

Abstract
Background
Early treatment with valproic acid (VPA) has demonstrated benefit in pre-clinical models of traumatic brain injury (TBI), including smaller brain lesion size, decreased edema, reduced neurologic disability, and faster recovery. Mechanisms underlying these favorable outcomes are not fully understood. We hypothesized that VPA treatment would upregulate genes involved in cell survival and proliferation and downregulate those associated with cell death and the inflammatory response.
Methods
Ten female swine were subjected to a protocol of TBI and hemorrhagic shock. They were assigned to two groups (n=5): normal saline (NS; 3X volume of shed blood), or NS + VPA (150 mg/kg). Following 6 hours of observation, brain tissue was harvested to evaluate lesion size and edema. Brain tissue was processed for RNA sequencing. Gene set enrichment and pathway analysis was performed to determine the differential gene expression patterns following injury.
Results
Animals treated with VPA were noted to have a 46% reduction in brain lesion size and a 57% reduction in ipsilateral brain edema. VPA significantly up-regulated genes involved in morphology of the nervous system, neuronal development and neuron quantity. VPA treatment downregulated pathways related to apoptosis, glial cell proliferation, and neuroepithelial cell differentiation. Ingenuity Pathway Analysis identified VPA as the top upstream regulator of activated transcription, supporting it as a direct cause of these transcriptional changes. Master transcriptional regulator NEUROD1 was also significantly upregulated, suggesting that VPA may induce additional transcription factors.
Conclusions
Administration of VPA attenuated brain lesion size, reduced brain edema, and induced significant changes in the transcriptome of injured brain within 6 hours. Patterns of differential expression were consistent with the proposed neurogenic and pro-survival effects of VPA treatment.
Keywords: valproic acid, histone deacetylase inhibitor, RNA sequencing, hemorrhage, traumatic brain injury
BACKGROUND
Valproic acid (VPA) has gained recent attention for use as a pharmacologic agent in trauma, and has undergone extensive preclinical testing in the setting of traumatic brain injury (TBI).(1, 2) After decades of use as a treatment for epilepsy, it was discovered that in higher doses it has action as a histone deacetylase inhibitor (HDACI).(3) Nuclear DNA is complexed with histones within chromatin – these histones are subject to post-translational modifications, which affects their association with DNA. This is broadly controlled by two classes of enzymes, histone acetyltransferases (HATs), and histone deacetylases (HDACs). Inhibition of HDACs results in increased acetylation of histones, disrupting the association with DNA and causing relaxation of the chromatin structure. DNA is therefore made more accessible to transcriptional machinery and gene transcription is up-regulated.
An imbalance in HAT/HDAC activity has been shown in both hemorrhagic shock (HS)(4) and neurodegeneration(5), which justifies the testing of VPA in this context. Our group has tested VPA treatment in various models of HS,(6) TBI,(7) and polytrauma,(2) and shown significant improvement in outcomes. These positive studies have led to phase I clinical testing of VPA (NCT01951560).(8) The beneficial effects of VPA are largely attributed to its action as an HDACI, but the exact mechanisms are still not fully characterized.
Mechanistic studies have demonstrated differential regulation of master transcription factors by VPA, and modulation of multiple pathways involved in the inflammatory response, cell survival, and neurogenesis.(9, 10) Our group and others have also shown pro-survival changes at the protein level, including quantitative differences in heat shock protein 70, hypoxia-inducible factor 1a, and beta-catenin.(6, 11, 12) However, evidence from cancer research demonstrates situation-specific divergence in VPA’s mechanism, and that it likely has differential effects depending on cell type and status. This highlights the need to investigate its effects in our specific area of interest, TBI and HS. The aim of this study was to define the transcriptomic changes induced by VPA in swine subjected to TBI and HSthis setting. RNA sequencing was performed on injured brain tissue in swine subjected to TBI and HS to allow for characterization of the transcriptome and differential gene expression analysis. This study was carried out in the injured brain to directly assess the effects on this tissue. Our hypothesis was that VPA treatment would up-regulate genes involved in cell survival and proliferation, and down-regulate those that are associated with cell death and inflammation.
METHODS
All experiments adhered to guidelines stipulated in the Animal Welfare Act and all federal statutes regarding animal experiments. These experiments were approved by the Institutional Animal Care and Use Committee and performed under supervision of veterinarian staff.
Injury Model
Swine were subjected to a well-described model of TBI and HS.(13, 14) In brief, female Yorkshire swine (40–50 kg; Michigan State University, East Lansing, MI) were anesthetized and instrumented for hemodynamic and intracranial monitoring. Hemodynamic, intracranial, and laboratory parameters were collected throughout the experiment. Hemodynamic data were compared between groups using unpaired t-tests with Holm-Sidak correction and laboratory parameters compared using t-tests with Welch’s correction. All statistical analyses for this study was performed using GraphPad Prism version 6.00 (GraphPad Software, San Diego CA). Animals were placed in the prone position and the head was secured in a stereotactic frame. A 20mm craniotomy was made anterior to the bregma. Next, a computer-controlled cortical impact device (custom made – University of Michigan Medical Innovation Center; Ann Arbor, MI) was used to induce a TBI (15mm cylindrical impactor, 4m/s velocity, 100ms dwell time, 12mm impact depth). Concurrently, animals were subjected to 40% total blood volume hemorrhage and maintained in a state of hypovolemic shock (mean arterial pressure, MAP; 30–35 mmHg) for two hours via titration of isoflurane. Resuscitation was delayed for 2 hours to simulate prolonged extrication in a pre-hospital setting.
Animals were then randomized to one of two groups: normal saline alone (NS) or NS + VPA (n = 5/cohort). All animals were resuscitated with NS (3x hemorrhage volume, infused over 1 hour) at the end of the shock period. Animals assigned to the NS + VPA group received a single 150mg/kg peripheral intravenous injection of VPA (concentration: 100mg/ml, total volume infused 55–67.5ml) infused over 90 minutes, starting one hour into shock, before NS resuscitation,. This schedule was chosen to simulate early treatment initiated by first responders a bridge to definitive care. At the end of the shock period, all the animals were resuscitated with NS (3x hemorrhage volume, infused over 1 hour). Following a six-hour observation period, animals were euthanized, and tissues were collected for further analysis.
Tissue Sampling Lesion Size and Edema
Our tissue sampling methods have been previously described. In brief, tThe brain was harvested and sliced into 5mm coronal sections. I, and images were obtained for the calculation of edema using ImageJ analysis software (National Institutes of Health, Bethesda, MD). The following formula was applied to calculate brain edema: brain edema = (volume of ipsilateral hemisphere/volume of contralateral hemisphere – 1) × 100%.(15) Sections were then stained with 2% 2,3,5-triphenyltetrazolium chloride (Sigma Chemical Co., St. Louis, MO) to detect viable tissue and further images were obtained for lesion size calculation using ImageJ software. Lesion sizes and edema were compared using unpaired t-test with Welch’s correction, and are expressed as mean ± standard deviation. Brain tissue from the region inferior to the most injured site was flash frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
Tissue Sampling and RNA Extraction
Brain tissue from the region inferior to the most injured site was flash frozen in liquid nitrogen and stored at −80°C for subsequent analysis. RNA preparation methods have been described previously by our laboratory.(9, 10) RNA extraction from the frozen specimens was conducted utilizing an RNeasy Mini Kit (Qiagen, Valencia, CA).
RNA Sequencing
RNA sequencing was performed by the DNA Sequencing Core. RNA quality was assessed using TapeStation (Agilent, Santa Clara, CA), and samples with an RNA Integrity Number (RIN) of 8 or greater were prepared using the Illumina TruSeq Stranded mRNA Library Prep kits (RS-122-2101 and RS-122-2102, Illumina, San Diego, CA) per manufacturer’s protocols. For each sample, 0.1–0.3μg of RNA was converted to messenger RNA (mRNA) using polyA purification methods. The mRNA was then fragmented and copied into first-strand complimentary DNA (cDNA) utilizing random primers and reverse transcriptase. Prime ends of cDNA were then adenylated, and adapters ligated. For multiplexed sequencing, a unique six-nucleotide barcode was identified in each sample. Final products were purified and enriched by polymerase chain reaction (PCR, 12 cycles) for the construction of a final cDNA library. Final libraries were again assessed using TapeStation and quantitative PCR (KAPA library quantification kit for Illumina Sequencing platforms, KK4835, KAPA Biosystems, Wilmington, MA), per manufacturer’s protocols. After pooling, samples were clustered on the cBot system (Illumina, San Diego, CA) and sequenced over three lanes on the HiSeq 4000 (Illumina, San Diego, CA) using paired-end 50-nucleotide reads.
Gene Set Enrichment, Pathway Analysis, and Network Analysis
Following alignment Reads were aligned to a reference utilizing STAR,(16) and multiple redundant methods for differential RNA sequencing expression analysis were utilized by two independent researchers, including DESeq2(17) and CuffDiff2(18) in R Bioconductor,(19) and as well as D-GEX(20) in Python.(21) Only the agreement between these programs was included in analysis. Based on a negative binomial data model,(17, 22) differentially expressed genes were selected based on a significance level corrected for multiple comparisons using statistical comparisons were made utilizing a false discovery rate with a stringent threshold of FDR ≤ 0.001. To ensure data normalization and bias exclusion, conditional quantile normalization was applied and genes with low-level expression were filtered out. and low count filter preprocessing were applied. MA plots were generated to visualize pairwise comparison of utilizing pair-wide comparisons at each time point of a log2-fold change versus mRNA isoform abundance were generated to examine for each differentially expressed transcript, and the distribution delineation of significantly (FDR ≤ 0.001) up- and down-regulated genes. Gene symbol, disease involvement, and gene function data were obtained from both Gene Ontology (GO)(23) and the gene set enrichment analysis (GSEA) module in Ingenuity Pathway Analysis® software (IPA).(24) Data was were curated by independent researchers who were blinded to each other’s correction. Pathway analysis was performed using IPA with filters specific to CNS tissue. The pathway results were compared to the M30 epilepsy network, which has been suggested as a gene network target of VPA.
RESULTS
Animals in both groups demonstrated comparable degrees of shock and response to resuscitation in terms of heart rate, MAP, cardiac output and intracranial pressure (summarized in Supplemental Digital Content 1). Laboratory parameters are summarized in Supplemental Digital Content 2. As has been seen previously, VPA treatment resulted in impaired lactate clearance.(7) There were no adverse events associated with treatment. Brain lesion sizes were significantly smaller in the NS + VPA group than the NS only group (mean lesion size, mm3: NS = 3107 ± 999 vs NS + VPA = 1690 ± 121, p = 0.03). Similarly, ipsilateral brain edema was significantly reduced in the NS + VPA group (edema, %: NS = 32.5 ± 6.6 vs NS + VPA = 13.9 ± 10.6, p = 0.01).
IPA analysis revealed an inter-connected subnetwork of 257 significantly up-regulated genes following NS + VPA treatment (Figure 1). Up-regulated genes included master transcription factors involved in astrocyte programming into glial differentiation, including NEUROD1, ASCL1 and SOX1; members of the circadian CLOCK gene family; and genes which code for hormone receptors and growth-related transcription factors (e.g., NR3C1, EGR1), ion channels (e.g., SCN1A, SCN8A, CACNB2, KCNB1), neurotransmitter receptors (e.g., ADRGB2, CHRNB2, OPRK1), synaptic molecules (e.g., SNAP25, SV2B, CHGA), neurofilament and associated cytoskeletal molecules (e.g., INA, NEFL, NEFM, NEFM), mRNA splicing regulators (e.g., RBFOX1, CELF4), chromatin factors, histone acetyl- and methyl-transferases (e.g., ARID1A, EP300), and the neuronal progenitor complex.
Figure 1.

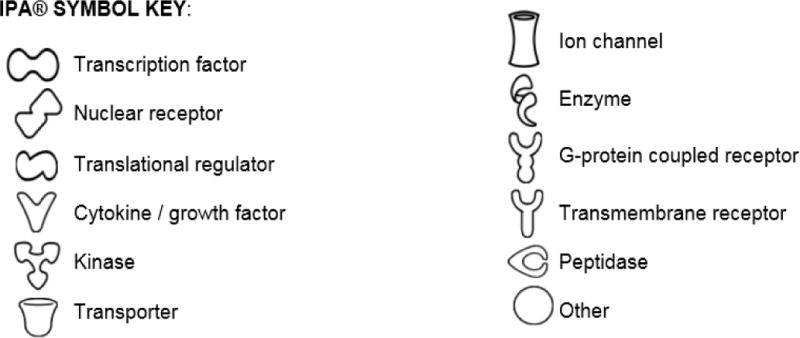
Molecular network connected using Ingenuity Pathway Analysis (IPA)® of up-regulated genes following VPA treatment in a swine model of traumatic brain injury and hemorrhagic shock (p=1xE-27; Fishers exact test). See IPA® Symbol Key for molecule definitions.
IPA also revealed a subnetwork of 156 genes which were significantly down-regulated (Figure 2). These genes referred to angiogenic factors and macrophage scavengers (e.g., MSR1, THBS1), inflammatory molecules and chemokines (e.g., CXCR, IL1A), epithelial cell proliferation, endothelial chemotactic molecules (e.g., FGF7, SELE), oligodendrocyte proliferation and myelin molecules (e.g., OPALIN, SOX10), cell death and apoptotic factors (e.g., BAD, IAPP, SMARCA2, SMARCB1) and cell cycle regulators (e.g., CCNB1, CDC7).
Figure 2.
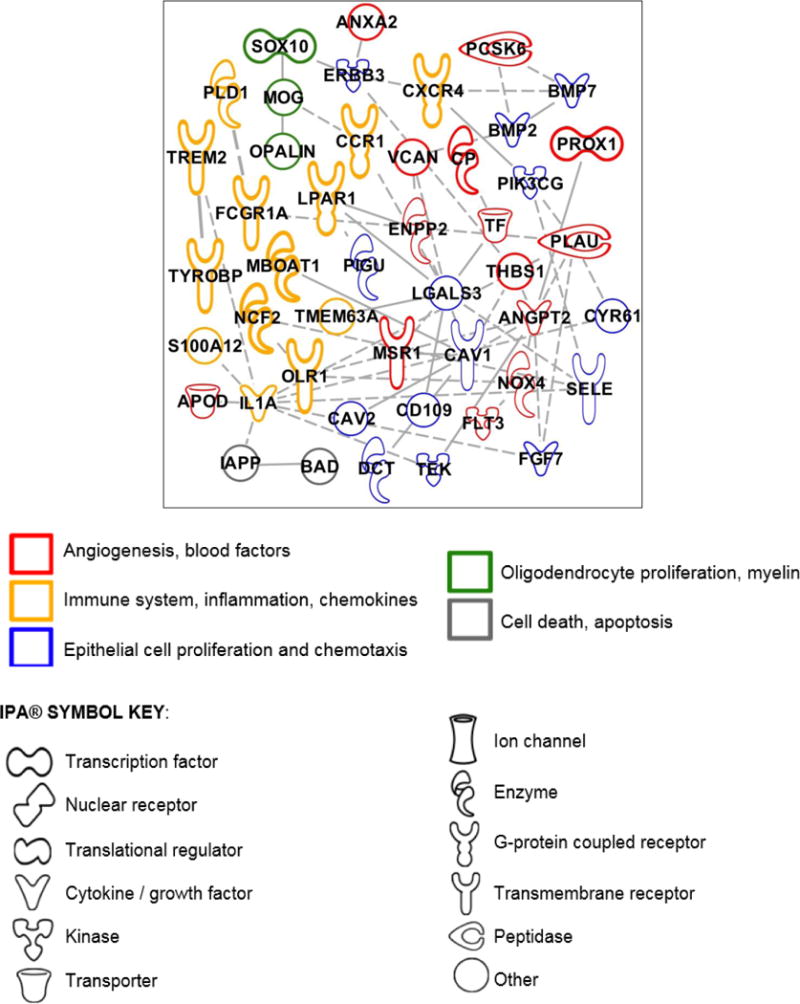
Molecular network connected using Ingenuity Pathway Analysis (IPA)® of down-regulated genes following VPA treatment in a swine model of traumatic brain injury and hemorrhagic shock (p=1xE-22; Fishers exact test). See IPA® Symbol Key for molecule definitions.
Network-based GSEA using IPA was utilized to identify the top 5 functional associations amongst genes affected by VPA treatment (Table 1). The top 5 functions of up-regulated genes were associated with differentiation of neurons (p = 6.03E-45), quantity of cells (p = 6.63E-42), circadian rhythm (p = 4.84E-32), CNS morphology (p = 1.01E-32) and neurogenesis (p = 4.42E-30). The top 5 functions of down-regulated genes were associated with apoptosis (p = 5.12E-28), glial cell proliferation (p = 6.07E-26), migration of cells (p = 1.1E-24), chemotaxis of endothelial cells (p = 1.15E-22), and movement of endothelial cells (p = 1.06E-18).
Table 1.
IPA® Gene Set Enrichment Analysis of Genes Modulated by VPA
| Genes | Top 5 functions (p value) | Top 5 Regulators (p value) | Top 5 canonical pathways (p value) |
|---|---|---|---|
| Up-regulated | 1. Differentiation of neurons (6.03E-45) 2. Quantity of cells (6.63E-42) 3. Circadian rhythm (4.84E-32) 4. CNS morphology (1.01E-31) 5. Neurogenesis (4.42E-30) |
1. VPA (3.94E-43) 2. HTT (1.17E-32) 3. NEUROD1 (1.66E-32) 4. ASCL1 (8.04E-30) 5. BDNF (1.01E-19) |
1. Corticotrophin releasing hormone signaling (1.17E-13) 2. Circadian rhythm signaling (2.81E-13) 3. Transcriptional regulatory networks in embryonic stem cells (3.15E-14) 4. Glucocorticoid receptor signaling (1.19E-12) 5. Nitric oxide signaling (2.77E-10) |
| Down-regulated | 1. Apoptosis (5.12E-28) 2. Glial cell proliferation (6.07E-26) 3. Migration of cells (1.1E-24) 4. Chemotaxis of endothelial cells (1.15E-22) 5. Cell movement of endothelial cells (1.06E-18) |
1. VPA (1.32E-23) 2. OLIGO2 (9.99E-21) 3. FGF7 (1.02E-15) 4. PROX1 (6.09E-11) 5. PRXX1 (2.31E-09) |
1. Apoptosis signaling (3.44E-22) 2. Cellular movement (1.21E-20) 3. LXR/RXR activation (3.93E-16) 4. RHOA signaling (2.65E-10) 5. Caveolar-mediated endocytosis signaling (7.99E-09) |
IPA = Ingenuity Pathway Analysis; VPA = valproic acid.
In addition, IPA identified VPA as the top upstream regulator of up-regulated genes (p = 1.15E-22). The master transcriptional regulator NEUROD1 was also significantly represented (p = 1.66E-32). Further, VPA was identified as the top upstream regulator of down-regulated genes (p = 1.32E-23).
Similar results were obtained using GO, showing up-regulated gene transcripts associated with neurogenesis, neuronal differentiation, CNS development and circadian regulation of gene expression (Table 2), and down-regulated genes demonstrating repression of those involved in immune function and inflammation, glial cell and leukocyte migration, epithelial cell proliferation, and behavioral attributes such as locomotion and the defense response (Table 3).
Table 2.
GO Gene Set Enrichment Analysis of Genes Up-regulated by VPA
| GO biological process complete | Fold Enrichment | p value |
|---|---|---|
| Nervous system development | 3.91 | 5.51E-34 |
| Generation of neurons | 4.51 | 1.12E-26 |
| Neurogenesis | 4.27 | 1.60E-25 |
| Regulation of nervous system development | 5.95 | 4.52E-24 |
| Regulation of neurogenesis | 6.01 | 2.45E-21 |
| Regulation of neuron differentiation | 6.54 | 6.75E-20 |
| Central nervous system development | 4.17 | 2.41E-12 |
| Circadian regulation of gene expression | 22.03 | 7.48E-12 |
| Positive regulation of neurogenesis | 6.2 | 1.04E-11 |
| Circadian rhythm | 11.37 | 1.43E-10 |
GO= Gene Ontology; VPA = valproic acid.
Table 3.
GO Gene Set Enrichment Analysis of Genes Down-regulated by VPA
| GO biological process complete | Fold Enrichment | p value |
|---|---|---|
| Immune system process | 2.83 | 3.28E-07 |
| Regulation of epithelial cell proliferation | 3.34 | 4.77E-06 |
| Locomotion | 3.71 | 1.34E-05 |
| Regulation of locomotion | 4.43 | 1.90E-05 |
| Glial cell migration | 4.23 | 4.49E-05 |
| Leukocyte migration | 5.89 | 5.02E-05 |
| Regulation of epithelial cell proliferation | 1.38 | 5.10E-05 |
| Regulated exocytosis | 6.19 | 7.32E-05 |
| Defense response | 4.23 | 1.00E-04 |
| Regulation of cell adhesion | 5.96 | 1.24E-04 |
GO = Gene Ontology; VPA = valproic acid.
The up-regulated genes were compared to a known set of 320 genes involved in epilepsy, called the ‘M30 network’.(25) Of the 257 genes in this set, 107 genes overlapped with the M30 network (Figure 3). These genes were associated with nervous system development (p = 5.42E-06), action potentials (p = 2.78E-05), trans-synaptic signaling (p = 3.09E-05), synaptic signaling (p = 3.09E-05) and regulation of membrane potential (p = 1.69E-04). The remaining 150 genes up-regulated by VPA following TBI and HS were associated with nervous system development (p = 6.79E-30), generation of neurons (p = 2.21E-23), neurogenesis (p = 6.21E-22), regulation of nervous system development (p = 2.56E-10), and regulation of neurogenesis (p = 3.03E-19).
Figure 3.
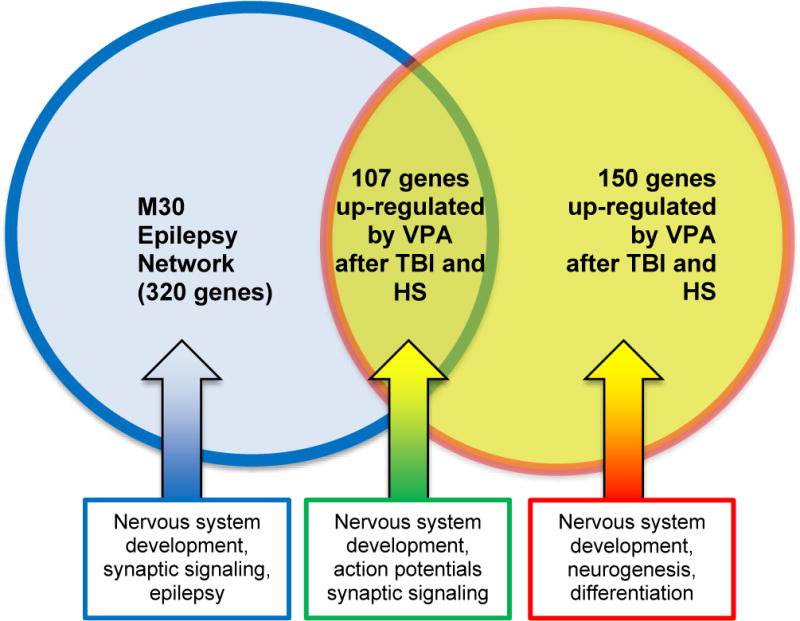
Overlap of up-regulated genes by valproic acid (VPA) at 6 hours following traumatic brain injury (TBI) and hemorrhagic shock (HS) in an animal model with the M30 epilepsy network in brain, a target of VPA containing genes with variants that have been significantly associated with different types of epilepsy in genome-wide association studies.
DISCUSSION
In this study, we utilized RNA sequencing to demonstrate changes in gene expression induced by VPA in swine subjected to TBI and HS. We found that VPA up-regulated genes associated with neurogenesis, neurotransmission, and neuroregulation, while down-regulating genes associated with cell death and inflammation. These changes correlate with reduced brain lesion size and edema. Overall, these findings are consistent with our prior work and continue to expand our knowledge of the mechanisms that are responsible for the observed benefits of VPA in TBI models.
Differential expression analysis revealed a total of 257 genes selectively up-regulated by VPA administration. GSEA using both IPA and GO, which groups individual genes according to biological function, demonstrated a pattern of increased neuronal proliferation and differentiation, regulation over CNS morphology, and control of circadian rhythm. This was confirmed by both IPA and GO. IPA also demonstrated that these genes map into an interconnected sub-network of genes expressed in brain tissue. This revealed additional functions in combination, including control of neurotransmission and regulation of cytoskeletal stability. This implies overall trends in increased neurogenesis, regulation of neurotransmission, and cytoskeletal stability associated with VPA treatment.
These data suggest VPA may up-regulate development of new neurons in the injured brain. Neurogenesis occurs in response to TBI,(26) mainly in the perilesional area(27). This is thought to be a component of remodeling after injury, which contributes to functional recovery.(28) VPA may therefore increase the rate of neurocognitive recovery by this mechanism, as seen in our long-term studies.(2, 7) VPA also increased expression of genes involved in neurotransmission and regulation of synaptic functions. Neurotransmission is shown to be disordered following brain injury, which is associated with potentiation of the primary injury.(29) The fact that these changes correlate with decreased lesion sizes suggests that VPA may help to restore normal functions.
GSEA also suggested that a single dose of VPA in this model up-regulates genes involved in the circadian rhythm. TBI is associated with acute dysregulation of circadian rhythm, and this has been suggested as a potential target for treatment.(30) Circadian rhythm and clock genes are connected with many cellular functions, including the cell cycle,(31) and deregulation of cell cycle progression may result in cell death(32). Thus, VPA may decrease cell death via modulation of clock genes.
Differential expression analysis revealed a total of 156 genes down-regulated by VPA. GSEA using both IPA and GO demonstrated a pattern of decreased apoptosis, immune activation, leukocyte migration, glial cell proliferation and proliferation and movement of endothelial cells. Overall this suggested a phenotype of attenuated cell death and inflammation, and decreased gliosis (responsible for post-injury scarring). Network mapping and functional analysis additionally showed decreased angiogenesis represented by these transcripts.
These data imply modulation of the immune response and inflammation by VPA treatment, which corroborates our previous work.(9) This may translate into attenuation of secondary brain injury, and decreased brain lesion size and edema. Along with the down-regulation of pro-apoptotic genes, this suggests that VPA treatment offers an element of neuroprotection. Down-regulation of gliosis by VPA could also explain functional recovery seen in other models,(2, 7) by allowing for remodeling in the place of scarring. Interestingly, although angiogenesis is thought to be paired with neurogenesis in mediating remodeling,(33) this study shows down-regulation of genes involved in angiogenesis with VPA treatment.
Our analysis also identified key mediators of VPA-induced transcriptomic changes. As anticipated, VPA was found to be the top regulator. In addition to this, VPA was shown to induce master transcription factors including NEUROD1, as has been shown in other studies.(34) Overall, these findings indicate that VPA has both direct and downstream effects on gene regulation.
VPA’s effects on gene transcription were compared with its mechanism involved in the treatment of epilepsy. Gene susceptibility for epilepsy has been studied extensively within recent years, revealing a common set of involved genes, known as the M30 network. This network involves 320 genes which are under tight developmental control and are highly involved in neural processes and synaptic function.(25) This network is thought to be targeted by VPA.
Our study demonstrates that 107 of the genes up-regulated by VPA are shared with the M30 network; such genes play key roles in neurogenesis and neurotransmission. In addition to this, a separate cohort of 150 genes was also up-regulated. These are also involved in neurogenesis and neurotransmission and only strongly induced following TBI.(34) Overall, this suggests that VPA possesses baseline effects on the M30 network as well as additional effects in the setting of TBI and HS. While this study focuses on VPA’s action as an HDACI, it is likely that its mechanism extends beyond that of simple acetylation of histones. Cell lines treated with VPA have demonstrated direct activation of chromatin-state remodeling complexes. Although a number of possible mechanisms and key mediators have been discovered within recent years, future research should continue to expand on these.
There are several limitations to this study. First, although swine are commonly utilized for human translation, they are an imperfect surrogate for humans. Second, our injury model is not representative of all forms of TBI. Third, we evaluated genetic changes at 6 hours following treatment. It is well known that transcriptional changes are dynamic and time-dependent.; Oour findings represent a single time point along a continuum of cellular alterations following treatment. Finally, we did not utilize a VPA-only cohort, as this was a lethal model which required resuscitation. However, we attempted to isolate its effects by comparing the NS and NS+VPA groups. We also do not have gene expression data from a sham group, so it is unclear if the effects of VPA on gene expression are restorative or de novo. Additional studies are already underway to fill in many of these gaps.
In conclusion, this study demonstrates VPA’s effects on the transcriptome in a clinically relevant large animal model of TBI and HS. Following injury, VPA up-regulates genes associated with neurogenesis, neurotransmission, and neuroregulation, and down-regulates genes associated with cell death and inflammation. Such changes may play a key role in VPA’s benefit following injury. This study provides further explanation of VPA’s effect in trauma; however, more work is required for a comprehensive understanding of its mechanism.
Supplementary Material
Acknowledgments
Grant funding – VCN, HBA. We would also like to acknowledge the work of Kiril Chtraklin and Celia Tafatia who assisted in animal experiments.
Funding: US Army Medical Research Materiel Command W81XWH-09-1-0520 (Alam, HB); Frederick A. Coller Society Research Fellowship Grant (Nikolian, VC).
Footnotes
Conflict of Interest: none to report.
Meeting presentation: this study was presented at the 76th Annual Meeting of AAST and Clinical Congress of Acute Care Surgery (Baltimore, MD, September, 2017.)
Level of evidence: not applicable (pre-clinical study.)
Study type: therapeutic.
AUTHOR CONTRIBUTIONS
Conception and design – VCN, ISD, HBA. Data acquisition – VCN, ISD, GAH, AMW, MW, PEG, HE, MHG, PC; Data interpretation – VCN, ISD, GAH, AMW, MW, HBA; Manuscript preparation – VCN, ISD, AMW; Critical revisions – all authors.
References
- 1.Chen S, Wu H, Klebe D, Hong Y, Zhang J. Valproic acid: a new candidate of therapeutic application for the acute central nervous system injuries. Neurochem Res. 2014;39(9):1621–33. doi: 10.1007/s11064-014-1241-2. [DOI] [PubMed] [Google Scholar]
- 2.Nikolian VC, Georgoff PE, Pai MP, Dennahy IS, Chtraklin K, Eidy H, Ghandour M, Han Y, Srinivasan A, Li Y, et al. Valproic acid decreases brain lesion size and improves neurologic recovery in swine subjected to traumatic brain injury, hemorrhagic shock, and polytrauma. J Trauma Acute Care Surg. 2017 doi: 10.1097/TA.0000000000001612. [DOI] [PubMed] [Google Scholar]
- 3.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20(24):6969–78. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin T, Alam HB, Chen H, Britten-Webb J, Rhee P, Kirkpatrick J, Koustova E. Cardiac histones are substrates of histone deacetylase activity in hemorrhagic shock and resuscitation. Surgery. 2006;139(3):365–76. doi: 10.1016/j.surg.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 5.Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler J-P, Boutillier A-L. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22(24):6537–49. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alam HB, Shuja F, Butt MU, Duggan M, Li Y, Zacharias N, Fukudome EY, Liu B, Demoya M, Velmahos GC. Surviving blood loss without blood transfusion in a swine poly-trauma model. Surgery. 2009;146(2):325–33. doi: 10.1016/j.surg.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Halaweish I, Bambakidis T, Chang Z, Wei H, Liu B, Li Y, Bonthrone T, Srinivasan A, Bonham T, Chtraklin K, et al. Addition of low-dose valproic acid to saline resuscitation provides neuroprotection and improves long-term outcomes in a large animal model of combined traumatic brain injury and hemorrhagic shock. J Trauma Acute Care Surg. 2015;79(6):911–9. doi: 10.1097/TA.0000000000000789. [DOI] [PubMed] [Google Scholar]
- 8.Georgoff PE, Nikolian VC, Bonham T, Pai MP, Tafatia C, Halaweish I, To K, Watcharotone K, Parameswaran A, Luo R, et al. Safety and Tolerability of Intravenous Valproic Acid in Healthy Subjects: A Phase I Dose-Escalation Trial. Clin Pharmacokinet. 2017 doi: 10.1007/s40262-017-0553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bambakidis T, Dekker SE, Sillesen M, Liu B, Johnson CN, Jin G, de Vries HE, Li Y, Alam HB. Resuscitation with Valproic Acid Alters Inflammatory Genes in a Porcine Model of Combined Traumatic Brain Injury and Hemorrhagic Shock. J Neurotrauma. 2016;33(16):1514–21. doi: 10.1089/neu.2015.4163. [DOI] [PubMed] [Google Scholar]
- 10.Dekker SE, Bambakidis T, Sillesen M, Liu B, Johnson CN, Jin G, Li Y, Alam HB. Effect of pharmacologic resuscitation on the brain gene expression profiles in a swine model of traumatic brain injury and hemorrhage. J Trauma Acute Care Surg. 2014;77(6):906–12. doi: 10.1097/TA.0000000000000345. discussion 12. [DOI] [PubMed] [Google Scholar]
- 11.Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, Leeds P, Chuang DM. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem. 2009;111(4):976–87. doi: 10.1111/j.1471-4159.2009.06385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang C, Zhu J, Zhang J, Li H, Zhao Z, Liao Y, Wang X, Su J, Sang S, Yuan X, et al. Neuroprotective and anti-apoptotic effects of valproic acid on adult rat cerebral cortex through ERK and Akt signaling pathway at acute phase of traumatic brain injury. Brain Res. 2014;1555:1–9. doi: 10.1016/j.brainres.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 13.Imam AM, Jin G, Duggan M, Sillesen M, Hwabejire JO, Jepsen CH, DePeralta D, Liu B, Lu J, deMoya MA, et al. Synergistic effects of fresh frozen plasma and valproic acid treatment in a combined model of traumatic brain injury and hemorrhagic shock. Surgery. 2013;154(2):388–96. doi: 10.1016/j.surg.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Jin G, Duggan M, Imam A, Demoya MA, Sillesen M, Hwabejire J, Jepsen CH, Liu B, Mejaddam AY, Lu J, et al. Pharmacologic resuscitation for hemorrhagic shock combined with traumatic brain injury. J Trauma Acute Care Surg. 2012;73(6):1461–70. doi: 10.1097/TA.0b013e3182782641. [DOI] [PubMed] [Google Scholar]
- 15.Jin G, Sun PZ, Singhal AB, Ayata C, Lo EH. First-order mathematical modeling of brain swelling in focal cerebral ischemia. Transl Stroke Res. 2010;1(1):65–70. doi: 10.1007/s12975-009-0009-5. [DOI] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotech. 2013;31(1):46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Li Y, Narayan R, Subramanian A, Xie X. Gene expression inference with deep learning. Bioinformatics. 2016;32(12):1832–9. doi: 10.1093/bioinformatics/btw074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobesova Z. Electrical and Control Engineering (ICECE). 2011 International Conference on 2011 Sep 16. IEEE; Programming language Python for data processing; pp. 4866–9. [Google Scholar]
- 22.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–D8. doi: 10.1093/nar/gkw1108. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delahaye-Duriez A, Srivastava P, Shkura K, Langley SR, Laaniste L, Moreno-Moral A, Danis B, Mazzuferi M, Foerch P, Gazina EV, et al. Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol. 2016;17(1):245. doi: 10.1186/s13059-016-1097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu TS, Zhang G, Liebl DJ, Kernie SG. Traumatic brain injury-induced hippocampal neurogenesis requires activation of early nestin-expressing progenitors. J Neurosci. 2008;28(48):12901–12. doi: 10.1523/JNEUROSCI.4629-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng W, ZhuGe Q, Zhong M, Chen G, Shao B, Wang H, Mao X, Xie L, Jin K. Neurogenesis in adult human brain after traumatic brain injury. J Neurotrauma. 2013;30(22):1872–80. doi: 10.1089/neu.2010.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun D, McGinn MJ, Zhou Z, Harvey HB, Bullock MR, Colello RJ. Anatomical integration of newly generated dentate granule neurons following traumatic brain injury in adult rats and its association to cognitive recovery. Exp Neurol. 2007;204(1):264–72. doi: 10.1016/j.expneurol.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 29.McAllister TW. Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci. 2011;13(3):287–300. doi: 10.31887/DCNS.2011.13.2/tmcallister. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boone DR, Sell SL, Micci M-A, Crookshanks JM, Parsley M, Uchida T, Prough DS, DeWitt DS, Hellmich HL. Traumatic Brain Injury-Induced Dysregulation of the Circadian Clock. PLoS ONE. 2012;7(10):e46204. doi: 10.1371/journal.pone.0046204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, Liu X, Atwood A, Huss JW, 3rd, Janes J, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell. 2009;139(1):199–210. doi: 10.1016/j.cell.2009.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pucci B, Kasten M, Giordano A. Cell Cycle and Apoptosis. Neoplasia. 2000;2(4):291–9. doi: 10.1038/sj.neo.7900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong Y, Mahmood A, Chopp M. Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs. 2010;11(3):298–308. [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins GA, Georgoff P, Nikolian V, Allyn-Feuer A, Pauls B, Higgins R, Athey BD, Alam HE. Network Reconstruction Reveals that Valproic Acid Activates Neurogenic Transcriptional Programs in Adult Brain Following Traumatic Injury. Pharm Res. 2017;34(8):1658–72. doi: 10.1007/s11095-017-2130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.