

Abstract
IgA nephropathy (IgAN) is the most common form of primary glomerulonephritis worldwide, exhibiting a variable clinical and pathological course and significantly contributing to the global burden of chronic kidney disease and end-stage renal disease. Current standards of care focus on optimization of antihypertensive and antiproteinuric therapies (typically renin- angiotensin system blockade) to reduce disease progression. Much recent attention has focused on whether additional immunosuppression confers better outcomes than supportive management alone, and indeed, several trials have demonstrated renoprotective effects following the use of oral corticosteroids. However, results have been inconsistent, and perceived benefits must be balanced against risks and adverse effects associated with generalized immunosuppression, as highlighted by the high-profile STOP-IgAN and TESTING clinical trials. Recent translational research in vitro and animal models of IgAN have generated greater insight into potential therapeutic targets for this complex autoimmune disease. Deeper understanding of the roles of the mucosal immune barrier, complement activation and deposition, T-cell dependent and independent mechanisms of B cell activation, and of the deposition and downstream inflammatory signaling pathways of nephritogenic polymeric IgA1 complexes (e.g., signaling of immune receptors via spleen tyrosine kinase) has formed the rationale for the development of novel agents and clinical trials of more targeted therapies. However, translating findings into clinical practice is challenging, with many immunopathological features of IgAN specific to humans. Recent comprehensive reviews outline current understanding of mechanisms of IgAN as well as ongoing and future clinical trials; it is not our aim to replicate this here. Instead, we take a mechanistic approach to current treatment strategies, outlining advantages and limitations of each before exploring ongoing research with potential translation into future targeted therapies for this complex disease.
Keywords: IgA nephropathy, glomerulonephritis, immunosuppression, chronic kidney disease, Syk, complement
Epidemiology and clinical features
IgA nephropathy (IgAN) is the most common form of glomerulonephritis (GN) worldwide, with an estimated incidence of at least 2.5/100,000/year in adults.1 There seems to be geographical variation in disease prevalence, with data from biopsy and organ replacement registries suggesting a higher burden of disease in East and Pacific Asian countries. However, estimates of global disease burden should be interpreted with caution, due to heterogeneity of data sources (biopsy, dialysis, and renal registry) and variability in local urine screening and biopsy practices. Patients with IgAN from these geographical regions may also have higher rates of progression toward end-stage renal disease (ESRD), as recently demonstrated in a North American cohort of 669 patients.2
The disease is variable in both its clinical and histopathological presentations. While presentation may occur at any age, peak incidence is in the second and third decades of life.3,4 Most patients present with either single or episodic macroscopic hematuria (characteristically following an upper respiratory tract infection – “synpharyngitic” – or athletic exertion) or following detection of microscopic hematuria and/or proteinuria on routine urine testing. A much smaller proportion present with acute kidney injury, usually as a result of crescentic IgAN or gross hematuria causing tubular obstruction. While the latter is often reversible, renal recovery may be incomplete. Nephrotic syndrome usually occurs only in more advanced stages of the disease.4
Definitive diagnosis is established by kidney biopsy, with immunofluorescence or immunoperoxidase studies for IgA deposits (Figure 1). Pathognomonic of the condition are prominent, globular deposits of IgA (often accompanied by C3 and IgG) in the mesangium and less prominently along the glomerular capillary wall.1,5 Important histopathological features that have been shown to predict long-term outcome according to the Oxford classification of IgAN include mesangial proliferation, endocapillary proliferation, segmental scarring, and tubular atrophy.6,7
Figure 1.
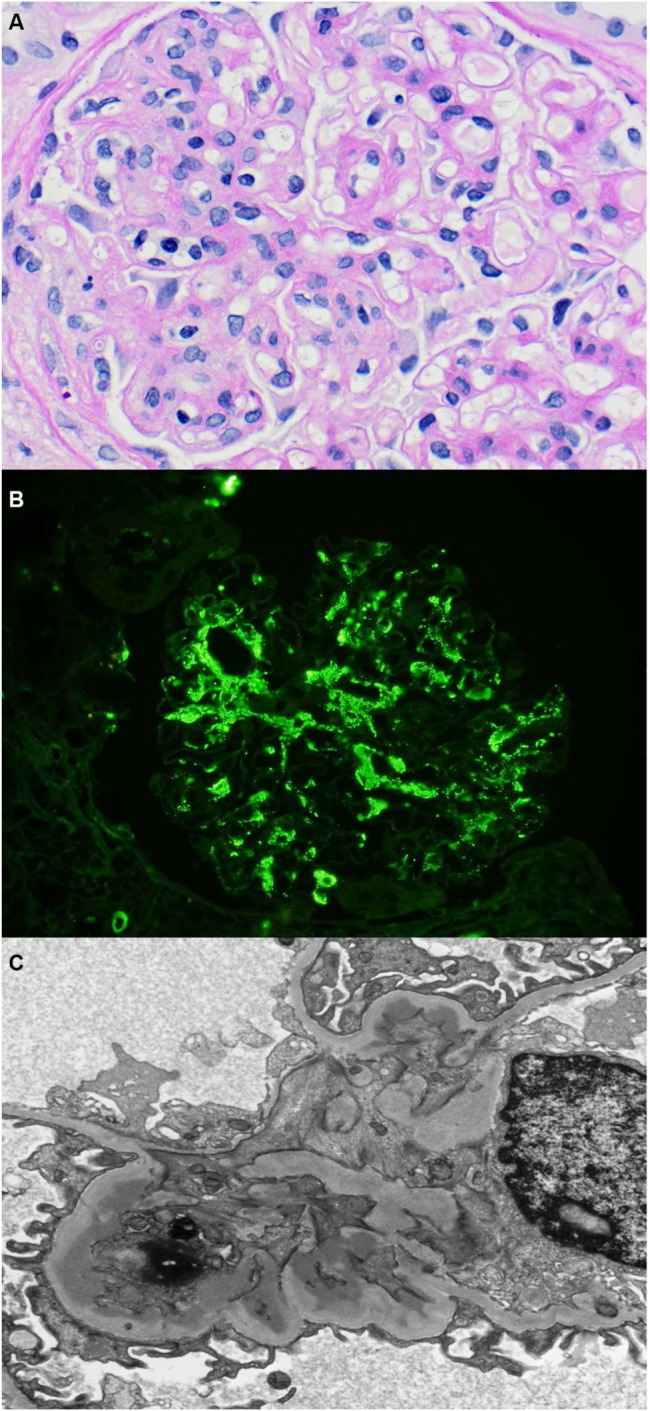
Histopathology of IgA nephropathy.
Notes: (A) Light microscopy demonstrating a segmental area of endocapillary proliferation within the glomerular tuft (hematoxylin and eosin, original magnification: ×200). (B) Immunofluorescence staining for IgA, showing predominantly mesangial IgA deposits. (C) Electron microscopy, demonstrating electron dense immune deposits in the glomerular mesangium.
Insights into pathological mechanisms
Here, we discuss IgAN as a single disease entity, defined by the presence of dominant or co-dominant mesangial IgA deposits on renal biopsy. However, this traditional understanding is increasingly being called into question, with observed heterogeneity in epidemiology, clinical presentation, histopathological response to IgA deposition, and genetic associations of the disease. It may be that mesangial IgAN merely represents a common end point for a disparate range of endophenotypes and diseases.8 This complicates discussions regarding pathogenesis and potential therapy, particularly when considering the external validity of studies undertaken in different regions of the world. As we further our understanding of this complex disease entity, it is likely that we will need to move beyond a simple pathological definition, perhaps subclassifying the disease to facilitate comparison between studies.
Most recent models of immunopathogenesis in IgAN propose a “multihit” process, with contribution of both genetic and environmental factors. A dysregulated mucosal immune system with defective immune tolerance to commensal or commonly encountered pathogens may be a key factor in triggering the disease. Characterization of both circulating and glomerular immune complexes in IgAN patients has revealed complexes comprising galactose-deficient IgA1 (Gd-IgA1) and C3. The pathophysiology underlying the formation and deposition of these is reviewed extensively elsewhere.9
The notion that IgAN is a systemic condition is supported by high rates of disease recurrence following renal transplant. Histologic evidence of recurrent IgAN is observed in over 35% of patients who receive renal allografts as treatment for ESRD due to IgAN despite these patients being on antirejection immunotherapy.10 The impact of recurrent IgAN on long-term renal graft failure has been controversial. This may be in part related to the limited number of cases in each series, variable follow-up duration, and different renal allograft biopsy policies. In the most recent study of data from the Australia and New Zealand Dialysis and Transplant Registry, the largest transplant registry worldwide over 30 years, recurrence of IgAN accounts for more than 50% of allograft loss among these recipients.11 GN recurrence rates appear to be significantly higher in living-related donor grafts compared with either living-unrelated or deceased donor grafts, shedding light on possible pathological mechanisms.12 Further understanding of the genetic basis of the disease may become important here; for instance, the human leukocyte antigen (HLA)-B8, DR3 haplotype has been associated with high immune reactivity, with the presence of the haplotype differentially affecting graft survival in IgAN patients versus controls.13
There is also evidence of subclinical deposition of IgA.14 The reported incidence of mesangial IgA deposition on renal biopsy in apparently healthy individuals with no clinical features of GN at the time of biopsy ranges from 3% to 16%.14 Whether the glycosylation pattern of this lanthanic glomerular IgA differs from that found in primary IgAN patients is yet to be determined. It is also not known whether some of these asymptomatic subjects may develop renal disease later. In a study of healthy Japanese allograft donors, IgA deposition on renal biopsy was associated with a mild degree of microhematuria, mesangial proliferation, and glomerular macrophage infiltration in some individuals, especially in those with combined IgA and C3 deposition. IgA deposition has also been observed in biopsies for other forms of GN including lupus nephritis, antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV), and membranous GN.15,16 While this may indicate overlapping of immunological disease mechanisms, the possibility that this observation is merely a result of the background prevalence of IgA deposits in the general population cannot be excluded. Since deposition of IgA alone does not appear to be sufficient for disease development and progression, the challenge comes in identifying additional risk factors. Accumulating evidence supports the notion that susceptibility may be in part hereditary, with genome-wide association studies identifying susceptibility loci mainly arising from the HLA chromosome region.17–19 These are present in both European and South-East Asian populations, although may differ between the two, and the putative role these loci play requires further elucidation.20 Advances in genetic techniques, such as next-generation sequencing, together with greater knowledge of the importance of copy number variants and epigenetics, create potential to enhance current understanding of the genetic basis of the disease. However, as discussed earlier, this is limited by lack of consistency in epidemiological clinicopathological studies both within and between continents and may be achieved only with more precise phenotyping of IgAN populations.
What triggers the production of aberrant Gd-IgA1 in susceptible individuals? It has been suggested that exposure to environmental triggers, such as pathogenic or commensal bacteria, may lead to excess production of aberrantly glycosylated IgA in mucosal-associated lymphoid tissue. IgA reacting with pathogens, including Haemophilus parainfluenzae, parvovirus, cytomegalovirus, Epstein–Barr virus, and Helicobacter pylori has been detected in both serum and glomerular samples from patients with IgAN. Furthermore, reported IgA deposition in patients with methicillin- resistant Staphylococcus aureus infection indicates a possible a role for microbial superantigens.21
There has been attempt to support these findings using evidence from experimental rodent models of IgAN. Persistent IgA deposits and microhematuria have been induced in mice by intranasal administration and systemic challenge of respiratory Sendai virus after experimental induction of defective mucosal tolerance.22 Recent attempts to characterize mediators of this response have focused on facilitators of B-cell differentiation and proliferation, such as B-cell activation factor of the TNF family (BAFF) and A proliferation- inducing ligand (APRIL); these may represent potential targets for therapy as discussed in detail below.23,24 Once formed, IgA–C3 immune complexes appear to contribute directly to glomerular inflammation and mesangial proliferation, triggering local and systemic renin-angiotensin systems (RAS) and activating complement pathways. IgA1 has been shown to activate both classical and lectin pathways of complement in vitro, with complement pathway components such as properdin, factor H, and mannan-binding lectin detected in mesangial immunodeposits.25 Complement factor H (CFH) is a major regulator of the alternative complement pathway. Circulating CFH-related proteins 1 and 5 (antagonists of factor H) correlate with disease activity in IgAN.26 Again, activators and modifiers of these complement pathways may be promising therapeutic targets.
Models of IgAN and challenges of translational research
Developing in vitro and in vivo animal models of IgAN is challenging. Believed to be a systemic disease, with heterogeneous clinical and histopathological manifestations, it is perhaps more realistic to model specific aspects of IgAN rather than the whole pathological process. Moreover, molecular features of human IgA1, the autoantigen thought to play a key role in the development of human IgAN, are very different from those of any rodent IgA.27 Lack of representative animal models may hinder testing of targeted therapies and progression to clinical trial, with quick dismissal of therapies following early negative results, when in fact the model may actually be unrepresentative of the human disease process.
A spontaneous animal model of IgAN – the ddY mouse – was first reported in 1985.28 It demonstrates glomerular IgA deposits but only mild proteinuria, with no hematuria and variable glomerular injury. More recently, an early-onset group of ddY mice was selectively intercrossed to establish a novel 100% early-onset grouped ddY mice, which overcomes the high degree of variability in age of onset and disease severity of the ddY model.27 These mice develop proteinuria at <8 weeks of age. Other animal models of IgAN include transgenic mice expressing human CD89 (an IgA receptor expressed by blood myeloid cells), a BAFF- transgenic mouse, a murine model of mucosal exposure to exogenous antigen, and a passive model, in which mice are injected with immune complexes preformed from human IgA1 with galactose-deficient O-glycans, mimicking complexes seen in IgAN patients.22,27–30 Findings generated from studies in these rodent models are explored more extensively elsewhere. While these models generate important insights into specific aspects of disease pathogenesis, the challenge comes in directly translating these findings to human IgAN and thereby developing targeted therapies for use in clinical practice.
Therapeutic approaches
If we consider IgAN to be an autoimmune disease with both systemic and local immunopathological mechanisms, it seems logical that both generalized and targeted immunosuppression may be effective. Of course, any therapy needs to be assessed against optimized supportive care, taking into account associated risks and side effects. Here, we outline current treatments, discuss evidence surrounding supportive and immunosuppressive approaches, and outline some of the current progress toward targeted therapies.
Current standards of care
Optimal management of IgAN remains a contentious topic. Latest Kidney Disease: Improving Global Outcomes (KDIGO) guidelines31 recommend a long-term RAS blockade (with angiotensin-converting enzyme inhibitors [ACE-I] or angiotensin receptor blocker [ARB]) for proteinuria >1 g/ day, with uptitration of the dose as far as tolerated, depending on blood pressure, to achieve proteinuria of <1 g/day. An RAS blocker is also suggested for proteinuria of 0.5–1 g/day. Recommended blood pressure targets are <130/80 mmHg or <125/75 mmHg when initial proteinuria is >1 g/day.
A 6-month course of corticosteroid therapy is suggested for patients with persistent proteinuria despite 3–6 months of optimized supportive care (including ACE-I/ARB and blood pressure control) and glomerular filtration rate (GFR) >50 mL/min/1.73 m2. Further immunosuppression with cyclophosphamide may be needed for those with crescentic IgAN with rapidly deteriorating renal function – analogous to the treatment of ANCA vasculitis.31,32
Supportive therapy
Supportive care, particularly antihypertensive and antiproteinuric therapy, remains a cornerstone of management for IgAN. As for other causes of chronic kidney disease (CKD), elevated blood pressure, proteinuria, and lower GFR have been associated with higher risk of underlying disease progression and adverse long-term outcomes.33–35 These features have been used to derive a prognostic scoring system that may be used to estimate progression toward ESRD in IgAN, although the authors note that other prognostic scoring systems exist.36
Meta-analysis of clinical trials has demonstrated that RAS blockers (ACE-Is or ARBs) have statistically significant effect on renal protection and reduction of proteinuria in IgAN.37,38 This association appears to be stronger amongst those demonstrating reduced estimated glomerular filtration rate (eGFR), proteinuria, acute inflammatory pathological findings, and those receiving methylprednisolone pulse therapy.39 ACE-I demonstrates variable therapeutic efficacy in slowing disease progression, which may in part be genetically determined.40 Mechanisms of secondary protection are likely similar to those in other causes of proteinuric CKD, mediated at least in part through lowering of both systemic blood pressure and intraglomerular pressure through inhibition of angiotensin II-mediated efferent arteriolar vasoconstriction. RAS blockade reduces proteinuria when compared with both placebo and calcium channel blockers in normo- and hypertensive patients, suggesting a proteinuria-reducing effect independent of antihypertensive effect.41 While earlier studies suggested that dual blockade with ACE-I and ARB may confer additional benefits over single agent alone in the reduction of proteinuria, more recent analyses have failed to consistently demonstrate this, with concerns over incidence of adverse effects and long-term outcomes limiting clinical applicability.42–45 Regulatory agencies warn against the dual-blockade approach, mainly due to concerns regarding hyperkalemia.46 Tight lipid control is also important. Statins, in addition to their well-documented cardioprotective effects in renal disease, may also delay progression of CKD.47–49
The therapeutic benefits of high-quality supportive care were very apparent in the recent STOP-IgAN trial, which assessed outcomes of supportive care versus supportive care plus immunosuppression in IgAN.50 The study employed a rigorous 6-month run-in phase, during which patients received intensive supportive care including RAS blocker (target blood pressure for all patients <125/75 mmHg), dietary counseling, statins if required (target total cholesterol <5.2 mmol/L), and advice to stop smoking and avoid nephrotoxic drugs. Target proteinuria was <0.75 g/day (in contrast to KDIGO guidelines of <1 g/day). The run-in phase involved visits at weeks 0, 4, 8, 16, 20, 23, and 24; after this, only patients still deemed to be at high risk (proteinuria >0.75 g/day) were randomly assigned to treatment groups. Of the 309 patients completing the run-in phase, 106 (34%) demonstrated a sufficient response to supportive care alone to render them ineligible for randomization. It is worth noting here that run-in blood pressure and proteinuria targets were more stringent than those of current KDIGO guidelines and patient monitoring more frequent than may be realistic in many outpatient settings. A “multimodal” approach with close monitoring and careful uptitration of supportive therapies has been shown to achieve remission or regression of proteinuria and stabilization of kidney function in other clinical settings and emphasizes the importance of high-quality supportive care.51 Ongoing work is needed to identify clinical biomarkers of patients demonstrating the best response to intensive supportive care and to offer maximally tolerated therapy to prevent or reduce progression toward ESRD.
Note should also be made here of reported beneficial effects of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA; omega-3 polyunsaturated fatty acids) as anti- inflammatory and inflammation-convergent drugs in the treatment of IgAN.52 Successful treatment of several cases of IgAN was reported recently with a combination of aspirin and EPA.53 While the precise mechanism for this combination is unknown, it is plausible that aspirin potentiates the effect of anti-inflammatory lipid mediators. Furthermore, addition of DHA to EPA was found to stabilize renal function of IgAN patients, with improvements in clinical parameters and possible further pleiotropic effects.54 Well-designed molecular experiments and clinical trials are necessary to better understand the mechanisms by which these agents may affect IgAN.
General immunosuppression
Much recent attention has focused on whether additional immunosuppression confers better outcomes than supportive management alone; the presence of inflammation and immune complexes on renal histologic examination would certainly suggest it may be beneficial. A generalized approach has the advantage that a fully comprehensive understanding of disease pathophysiology is not required, and immunosuppression can simultaneously or even synergistically exert effects via different mechanisms.
Many studies including several high-profile randomized controlled trials (RCTs) have demonstrated renoprotective effects and reduction of proteinuria following a short course of oral corticosteroids.55–57 However, some of these have been criticized due to small size and suboptimal methodological quality, with short run-in periods, inconsistent blockade of the RAS system, reporting only favorable or surrogate outcomes, and underreporting adverse effects. The recent STOP-IgAN trial employed a rigorous run-in phase for standardization and optimization of supportive care. Researchers found that addition of immunosuppressive therapy (oral corticosteroids, plus cyclophosphamide and azathioprine for those with eGFR 30–59 mL/min/1.73 m2) to intensive supportive care in high-risk IgAN did not confer substantial renal benefits. Moreover, during the 3-year study phase, more adverse effects were observed amongst patients who received immunosuppressive therapy, including one steroid-related death due to sepsis. However, a major criticism levied against this trial is that patients with a urinary protein excretion rate >3.5 g/day, eGFR <30 mL/min/1.73 m2, or decrease in eGFR of more than 30% were excluded at the end of the 6-month run-in phase. Such patients have a relatively high risk of underlying disease progression and in previous studies have demonstrated a particularly favorable response to steroids.55,58,59
Recently, results of the TESTING study, an international, multicenter, double-blind RCT that sought to explore efficacy and safety of corticosteroids in IgAN patients at higher risk of progression, were published.60 The study aimed to recruit 750 participants with proteinuria >1 g/day and eGFR of 20–120 mL/min/1.73 m2 after 3 months of blood pressure control with RAS blockade. Primary composite outcome was ESRD, death (due to renal failure), or 40% reduction in eGFR, and safety outcomes were predefined relating to steroid adverse effects. The most significant finding was that the trial was terminated after randomization of only 262 participants and 2.1 years median follow-up, due to excess serious adverse events in the methylprednisolone group (28 serious adverse events in 20 participants [14.7%] vs 4 in the placebo group [3.2%]; p=0.001), mostly related to serious infections and including two infection-related deaths. Although early results may be indicative of potential renal benefits with steroid therapy, definitive conclusions regarding treatment efficacy could not be drawn due to early trial termination. Furthermore, steroids were administered to patients in both active and chronic phases of disease, and kidney biopsies were not analyzed. The TESTING trial supports safety concerns raised by STOP-IgAN; both highlight the importance of a cautious approach to steroid use (calling perhaps for revision of current KDIGO guidelines), intensive monitoring of steroid therapy, and the need to pursue safer and more effective alternatives. All of these studies have highlighted the need to identify both clinically and pathologically those patients who will respond favorably to steroid therapy. Meta-analysis has shown that IgAN patients with more serious pathological features according to the Oxford classification system (outlined earlier) may be more resistant to steroid than those with slight pathological changes.61
The role of other immune-modulating therapies should also be considered. Prognosis in crescentic and other rapidly progressive forms of IgAN is usually poor, despite aggressive therapy. However, steroids and intravenous cyclophosphamide have been demonstrated to induce both clinical and histological responses in these patients, with reduction in proliferative lesions, proteinuria, and stabilization of renal function.62 Some patients with crescentic IgA in the Oxford cohort had improvements in outcomes following enhanced immunosuppression.63 These findings are reflected in latest KDIGO recommendations, with enhanced immunosuppression generally warranted in a disease of such aggressive nature and time course. Furthermore, plasma exchange may also play a therapeutic role, and it has been shown to reduce proteinuria and serum creatinine levels, as well as dialysis dependency and kidney survival, when added to steroid and cyclophosphamide immunosuppression.64 Use as a sole treatment for rapidly progressive forms of the disease is supported by evidence from case series; children with severe IgAN or Henoch–Schönlein purpura have demonstrated recovery with plasmapheresis alone, negating need for immunosuppressive therapy.65,66 However, the use of plasma exchange remains less well defined in the adult population and certainly warrants further assessment in large RCTs.
On the other hand, benefits of intensive immunosuppression in halting progressive IgAN must be more carefully weighed against well-documented adverse cytotoxic effects. Mycophenolic acid inhibits inosine 5′-monophosphate dehydrogenase, a key enzyme in glycosylation of B and T lymphocyte adhesion molecules; it has therefore been speculated that this agent may reduce the inflammatory response in immune-mediated GN. Although some studies have generated promising results regarding remission of proteinuria and renoprotection with mycophenolate mofetil (MMF) therapy, these have been inconsistent. Certainly, further evaluation of adverse effects and long-term outcomes are needed before use could be incorporated into routine practice.67–71 Assessment of histological changes will be valuable in assessing the clinical outcome. Recently, MMF therapy was shown to reduce mesangial IgA deposition, endocapillary hypercellularity, and cellular/fibrocellular crescents in a retrospective case series; however, it is notable that this series included only those with endocapillary hypercellularity seen on biopsy.72 Trials of azathioprine have also proved equivocal.73–75 A role for calcineurin inhibitors (CNIs) has also been postulated, with a recent meta-analysis demonstrating that CNIs such as Tacrolimus may reduce proteinuria when combined with medium-dose steroid.76 However, no significant benefits for kidney function or decline in eGFR was seen, and there was increased risk of adverse effects. No large high-quality trial with sufficient sample size has yet been conducted to reliably evaluate safety and efficacy of CNIs in IgAN.
Studying clinical and histological disease recurrence following transplant for end-stage IgAN may also allow comparison of efficacy of immunosuppressants, shedding light on putative disease mechanisms. Transplant is the treatment of choice for ESRD secondary to IgAN but disease recurrence is frequent, with reported rates from 13% to 50%.10,77 A recent US-based transplant registry analysis indicates that steroid use may be associated with decreased risk of recurrence, although of note there were no significant associations between steroid use and patient survival or death-censored graft survival.78 Many studies indicate that MMF alone or in combination does not lessen recurrence or clinical impact of IgAN when compared with azathioprine or other immunosuppressive agents.79,80 In another study, initiation of maintenance immunosuppression with MMF and/or prednisolone within the first year posttransplant reduced risk of IgAN recurrence in a cohort of 133 patients.81 Notably, disease recurrence did not affect graft survival for this cohort.
Of course, a major disadvantage of a generalized approach is the well-documented side effects of immunosuppression, especially from steroids. In the STOP-IgAN trial, as well as one patient died with pneumogenic sepsis in the immunsuppression group, there was a numerically higher number of malignant neoplasms and higher rates of impaired glucose metabolism and bodyweight gain in the immunosuppression group. Serious adverse events resulted in premature termination of the TESTING trial, with an excess of serious infections (two fatal) in the steroid group. Another consideration is that prior immunosuppression may complicate subsequent choice of immunosuppression following transplant for those that reach ESRD.
Furthermore, if underlying mechanisms of drug action are not fully understood and there are no known biomarkers to monitor therapeutic efficacy, it is difficult to make decisions regarding optimal duration and safe withdrawal of immunosuppressive therapy. For some glomerulonephritides such as anti-glomerular basement membrane (anti-GBM) disease or AAV, which are rapidly progressive with high rates of permanent renal failure and fatality, an aggressive short-term regimen is necessary and warranted. For IgAN, which most often exhibits a chronic and insidious course, risks of long- term immunosuppression must be carefully balanced against benefits. It seems likely that as understanding of pathogenesis increases, differential therapeutic approaches may be warranted based on clinical and histological disease features, with optimization of supportive care a goal for all patients.
Targeted immunosuppression
As we gain insight into mechanisms underpinning this complex immunopathological disease, further therapeutic targets become apparent (Figure 2). Selective immunosuppressive regimens have the advantage that they target specific mechanisms of disease and thereby avoid some of the off-target effects of generalized immunosuppression. As described earlier, development of IgG and/or IgA autoantibodies against polymeric Gd-IgA1 is thought to be a key step in IgAN pathogenesis. It may be possible to reduce production of antibody or antigen or prevent interaction between the two to form nephritogenic immune complexes. Circulating levels of Gd-IgA1 are frequently elevated in IgAN patients, with correlation between serum levels and disease progression.82,83 Depleting B cells that may theoretically lead to a reduction in the antibody-producing plasma cells which are presumed to drive this pathology is an appealing prospect. B-cell depletion has been shown to be effective in other autoantibody-mediated renal diseases including membranous nephropathy, lupus nephritis, and AAV.84–86 However, in a recent open-label, multicenter RCT, B-cell depletion with rituximab treatment did not have a significant effect on proteinuria or renal function in comparison to the baseline or the control group after 1-year follow-up.87
Figure 2.

Pathogenesis and potential therapeutic targets in IgA nephropathy.
Notes: (A) Development of IgG and/or IgA autoantibodies against polymeric galactose-deficient IgA1. (B) B-cell depletion of autoantibody-producing plasma cells using rituximab. (C) Targeting BAFF or APRIL. These may be involved in promoting B-cell class switch to IgA1-producing plasma cells. (D) Using budesonide to target dysregulation of the mucosal immune system. (E) Targeting mesangial cell activation and production of inflammatory mediators such as via Syk or NF-kB. (F) Targeting elements of the complement cascade such as properdin or C3.
Abbreviations: APRIL, a proliferation-inducing ligand; BAFF, B-cell activation factor of the TNF family; MAC, membrane attack complex; NF-kB, nuclear factor-kappa B; Syk, spleen tyrosine kinase.
Production of IgA by plasma cells is thought to occur by both T-cell dependent and independent processes. There have been attempts to characterize some of the T-cell cytokines which promote the B-cell class switch to IgA1-producing plasma cells, with recent attention on BAFF and APRIL.88 Serum BAFF levels in IgAN patients are elevated and associated with disease severity clinically and histopathologically.23 In vivo, transgenic mice overexpressing BAFF exhibit features of autoimmune disease and develop fatal nephritis with age. The nephritic mice demonstrate mesangial IgA deposition, along with high circulating levels of aberrantly glycosylated polymeric IgA.30 In addition, elevated serum levels of APRIL (a cytokine structurally related to BAFF) have been observed in an IgAN cohort, with correlation between APRIL expression and elevated circulating levels of galactose-deficient IgA and more severe clinical phenotype (high proteinuria and low eGFR).89 These cytokines represent exciting targets for future therapy; indeed, anti-APRIL antibody treatment has already been shown to significantly decrease albuminuria, tissue damage, serum IgA levels, and deposition of glomerular IgA in a murine model of disease.24
Another option is to target the mucosal immune system, dysregulation of which may contribute to aberrant IgA formation. Results of the recent Phase IIb trial of modified- release oral budesonide have recently been published.90 Administered in a form targeted to the enteric region, the drug acts on the intestinal mucosa before undergoing high first- pass metabolism, resulting in low systemic exposure when compared with oral corticosteroids. Following a 6-month run-in phase, patients treated with targeted release formation (TRF)-budesonide 16 mg/day demonstrated greater reduction in proteinuria and stabilization of eGFR (a secondary end point) versus placebo-treated controls. Both groups received maximally tolerated RAS blockade. Concerns had previously been raised regarding safety of this treatment, with higher rates of adverse events in the budesonide-treated group in Phase IIa trial.91 Most recent results indicate that TRF-budesonide is safe and generally well tolerated, without serious adverse events, although there was a dose-dependent trend in the incidence of corticosteroid-related adverse events and resultant discontinuations. Certainly, modified-release budesonide is a promising therapy, offering an alternative to high-dose systemic steroids, which may be safer and better tolerated. However, larger studies are needed to confirm this.
Instead of targeting immune complex formation, it may be possible to prevent deposition of nephritogenic polymeric IgA1 complexes and/or their downstream inflammatory signaling pathways. Deposition of complexes drives activation of mesangial cells and excessive inflammatory mediator production. An emerging and promising target in this regard is the enzyme spleen tyrosine kinase (Syk), which plays an important immunoreceptor signaling role in a range of hematological and immune-mediated disorders. A role in IgAN is supported by evidence of increased Syk staining in biopsies from patients with IgAN versus minimal change glomerulopathy controls.92 In vitro, IgA1 from serum of patients with IgAN has been shown to induce activation (i.e., phosphorylation) of Syk in human mesangial cells (HMCs), and addition of R406, the active metabolite of the selective Syk-inhibitor fostamatinib, inhibited production of multiple inflammatory mediators.92 Translating these findings to an experimental autoimmune glomerulonephritis (EAG) rodent model, late treatment with oral fostamatinib was shown to reduce hematuria, proteinuria, and autoantibody production and to reverse histopathological kidney damage when compared with vehicle.93 We eagerly await to see whether these preclinical findings can be successfully translated into clinical trial. Fostamatinib has undergone clinical testing in rheumatoid arthritis, refractory immune thrombocytopenic purpura, leukemia, and lymphoma, and in animal models of renal disease, including the EAG and nephrotoxic nephritis models of anti-GBM disease, lupus nephritis, and renal fibrosis models. The use of fostamatinib in IgAN patients demonstrating proliferative lesions on renal biopsies is currently being investigated in a Phase II RCT (ClinicalTrials. gov NCT02112838), with primary outcome being proteinuria at 24 weeks. This study also aims to examine histological changes on repeat renal biopsy after a 6-month treatment period.
Mesangial cell activation is thought to be amplified by complement activation, with both alternative and lectin pathways implicated. The role of complement in IgAN has been outlined earlier and forms the subject of a recent review.94 C3, properdin, and CFH have all been detected in IgAN glomerular immune deposits. Absence of CFH-related (CFHR) genes CFHR1 and CFHR3 is associated with altered CFH levels and enhanced CFH activity in IgAN.95 In a recent study, C3aR and C5aR antagonists repressed IgA-induced cell proliferation and IL-6 and MCP-1 production in cultured HMCs, while in the Sendai virus-induced mouse model, C3aR/C5aR-deficient mice demonstrated reduced proteinuria, lower renal IgA and C3 deposition, less histologic damage, and reduced mesangial proliferation compared with wild-type mice.96 However, C3aR/ C5aR-deficient and wild-type mice with IgAN did not differ with respect to their serum urea or creatinine levels. Given the supposed importance of complement deposition in IgAN, use of the humanized anti-C5 monoclonal antibody eculizumab has been proposed as a rational treatment. This has been trialed only in isolated cases thus far, with inconclusive outcomes.97,98 Developing further therapies to target mediators and regulators of complement signaling remains an attractive goal.
Nuclear factor-kappa B (NF-kB) has also been studied in the context of GN. NF-kB expression has been shown to correlate with clinical and histological parameters of IgAN, with purported activation and a role in tubulointerstitial injury, particularly in proteinuric states. Increased tubular NF-kB expression may be predictive of poor prognosis in IgAN. In one rat model of IgAN, treatment with the flavonoid icariin was shown to block NF-kB and reduce downstream cytokine production with reduction in proteinuria, improved renal function, and decreased glomerular fibrosis.99 However, in a mouse model of crescentic GN, inhibition of NF-kB prior to disease induction resulted in improved disease phenotype, but treatment after onset of disease resulted in marked worsening of disease with higher proteinuria, lower creatinine clearance, and greater severity of histology.100 Further work investigating the role of this complex pathway is needed before translation to clinical studies.
Other pilot clinical studies include trials of Acthar gel, a purified form of adrenocorticotropic hormone, for patients with IgAN and nephrotic syndrome who have failed previous immunosuppression (NCT02382523), of bortezomib, a proteasome inhibitor currently used in multiple myeloma and mantle cell lymphoma (NCT01103778), and of Blisibimod, a fusion between the Fc portion of IgG and a peptide with high affinity to both soluble and membrane-bound BAFF (NCT02062684).101
Conclusion and future directions
IgAN remains a common cause of glomerular disease and a significant contributor to the global burden of CKD and ESRD. Studies in vitro and in vivo animal models continue to shed light on mechanisms of disease, identifying key players in the immunopathological interaction between Gd-IgA1 antigen and circulating antiglycan antibodies and formation and deposition of immune complexes.
International treatment guidelines, supported by multicenter clinical trials, advocate high-quality supportive care, including RAS blockers and carefully titrated blood pressure control. Steroids and further immunosuppressive therapies are considered for those with persistently high proteinuria or rapidly progressive disease, respectively. Notably, the recent STOP-IgAN trial did not demonstrate benefits of broad immunosuppressive therapy, including steroid, in IgAN to justify use in routine care (although note that patients with >3.5 g/ day proteinuria were excluded from the study). Trials of other immunosuppressants have not provided conclusive evidence of overall benefits for disease progression and patient outcomes when balanced against risks associated with these therapies.
Recent research has generated greater insight into potential therapeutic targets for IgAN. Deeper understanding of the roles of the mucosal immune barrier, complement activation and deposition, and T-cell dependent and independent mechanisms of B-cell activation and downstream signaling of immune receptors via Syk has formed the rationale for the development of novel agents and clinical trials of more targeted therapies. Results of a Phase IIb trial of targeted oral budesonide appear promising. Meanwhile, results of the Phase II RCT of oral Syk inhibitor fostamatinib are eagerly awaited. It is hoped that continuing research will generate further putative therapeutic targets to change the future landscape of treatment strategies for IgAN.
Acknowledgments
We would like to thank Dr Candice Roufosse for providing the images in Figure 1. This research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London.
Footnotes
Disclosure
M Prendecki is supported by a Medical Research Council (UK) Clinical Research Training Fellowship. S McAdoo is supported by NIHR Clinical Lectureship. FWK Tam is supported by the Diamond Fund from Imperial College Healthcare Charity, Ken and Mary Minton Chair of Renal Medicine, and Make Every Kidney Count programme grant from Kidney Research UK. He has received research project grants from AstraZeneca Limited, Baxter Biosciences, Boehringer Ingelheim, and MedImmune and is the Principal Investigator of an ongoing international clinical trial of a Syk inhibitor in IgA nephropathy (ClinicalTrials.gov NCT02112838), funded by Rigel Pharmaceuticals. He has consultancy agreements with Rigel Pharmaceuticals, Novartis, and Baxter Biosciences. RS Penfold reports no conflicts of interest in this work.
References
- 1.Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368(25):2402–2414. doi: 10.1056/NEJMra1206793. [DOI] [PubMed] [Google Scholar]
- 2.Barbour SJ, Cattran DC, Kim SJ, et al. Individuals of Pacific Asian origin with IgA nephropathy have an increased risk of progression to end-stage renal disease. Kidney Int. 2013;84(5):1017–1024. doi: 10.1038/ki.2013.210. [DOI] [PubMed] [Google Scholar]
- 3.D’Amico G, Imbasciati E, Di Belgioioso GB, et al. Idiopathic IgA mesangial nephropathy: clinical and histological study of 274 patients. Medicine. 1985;64(1):49–60. [PubMed] [Google Scholar]
- 4.Ibels LS, Gyory AZ. IgA nephropathy: analysis of the natural history, important factors in the progression of renal disease, and a review of the literature. Medicine (Baltimore) 1994;73(2):79–102. [PubMed] [Google Scholar]
- 5.Emancipator SN. IgA nephropathy: morphologic expression and pathogenesis. Am J Kidney Dis. 1994;23(3):451–462. doi: 10.1016/s0272-6386(12)81011-0. [DOI] [PubMed] [Google Scholar]
- 6.Cattran DC, Coppo R, Cook HT, et al. The oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76(5):534–545. doi: 10.1038/ki.2009.243. [DOI] [PubMed] [Google Scholar]
- 7.Roberts IS, Cook HT, Troyanov S, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76(5):546–556. doi: 10.1038/ki.2009.168. [DOI] [PubMed] [Google Scholar]
- 8.Cheung CK, Barratt J. Is IgA nephropathy a single disease? In: Tomino T, editor. Pathogenesis and Treatment in IgA Nephropathy. Tokyo: Springer; [Google Scholar]
- 9.Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22(10):1795–1803. doi: 10.1681/ASN.2011050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ponticelli C, Traversi L, Feliciani A, Cesana BM, Banfi G, Tarantino A. Kidney transplantation in patients with IgA mesangial glomerulonephritis. Kidney Int. 2001;60(5):1948–1954. doi: 10.1046/j.1523-1755.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- 11.Allen PJ, Chadban SJ, Craig JC, et al. Recurrent glomerulonephritis after kidney transplantation: risk factors and allograft outcomes. Kidney Int. 2017;92(2):461–469. doi: 10.1016/j.kint.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Kennard AL, Jiang SH, Walters GD. Increased glomerulonephritis recurrence after living related donation. BMC Nephrol. 2017;18(1):25. doi: 10.1186/s12882-016-0435-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andredottir MB, Haasnoot GW, Persijn GG, Claas FH. HLA-B8, DR3: a new risk factor for graft failure after renal transplantation in patients with underlying immunoglobulin A nephropathy. Clin Transplant. 2009;23(5):660–5. 5. doi: 10.1111/j.1399-0012.2009.01059.x. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003;63(6):2286–6. 94. doi: 10.1046/j.1523-1755.63.6s.2.x. [DOI] [PubMed] [Google Scholar]
- 15.Haas M, Jafri J, Bartosh SM, Karp SL, Adler SG, Meehan SM. ANCA- associated crescentic glomerulonephritis with mesangial IgA deposits. Am J Kidney Dis. 2000;36(4):709–718. doi: 10.1053/ajkd.2000.17615. [DOI] [PubMed] [Google Scholar]
- 16.Stokes MB, Alpers CE. Combined membranous nephropathy and IgA nephropathy. Am J Kidney Dis. 1998;32(4):649–656. doi: 10.1016/s0272-6386(98)70031-9. [DOI] [PubMed] [Google Scholar]
- 17.Kirmizis D, Papagianni A, Schena FP. IgA nephropathy: insights into genetic basis and treatment options. In: Prabhakar S, editor. An Update on Glomerulopathies – Clinical and Treatment Aspects. InTech16; USA: 2011. Edited by Sharma S. Prabhakar, ISBN 978-953-307-673-7, 482 pages, Publisher: InTech, Chapters published November 02, 2011 under CC BY 3.0 license. [Google Scholar]
- 18.Schena FP, Coppo R. IgA nephropathies. In: Davison AM, Cameron JS, Grünfeld JP, et al., editors. Oxford Textbook of Clinical Nephrology. 3rd. Oxford: Oxford University Press; 2005. pp. 469–501. [Google Scholar]
- 19.Gharavi AG, Kiryluk K, Choi M, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–327. doi: 10.1038/ng.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feehally J, Farrall M, Boland A, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21(10):1791–1797. doi: 10.1681/ASN.2010010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kai H, Shimizu Y, Hagiwara M, et al. Post-MRSA infection glomerulonephritis with marked Staphylococcus aureus cell envelope antigen deposition in glomeruli. J Nephrol. 2006;19(2):215–219. [PubMed] [Google Scholar]
- 22.Amore A, Coppo R, Nedrud JG, Sigmund N, Lamm ME, Emancipator SN. The role of nasal tolerance in a model of IgA nephropathy induced in mice by Sendai virus. Clin Immunol. 2004;113(1):101–108. doi: 10.1016/j.clim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 23.Xin G, Shi W, Xu LX, Su Y, Yan LJ, Li KS. Serum BAFF is elevated in patients with IgA nephropathy and associated with clinical and histopathological features. J Nephrol. 2013;26(4):683–690. doi: 10.5301/jn.5000218. [DOI] [PubMed] [Google Scholar]
- 24.Kim YG, Alvarez M, Suzuki H, et al. Pathogenic role of a proliferation- inducing ligand (APRIL) in murine IgA nephropathy. PLoS One. 2015;10(9):e0137044. doi: 10.1371/journal.pone.0137044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maillard N, Wyatt RJ, Julian BA, et al. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol. 2015;26(7):1503–1512. doi: 10.1681/ASN.2014101000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medjeral-Thomas NR, Lomax-Browne HJ, Beckwith H, et al. Circulating complement factor H-related proteins 1and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017;92(4):942–952. doi: 10.1016/j.kint.2017.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki H, Suzuki Y, Novak J, Tomino Y. Development of animal models of human IgA nephropathy. Drug Discov Today Dis Models. 2014;11:5–11. doi: 10.1016/j.ddmod.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai H, Nakamoto Y, Asakura K, Miki K, Yasuda T, Miura AB. Spontaneous glomerular IgA deposition in ddY mice: an animal model of IgA nephritis. Kidney Int. 1985;27(5):756–761. doi: 10.1038/ki.1985.76. [DOI] [PubMed] [Google Scholar]
- 29.Launay P, Grossetête B, Arcos-Fajardo M, et al. Fcα receptor (Cd89) mediates the development of immunoglobulin a (Iga) nephropathy (Berger’s disease) J Exp Med. 2000;191(11):1999–2009. doi: 10.1084/jem.191.11.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy DD, Kujawa J, Wilson C, et al. Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J Clin Invest. 2011;121(10):3991–4002. doi: 10.1172/JCI45563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.KDIGO Clinical Practice Guideline for Glomerulonephritis [Accessed July 27, 2017];Kidney International Supplements. 2012 Jun;2(2) Available from: http://www.kdigo.org/clinical_practice_guidelines/pdf/KDIGO-GN-Guideline.pdf. 2012. [Google Scholar]
- 32.McIntyre CW, Fluck RJ, Lambie SH. Steroid and cyclophosphamide therapy for IgA nephropathy associated with crescenteric change: an effective treatment. Clin Nephrol. 2001;56(3):193–198. [PubMed] [Google Scholar]
- 33.Bartosik LP, Lajoie G, Sugar L, Cattran DC. Predicting progression in IgA nephropathy. Am J Kidney Dis. 2001;38(4):728–735. doi: 10.1053/ajkd.2001.27689. [DOI] [PubMed] [Google Scholar]
- 34.Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L. Predicting the risk for dialysis or death in IgA nephropathy. J Am Soc Nephrol. 2011;22(4):752–761. doi: 10.1681/ASN.2010040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moriyama T, Tanaka K, Iwasaki C, et al. Prognosis in IgA nephropathy: 30-year analysis of 1,012 patients at a single center in Japan. PLoS One. 2014;9(3):e91756. doi: 10.1371/journal.pone.0091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakai K, Kawamura T, Endoh M, et al. A scoring system to predict renal outcome in IgA nephropathy: from a nationwide prospective study. Nephrol Dial Transplant. 2006;21(10):2800–2808. doi: 10.1093/ndt/gfl342. [DOI] [PubMed] [Google Scholar]
- 37.Cheng J, Zhang W, Zhang XH, He Q, Tao XJ, Chen JH. ACEI/ARB therapy for IgA nephropathy: a meta analysis of randomised controlled trials. Int J Clin Pract. 2009;63(6):880–888. doi: 10.1111/j.1742-1241.2009.02038.x. [DOI] [PubMed] [Google Scholar]
- 38.Reid S, Cawthon PM, Craig JC, Samuels JA, Molony DA, Strippoli GF. Non-immunosuppressive treatment for IgA nephropathy. Cochrane Database Syst Rev. 2011;3:CD003962. doi: 10.1002/14651858.CD003962.pub2. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka S, Ninomiya T, Katafuchi R, et al. The effect of renin-angiotensin system blockade on the incidence of end-stage renal disease in IgA nephropathy. Clin Exp Nephrol. 2016;20(5):689–698. doi: 10.1007/s10157-015-1195-y. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida H, Mitarai T, Kawamura T, et al. Role of the deletion of polymorphism of the angiotensin converting enzyme gene in the progression and therapeutic responsiveness of IgA nephropathy. J Clin Invest. 1995;96(5):2162–2169. doi: 10.1172/JCI118270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maschio G, Cagnoli L, Claroni F, et al. ACE inhibition reduces proteinuria in normotensive patients with IgA nephropathy: a multicentre, randomized, placebo-controlled study. Nephrol Dial Transplant. 1994;9(3):265–269. [PubMed] [Google Scholar]
- 42.Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008;148(1):30–48. doi: 10.7326/0003-4819-148-1-200801010-00190. [DOI] [PubMed] [Google Scholar]
- 43.Cheng J, Zhang X, Tian J, Li Q, Chen J. Combination therapy an ACE inhibitor and an angiotensin receptor blocker for IgA nephropathy: a meta-analysis. Int J Clin Pract. 2012;66(10):917–923. doi: 10.1111/j.1742-1241.2012.02970.x. [DOI] [PubMed] [Google Scholar]
- 44.Russo D, Minutolo R, Pisani A, et al. Coadministration of losartan and enalapril exerts additive antiproteinuric effect in IgA nephropathy. Am J Kidney Dis. 2001;38(1):18–25. doi: 10.1053/ajkd.2001.25176. [DOI] [PubMed] [Google Scholar]
- 45.Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin-angiotensin system: meta- analysis of randomised trials. BMJ. 2013;346:f360. doi: 10.1136/bmj.f360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Press release. Combined use of medicines affecting the renin- angiotensin system (RAS) to be restricted – CHMP endorses PRAC recommendation. European Medicines Agency. May 23, 2014. [Accessed July 27, 2017]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2014/05/news_detail_002107.jsp&mid=WC0b01ac058004d5c1. 2014.
- 47.Strippoli GF, Navaneethan SD, Johnson DW, et al. Effects of statins in patients with chronic kidney disease: meta-analysis and meta-regression of randomised controlled trials. BMJ. 2008;336(7645):645–651. doi: 10.1136/bmj.39472.580984.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su X, Zhang L, Lv J, et al. Effect of statins on kidney disease outcomes: a systematic review and meta-analysis. Am J Kidney Dis. 2016;67(6):881–892. doi: 10.1053/j.ajkd.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Moriyama T, Oshima Y, Tanaka K, et al. Statins stabilize the renal function of IgA nephropathy. Ren Fail. 2014;36(3):356–360. doi: 10.3109/0886022X.2013.866512. [DOI] [PubMed] [Google Scholar]
- 50.Rauen T, Eitner F, Fitzner C, et al. Intensive supportive care plus immunosuppression in IgA nephropathy. N Engl J Med. 2015;373(23):2225–2236. doi: 10.1056/NEJMoa1415463. [DOI] [PubMed] [Google Scholar]
- 51.Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol. 2008;19(6):1213–1224. doi: 10.1681/ASN.2007090970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirahashi J. Omega-3 polyunsaturated fatty acids for the treatment of IgA nephropathy. J Clin Med. 2017;6(7):E70. doi: 10.3390/jcm6070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirahashi J, Hanafusa N, Wada T, et al. Aspirin and eicosapentaenoic acid may arrest progressive IgA nephropathy: a potential alternative to immunosuppression. Intern Med. 2015;54(18):2377–2382. doi: 10.2169/internalmedicine.54.4623. [DOI] [PubMed] [Google Scholar]
- 54.Moriyama T, Kumon S, Kamiyama T, Karasawa K, Uchida K, Nitta K. The renoprotective effects of docosahexaenoic acid as an add-on therapy in patients receiving eicosapentaenoic acid as treatment for IgA nephropathy: a Pilot Uncontrolled Trial. Intern Med. 2018;57(2):173–179. doi: 10.2169/internalmedicine.9155-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pozzi C, Andrulli S, Del Vecchio L, et al. Corticosteroid effectiveness in IgA nephropathy: long-term results of a randomized, controlled trial. J Am Soc Nephrol. 2004;15(1):157–163. doi: 10.1097/01.asn.0000103869.08096.4f. [DOI] [PubMed] [Google Scholar]
- 56.Pozzi C, Bolasco PG, Fogazzi GB, et al. Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet. 1999;353(9156):883–887. doi: 10.1016/s0140-6736(98)03563-6. [DOI] [PubMed] [Google Scholar]
- 57.Lv J, Zhang H, Chen Y, et al. Combination therapy of prednisone and ACE inhibitor versus ACE-inhibitor therapy alone in patients with IgA nephropathy: a randomized controlled trial. Am J Kidney Dis. 2009;53(1):26–32. doi: 10.1053/j.ajkd.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 58.Tesar V, Troyanov S, Bellur S, et al. Corticosteroids in IgA nephropathy: a Retrospective Analysis from the VALIGA Study. J Am Soc Nephrol. 2015;26(9):2248–2258. doi: 10.1681/ASN.2014070697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim JK, Kim JH, Lee SC, et al. Clinical features and outcomes of IgA nephropathy with nephrotic syndrome. Clin J Am Soc Nephrol. 2012;7(3):427–436. doi: 10.2215/CJN.04820511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lv J, Zhang H, Wong MG, et al. TESTING Study Group Effect of oral methylprednisolone on clinical outcomes in patients with IgA nephropathy: the TESTING Randomized Clinical Trial. JAMA. 2017;318(5):432–442. doi: 10.1001/jama.2017.9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang P, Chen X, Zeng L, Hao H, Xu G. The response of the Oxford classification to steroid in IgA nephropathy: a systematic review and meta-analysis. Oncotarget. 2017;8(35):59748–59756. doi: 10.18632/oncotarget.19574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tumlin JA, Lohavichan V, Hennigar R. Crescentic, proliferative IgA nephropathy: clinical and histological response to methylprednisolone and intravenous cyclophosphamide. Nephrol Dial Transplant. 2003;18(7):1321–1329. doi: 10.1093/ndt/gfg081. [DOI] [PubMed] [Google Scholar]
- 63.Haas M, Verhave JC, Liu ZH, et al. A Multicenter Study of the predictive value of crescents in IgA nephropathy. J Am Soc Nephrol. 2017;28(2):691–701. doi: 10.1681/ASN.2016040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie X, Lv J, Shi S, et al. Plasma exchange as an adjunctive therapy for crescentic IgA nephropathy. Am J Nephrol. 2016;44(2):141–2. 9. doi: 10.1159/000448767. [DOI] [PubMed] [Google Scholar]
- 65.Shenoy M, Ognjanovic MV, Coulthard MG. Treating severe HenochSchlticenter and IgA nephritis with plasmapheresis alone. Pediatr Nephrol. 2007;22(8):1167–1671. doi: 10.1007/s00467-007-0498-y. [DOI] [PubMed] [Google Scholar]
- 66.Hattori M, Ito K, Konomoto T, Kawaguchi H, Yoshioka T, Khono M. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis. 1999;33(3):427–433. doi: 10.1016/s0272-6386(99)70178-2. [DOI] [PubMed] [Google Scholar]
- 67.Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA nephropathy: results of a 3-year prospective placebo-controlled randomized study. Kidney Int. 2004;65(5):1842–1849. doi: 10.1111/j.1523-1755.2004.00588.x. [DOI] [PubMed] [Google Scholar]
- 68.Frisch G, Lin J, Rosenstock J, et al. Mycophenolate mofetil (MMF) vs placebo in patients with moderately advanced IgA nephropathy: a double-blind randomized controlled trial. Nephrol Dial Transplant. 2005;20(10):2139–2145. doi: 10.1093/ndt/gfh974. [DOI] [PubMed] [Google Scholar]
- 69.Chen X, Chen P, Cai G, et al. A randomized control trial of mycophenolate mofeil treatment in severe IgA nephropathy. Zhonghua Yi Xue Za Zhi. 2002;82(12):796–801. Chinese. [PubMed] [Google Scholar]
- 70.Tang SC, Tang AW, Wong SS, Leung JC, Ho YW, Lai KN. Long-term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney Int. 2010;77(6):543–549. doi: 10.1038/ki.2009.499. [DOI] [PubMed] [Google Scholar]
- 71.Hogg RJ, Bay RC, Jennette JC, et al. Randomized controlled trial of mycophenolate mofetil in children, adolescents, and adults with IgA nephropathy. Am J Kidney Dis. 2015;66(5):783–791. doi: 10.1053/j.ajkd.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 72.Beckwith H, Medjeral-Thomas N, Galliford J, et al. Mycophenolate mofetil therapy in immunoglobulin A nephropathy: histological changes after treatment. Nephrol Dial Transplant. 2017;32(Suppl1):i123–i128. doi: 10.1093/ndt/gfw326. [DOI] [PubMed] [Google Scholar]
- 73.Pozzi C, Andrulli S, Pani A, et al. Addition of azathioprine to corticosteroids does not benefit patients with IgA nephropathy. J Am Soc Nephrol. 2010;21(10):1783–1790. doi: 10.1681/ASN.2010010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pozzi C, Andrulli S, Pani A, et al. IgA nephropathy with severe chronic renal failure: a randomized controlled trial of corticosteroids and azathioprine. J Nephrol. 2013;26(1):86–93. doi: 10.5301/jn.5000110. [DOI] [PubMed] [Google Scholar]
- 75.Stangou M, Ekonomidou D, Giamalis P, et al. Steroids and azathioprine in the treatment of IgA nephropathy. Clin Exp Nephrol. 2011;15(3):373–380. doi: 10.1007/s10157-011-0415-3. [DOI] [PubMed] [Google Scholar]
- 76.Song YH, Cai GY, Xiao YF, et al. Efficacy and safety of calcineurin inhibitor treatment for IgA nephropathy: a meta-analysis. BMC Nephrol. 2017;18(1):61. doi: 10.1186/s12882-017-0467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lionaki S, Panagiotellis K, Melexopoulou C, Boletis JN. The clinical course of IgA nephropathy after kidney transplantation and its management. Transplant Rev (Orlando) 2017;31(2):106–114. doi: 10.1016/j.trre.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 78.Leeaphorn N, Garg N, Khankin EV, Cardarelli F, Pavlakis M. Recurrence of IgA nephropathy after kidney transplantation in steroid continuation versus early steroid withdrawal: a retrospective analysis of the UNOS/OPTN database. Transpl Int. 2018;31(2):175–186. doi: 10.1111/tri.13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pham PT, Pham PC. The impact of mycophenolate mofetil versus azathioprine as adjunctive therapy to cyclosporine on the rates of renal allograft loss due to glomerular disease recurrence. Nephrol Dial Transplant. 2012;27(7):2965–2971. doi: 10.1093/ndt/gfr731. [DOI] [PubMed] [Google Scholar]
- 80.Chandrakantan A, Ratanapanichkich P, Said M, Barker CV, Julian BA. Recurrent IgA nephropathy after renal transplantation despite immunosuppressive regimens with mycophenolate mofetil. Nephrol Dial Transplant. 2005;20(6):1214–1221. doi: 10.1093/ndt/gfh773. [DOI] [PubMed] [Google Scholar]
- 81.Koutroutsos K, Kousios A, Leighton L, et al. The effect of immunosuppression regimen on IgA nephropathy recurrence post kidney transplantation [Abstract] Am J Transplant. 2016;16(Suppl 3) [Google Scholar]
- 82.Berthoux F, Suzuki H, Thibaudin L, et al. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol. 2012;23(9):1579–1587. doi: 10.1681/ASN.2012010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao N, Hou P, Lv J, et al. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 2012;82(7):790–796. doi: 10.1038/ki.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mansfield N, Hamour S, Habib AM, et al. Prolonged disease-free remission following rituximab and low-dose cyclophosphamide therapy for renal ANCA-associated vasculitis. Nephrol Dial Transplant. 2011;26(10):3280–3286. doi: 10.1093/ndt/gfr127. [DOI] [PubMed] [Google Scholar]
- 85.van den Brand J, Ruggenenti P, Chianca A, et al. Safety of rituximab compared with steroids and cyclophosphamide for idiopathic membranous nephropathy. J Am Soc Nephrol. 2017;28(9):2729phrol. doi: 10.1681/ASN.2016091022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Condon MB, Ashby D, Pepper RJ, et al. Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann Rheum Dis. 2013;72(8):1280–1286. doi: 10.1136/annrheumdis-2012-202844. [DOI] [PubMed] [Google Scholar]
- 87.Lafayette RA, Canetta PA, Rovin BH, et al. A randomized, controlled trial of rituximab in IgA nephropathy with proteinuria and renal dysfunction. J Am Soc Nephrol. 2017;28(4):1306–1313. doi: 10.1681/ASN.2016060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chintalacharuvu SR, Nagy NU, Sigmund N, Nedrud JG, Amm ME, Emancipator SN. T cell cytokines determine the severity of experimental IgA nephropathy by regulating IgA glycosylation. Clin Exp Immunol. 2001;126(2):326–333. doi: 10.1046/j.1365-2249.2001.01678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhai YL, Zhu L, Shi SF, Liu LJ, Lv JC, Zhang H. Increased APRIL expression induces IgA1 aberrant glycosylation in IgA nephropathy. Medicine (Baltimore) 2016;95(11):e3099. doi: 10.1097/MD.0000000000003099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fellstrom BC, Barratt J, Cook H, et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): a double-blind, randomised, placebo-controlled phase 2b trial. Lancet. 2017;389(10084):2117–2127. doi: 10.1016/S0140-6736(17)30550-0. [DOI] [PubMed] [Google Scholar]
- 91.Smerud HK, Bárány P, Lindström K, et al. New treatment for IgA nephropathy: enteric budesonide targeted to the ileocecal region ameliorates proteinuria. Nephrol Dial Transplant. 2011;26(10):3237–3342. doi: 10.1093/ndt/gfr052. [DOI] [PubMed] [Google Scholar]
- 92.Kim MJ, McDaid JP, McAdoo SP, et al. Spleen tyrosine kinase is important in the production of proinflammatory cytokines and cell proliferation in human mesangial cells following stimulation with IgA1 isolated from IgA nephropathy patients. J Immunol. 2012;189(7):3751–3758. doi: 10.4049/jimmunol.1102603. [DOI] [PubMed] [Google Scholar]
- 93.McAdoo SP, Reynolds J, Bhangal G, et al. Spleen tyrosine kinase inhibition attenuates autoantibody production and reverses experimental autoimmune GN. J Am Soc Nephrol. 2014;25(10):2291–2302. doi: 10.1681/ASN.2013090978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Daha MR, van Kooten C. Role of complement in IgA nephropathy. J Nephrol. 2016;29:1–4. doi: 10.1007/s40620-015-0245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu L, Zhai YL, Wang FM, et al. Variants in complement factor H and complement factor H-related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am SocNephrol. 2015;26(5):1195–1204. doi: 10.1681/ASN.2014010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang Y, Yan X, Zhao T, et al. Targeting C3a/C5a receptors inhibits human mesangial cell proliferation and alleviates immunoglobulin A nephropathy inmice. ClinExpImmunol. 2017;189(1):60–70. doi: 10.1111/cei.12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ring T, Pedersen BB, Salkus G, Goodship TH. Use of eculizumab in crescentic IgA nephropathy: proof of principle and conundrum? Clin Kidney J. 2015;8(5):489–491. doi: 10.1093/ckj/sfv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Herzog AL, Wanner C, Amann K, Lopau K. First treatment of relapsing rapidly progressive IgA nephropathy with Eculizumab after living kidney donation: a case report. Transplant Proc. 2017;49(7):1574–1577. doi: 10.1016/j.transproceed.2017.02.044. [DOI] [PubMed] [Google Scholar]
- 99.Zhang L, Wang X, Li YS, Zhang L, Hao LR. Icariin ameliorates IgA nephropathy by inhibition of nuclear factor kappa b/Nlrp3 pathway. FEBS Open Bio. 2016;7(1):54–63. doi: 10.1002/2211-5463.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gotot J, Piotrowski E, Otte MS, et al. Inhibitor of NFκB kinase subunit 2 blockade hinders the initiation but aggravates the progression of crescentic GN. J Am Soc Nephrol. 2016;27(7):1917–1924. doi: 10.1681/ASN.2015060699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Madan A, Mijovic-Das S, Stankovic A, Teehan G, Milward AS, Khastgir A. Acthar gel in the treatment of nephrotic syndrome: a multicenter retrospective case series. BMC Nephrol. 2016;17:37. doi: 10.1186/s12882-016-0241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]