C signaling shifts the balance. Stem Cells. 2011;29:618–628. doi: 10.1002/stem.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimizu T, et al. Dual inhibition of Src and GSK3 maintains mouse embryonic stem cells, whose differentiation is mechanically regulated by Src signaling. Stem Cells. 2012;30:1394–1404. doi: 10.1002/stem.1119. [DOI] [PubMed] [Google Scholar]
- 22.Kolodziejczyk AA, et al. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 2015;17:471–485. doi: 10.1016/j.stem.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theunissen TW, et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell. 2014;15:471–487. doi: 10.1016/j.stem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takashima Y, et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastor WA, et al. Naive Human Pluripotent Cells Feature a Methylation Landscape Devoid of Blastocyst or Germline Memory. Cell Stem Cell. 2016;18:323–329. doi: 10.1016/j.stem.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takashima Y, et al. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stadtfeld M, Maherali N, Borkent M, Hochedlinger K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat Methods. 2010;7:53–55. doi: 10.1038/nmeth.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaneda M, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 29.Dodge JE, et al. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J Biol Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 30.Boyle P, et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 2012;13:R92. doi: 10.1186/gb-2012-13-10-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 34.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keane TM, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Treff NR, et al. Next Generation Sequencing-Based Comprehensive Chromosome Screening in Mouse Polar Bodies, Oocytes, and Embryos. Biol Reprod. 2016;94:76. doi: 10.1095/biolreprod.115.135483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao A, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song Q, Smith AD. Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics. 2011;27:870–871. doi: 10.1093/bioinformatics/btr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eggan K, et al. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci USA. 2001;98:6209–6214. doi: 10.1073/pnas.101118898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 48.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the raw data files for the RRBS analysis reported in this paper is NCBI GEO: GSE97995.
The accession number for the raw data files for the RRBS analysis on female ESC lines is NCBI GEO: GSM1679872, GSM1679873, GSM1679874.
The accession number for the raw data files for the RNA-seq analysis reported in this paper is NCBI GEO: GSE97954, GSE93103.
The accession number for the raw data files for the Genome-sequencing analysis reported in this paper is NCBI SRA: SRP102419.
The accession number for the raw data files for the ChIP-seq analysis reported in this paper is NCBI GEO: GSE98771.