

Abstract
Background
Inflammation is an important cause of COPD. Alveolar macrophages are the major innate immune cells that have an important role in COPD pathology. Class A scavenger receptor (SR-A) is a pattern recognition receptor expressed on macrophages. This study investigates the role of SR-A in COPD progression via regulation of inflammation.
Patients and methods
SR-A expression in COPD patients and control subjects (smokers and nonsmokers without COPD) was measured by immunohistochemistry, immunofluorescence, and real-time PCR. The cytokine levels in BAL were measured by enzyme-linked immunosorbent assay. To further prove our hypothesis, we treated RAW264.7 cells that overexpress SR-A with lipopolysaccharides, poly(I:C), cigarette smoke extract, and H1N1 influenza separated from patients for 24 h and examined the levels of inflammatory cytokines.
Results
In both groups, COPD and smokers without COPD, SR-A expression level was upregulated in alveolar macrophages. SR-A mRNA level was positively correlated with inflammatory cytokines and negatively correlated with FEV1% predicted in COPD patients. In RAW-SR-A cells, level of inflammatory cytokines was significantly higher when compared with control ones.
Conclusion
SR-A could increase inflammation stimulated by cigarette smoke extracts, bacteria, and virus, leading to long-term inflammation in COPD, and thus might be used as a new therapeutic target for COPD treatment.
Keywords: chronic obstructive pulmonary disease, class A scavenger receptor, inflammation, cigarette smoke extract, lipopolysaccharides, poly(I:C), H1N1 influenza
Introduction
COPD is characterized by chronic inflammation, fibrotic remodeling of small airways, and irreversible emphysematous destruction of lung parenchyma.1 According to the World Health Organization, COPD is supposed to be ranked as the third cause of death by 2030, thus becoming a significant social and economic burden worldwide.2
Both innate and adaptive immune systems are involved in the progression of COPD. Macrophages are the major innate immune cells in COPD pathology. They are derived from blood monocytes, and they differentiate locally after traffic to the lungs. Alveolar macrophages (AMs) are localized to sites of airway wall destruction in patients with emphysema and are markedly increased in airways, lung parenchyma, and bronchoalveolar lavage (BAL) of COPD patients. The quantity of AMs is closely related to the severity of emphysema.3
Class A scavenger receptor (SR-A), a pattern recognition receptor, is mainly expressed on macrophages. SR-A is involved in the recognition and clearance of modified lipids, apoptotic cells, inhaled particles, and microorganisms, and has a critical role in metabolic processes.4 Recently, SR-A has been reported to be involved in innate immune defense. The collagen-like domain allows recognition of Gram-positive and Gram-negative bacteria.5 SR-A also participates in the regulation of inflammation. Once triggered, SR-A, in combination with other membrane receptors, leads to the activation of downstream pathways, initiating a signaling cascade that leads to the activation and nuclear translocation of the transcription factors. This process activates intracellular signaling pathways that cause the release of proinflammatory cytokines.6 However, the published data have given rise to some controversy. On one hand, SR-A is clearly beneficial and host protective in some models of disease, like atherosclerosis.7–9 On the other hand, SR-A has been reported to participate in the propagation of certain diseases, such as endotoxin shock and septic shock.10,11 The endoplasmic reticulum stress response may provide important insights into the conflicting effects of SR-A, but the exact mechanism of SR-A in disease progression still remains to be discovered.12
Since COPD is a chronic disease and the mechanism of SR-A in innate immune system remains unclear, it is possible that SR-A has a critical role in COPD progression via regulation of inflammation. In this study, we measured expression levels of SR-A in AMs and bronchial tissues of COPD patients. We treated RAW264.7 cells that overexpress SR-A with lipopolysaccharides (LPS), Poly(I:C), CSE (cigarette smoke extract), and the influenza A virus (H1N1) separated from patients with flu for 24 h, and then we examined the inflammatory responses. Our results suggested that patients with COPD have significantly higher levels of SR-A, while the levels of inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), IL-6, and IL-1β were much higher in RAW-SR-A cells compared with the control ones. These findings somewhat clarify a critical functional role of SR-A in COPD airway inflammation.
Patients and methods
Subject characteristics
Lung tissues were obtained from 6 patients with COPD and 12 control subjects without COPD (6 smokers and 6 nonsmokers, respectively) from the ward of Pulmonary and Critical Care Medicine, Ruijin Hospital, affiliated to School of Medicine, Shanghai Jiaotong University. The lung tissue samples were collected during thoracic surgery. COPD patients and the control subjects underwent resection of lung cancer. In addition, BAL from 10 COPD patients and 20 non-COPD control subjects (10 smokers and 10 nonsmokers, respectively) were collected from the middle or lingula lobe. All of the patients with COPD met the criteria proposed by the Global Initiative for Chronic Obstructive Lung Disease.1 All of the control subjects had normal lung function. Not even one patient suffered from respiratory tract infection recently or currently (8 weeks). Patients were studied without any exacerbations requiring oral steroid or antibiotic treatment.
This research was approved by Ruijin Hospital Ethics Committee, Shanghai Jiaotong School of Medicine. All patients provided their written informed consent to participate in this research. Characteristics of subjects included in this study are listed in Table 1.
Table 1.
Characteristics of all subjects
| Characteristics | Nonsmokers | COPD | Smokers |
|---|---|---|---|
| Lung tissues | N=6 | N=6 | N=6 |
| Age (years) | 59.83±1.70 | 67.83±2.30 | 48.67±1.05 |
| Gender (M:F) | 4:2 | 5:1 | 4:2 |
| FEV1 % predicted | 91.95±1.72 | 76.28±1.98 | 90.68±1.41 |
| FEV1/FVC | 84.93±2.41 | 62.82±2.33 | 85.10±2.18 |
| Smoking status | |||
| Never (%) | 100 | 0 | 0 |
| Former (%) | 0 | 0 | 0 |
| Current (%) | 0 | 100 | 100 |
| Pack-years | 0 | 49.00±6.04 | 42.17±4.00 |
| BALF | N=10 | N=10 | N=10 |
| Age (years) | 53.30±2.50 | 68.34±3.77 | 49.52±6.14 |
| Gender (M:F) | 5:5 | 6:4 | 9:1 |
| FEV1 % predicted | 87.13±4.24 | 53.56±8.73 | 86.78±1.65 |
| FEV1/FVC | 84.03±1.81 | 57.11±2.56 | 88.15±2.09 |
| Smoking status | |||
| Never (%) | 100 | 0 | 0 |
| Former (%) | 0 | 20 | 50 |
| Current (%) | 0 | 80 | 50 |
| Pack-years | 0 | 41.35±6.12 | 37.80±2.94 |
Notes: Data are represented as mean ± standard error of the mean. Nonsmokers, nonsmokers without COPD. Smokers, smokers without COPD.
Abbreviation: BALF, bronchoalveolar lavage fluid.
Harvesting of AMs from BAL
We obtained about 30 mL of BAL from each subject by infusing 50 mL of sterile sodium chloride in either the middle or lingula lobe. BAL was centrifuged for 20 min at the speed of 1,500 rpm, and then the supernatant was discarded. The deposit was resuspended in DMEM (Corning, New York, NY, USA) containing 10% fetal bovine serum (FBS), supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin. Cells were incubated on 3.5 cm untreated plates for 3 h, and then washed three times with PBS. AMs were obtained as attached cells on the dish.
Cell line
The mouse macrophage cell lines RAW264.7 that overexpress SR-A were constructed by lentivirus transduction. The control RAW264.7 cells were transfected by mock lentivirus. Both the RAW-SR-A and control cells were kindly offered by Department of Immunology and Microbiology (the Department of Microbiology and Parasitology, Institutes of Medical Sciences, Shanghai Jiaotong University School of Medicine), which approved the use of the gifted cell lines in this research. Cells were cultured in DMEM (Corning) supplemented with 10% FBS, Puromycin (2.5 mg/mL), and incubated in 37°C 5% CO2 under humidified conditions.
Preparation of CSE and cell stimulation
The commercial filtered cigarettes (11 mg of tar and 1.0 mg of nicotine in one cigarette, Double Happiness Cigarettes, Shanghai Tobacco Corporation, Shanghai, China) were used for CSE collection. Briefly, 10 cigarettes were smoked consecutively through an experimental apparatus with a constant airflow driven by an air compressor. The smoke was bubbled through 300 mL of DMEM (Corning). The CSE–DMEM was a 100% stock solution that was ready for use after adjusting the pH to 7.4 and sterile filtration (0.2 μm). In this case, we used a concentration of 20% to emulate the environment of COPD. The cells were cocultured with CSE for 24 h before total RNA isolation and enzyme-linked immunosorbent assay (ELISA) analysis.
Cell stimulation with LPS and poly(I:C)
RAW264.7 cells were plated as 106 cells/well in a six-well plate and cultured to 70%–80% confluence, and 1 μg/mL concentration of LPS (from Escherichia coli O55:B5; Sigma-Aldrich, St. Louis, MO, USA) and 50 μg/mL concentration of poly(I:C) (polyinosinic acid potassium salt; Sigma-Aldrich) were added respectively. The cells were cultured for 24 h before total RNA isolation and ELISA analysis.
In vitro viral infection
The influenza A virus (H1N1) was separated from outpatients infected with flu in Shanghai Ruijin Hospital who provided written informed consent to participate in the research. The virus we used in the present study was identified as the most popular strain in Eastern China from December 2015 to March 2016. The sequences of H1N1 were 99% matched with A/British Columbia/1/2016 (H1N1). RAW264.7 cells were plated at 106 cells/well in a six-well plate. After overnight incubation, the culture medium was replaced with serum-free DMEM containing H1N1 influenza virus (multiplicity of infection [MOI]=5, 10, 20) for 24 h with 2 μg/mL Trypsin-TPCK and 25 mM HEPES buffer. The supernatant was collected for ELISA analysis and the adherent cells were collected for real-time PCR.
Immunohistochemistry
Immunohistochemistry was used to analyze the protein levels and distribution of SR-A in lung tissues of COPD and control subjects. Lung tissues in paraffin were embedded and sectioned to 5 μm on the slides, and then dewaxed in xylene and rehydrated. Endogenous peroxidases were inhibited with 0.5% hydrogen peroxide in methanol for 10 min, followed by incubating overnight at 4°C with a rabbit monoclonal IgG antibody against SR-A (1:100, ab123946; Abcam, Cambridge, UK). Immunodetection was performed with diaminobenzidine, biotinylated goat anti-rabbit IgG reagent, and horseradish peroxidase (HRP) reagent.
Confocal microscopy
Cells were harvested into cover-glass chambers and then cultured in DMEM with 10% bovine serum for 37°C and 5% CO2 for 24 h. Unattached cells were removed by gentle medium exchange, and the attached cells were washed twice with PBS. Cells were fixed with 4% paraformaldehyde. Next, the cells were incubated for 30 min at room temperature with rabbit anti-SR-A antibody (1:100; Abcam), then incubated with FITC-conjugated goat anti-rabbit IgG antibody (1:500; Proteintech Group Inc., Rosemont, IL, USA) for 30 min at 37°C. After staining with DAPI (1:1,000; Thermo Fisher Scientific, Waltham, MA, USA), cells were observed with Laser Scanning Confocal Microscope (LSM-510 Meta; Leica, Frankfurt, Germany).
RNA extraction and real-time PCR
Total RNA isolated from AMs, bronchial tissues, and cells were extracted using an RNeasy Kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. Single-strand cDNA was synthesized for each sample with oligo (dT) as primers using RevertAid™ First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer’s manual. Five hundred nanograms of total RNA was performed with a 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) with Fast Start Universal SYBR Green (Roche, Branchburg, NJ, USA) after cDNA synthesis. Fold changes of the gene expression were calculated by 2−∆∆Ct relative to the internal reference gene (GAPDH). The sequences of all primers are shown in Table 2. The results were repeated at least three times. The accession number of SR-A cDNA in humans is NM_138715.2. The accession number of SR-A cDNA in mouse is NM_001113326.1.
Table 2.
Primer sequences of real-time PCR
| Genes of interest | Primer sequences (from 5′ to 3′) | Length (bp) | Efficiency (%) |
|---|---|---|---|
| Human | |||
| GAPDH | Forward primer: GAAGGTGAAGGTCGGAGTC | 226 | 99.30 |
| Reverse primer: GAAGATGGTGATGGGATTTC | |||
| TNF-α | Forward primer: ATGAGCACTGAAAGCATGATCC | 217 | 98.90 |
| Reverse primer: GAGGGCTGATTAGAGAGAGGTC | |||
| IL-8 | Forward primer: ATGACTTCCAAGCTGGCCGTGGCT | 292 | 99.50 |
| Reverse primer: TCTCAGCCCTCTTCAAAAACTTCTC | |||
| SR-A | Forward primer: TAGGCACTTGGGATGTCTGA | 200 | 98.30 |
| Reverse primer: GTCCTCAATTTGTATTGGTGCT | |||
| Mouse | |||
| GAPDH | Forward primer: TGTTCCTACCCCCAATGTG | 95 | 99.70 |
| Reverse primer: ATGCCTGCTTCACCACCTTC | |||
| SR-A | Forward primer: CAACATCACCAACGACCTC | 110 | 99.20 |
| Reverse primer: CTCTGTCTCCCTTTTCACCTT | |||
| TNF-α | Forward primer: CCCCAGTCTGTATCCTTCTA | 101 | 99.40 |
| Reverse primer: CCCAGCATCTTGTGTTTCT | |||
| IL-6 | Forward primer: CCAATGCTCTCCTAACAGAT | 161 | 99.50 |
| Reverse primer: TGTCCACAAACTGATATGCT | |||
| IL-1β | Forward primer: GTACAAGGAGAACCAAGCAA | 89 | 99.10 |
| Reverse primer: CCGTCTTTCATTACACAGGA |
Abbreviation: PCR, polymerase chain reaction.
Cytokine ELISAs
The concentrations of IL-8, TNF-α in the BAL, and TNF-α, IL-6, IL-1β in the cell supernatant were detected using DuoSet ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Western blots
RAW264.7 cells were lysed with 100 μL radio immuno-precipitation assay lysis buffer (Sigma-Aldrich). Protein concentrations were measured with bicinchoninic acid assay kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Equal amounts of proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a poly (vinylidene fluoride) membrane. The membrane was blocked with PBS containing 5% nonfat milk for 2 h at room temperature, then incubated overnight at 4°C with an anti-SR-A antibody (1:1,000; Abcam), or an anti-β-actin antibody (1:1,000; Cell Signaling, Danvers, MA, USA). The membrane was then washed three times for 5 min with 15 mL Tris-buffered saline Tween-20 and incubated with HRP-conjugated goat anti-rabbit IgG antibody (1:1,000 dilution; Sigma-Aldrich) for 1 h at room temperature. After washing, 1 mL chemiluminescent substrate (Thermo Fisher Scientific) was added to the membrane. The signal was detected and quantified with an enhanced chemiluminescence system (ImageQuant LAS-4000 MINI; GE, Fairfield, CT, USA). All samples were normalized to β-actin. Data were represented as mean ± standard error of the mean (SEM).
Statistical analysis
Results are presented as mean ± SEM. Two-tailed t-test was performed. A linear regression was adopted for associations between variables using Spearman’s rank correlation test. Data analysis was performed as described in each figure legend via GraphPad Prism6.0, and the results were considered significant when p<0.05.
Results
SR-A mRNA expression in AMs
From BAL of 10 COPD patients and 20 control subjects (10 smokers and 10 nonsmokers), we separated the AMs from BAL and then analyzed the mRNA level of SR-A in the AMs. Relative expression of SR-A compared with the housekeeping gene GAPDH was significantly higher in COPD patients than nonsmoker control subjects (13.65±2.24 vs 1.75±0.22, p<0.0001; Figure 1). In addition, the relative expression of SR-A was significantly higher in smoker subjects without COPD compared with nonsmokers (5.20±0.43 vs 1.75±0.22, p<0.0001; Figure 1), while the relative expression of SR-A was significantly higher in COPD patients compared with smokers (13.65±2.24 vs 5.20±0.43, p<0.0001; Figure 1).
Figure 1.
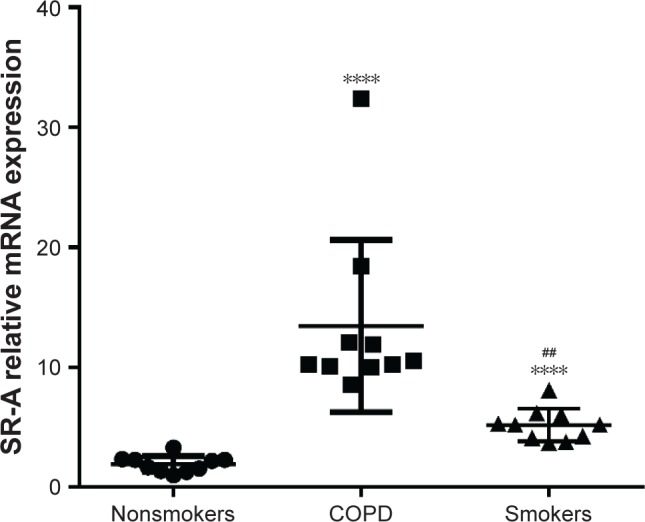
SR-A mRNA expression in AMs.
Notes: RT-qPCR detection of SR-A mRNA expression of alveolar macrophages. Compared with the control samples, COPD patients and smokers without COPD showed significantly higher levels of SR-A mRNA (1.75±0.22 vs 13.65±2.24, P<0.0001 and 1.75±0.22 vs 5.20±0.43, P<0.0001, respectively). Relative expression of SR-A was significantly higher in COPD patients than in smokers (13.65±2.24 vs 5.20±0.43, P<0.0001). The results are presented as mean ± SEM. ****Compared with nonsmoker group, p<0.0001; ##Compared with COPD group, p<0.01.
Abbreviations: AMs, alveolar macrophages; RT-qPCR, real-time reverse transcription quantitative polymerase chain reaction; SEM, standard error of the mean.
Immunohistochemistry analysis of SR-A in the lung tissues of COPD patients and control subjects
SR-A protein is mainly located in the membrane of macrophages.13 Some of the AMs were lost when lung tissues were sectioned to immunofluorescence slides. As a result, the level of SR-A was low. Nonetheless, immunohistochemistry staining still showed that SR-A protein in COPD patients was much higher compared with the control subjects (both smokers and nonsmokers) which can be seen in Figure 2A–F. The quantified analysis of immunofluorescence results is shown in Figure 2G.
Figure 2.

The distribution and expression levels of SR-A in lung tissue.
Notes: Immunohistochemistry of lung tissues with antibodies against SR-A. Representative SR-A expression (brown staining) in sections from control subjects (A, D) and COPD patients (B, E) and smokers without COPD subjects (C and F). (G) Quantification of the immunohistochemistry results expressed as integral optical density of brown staining in the different view of COPD and controls. The results are presented as mean ± SEM (n=18). Original magnification: ×200 or ×400. **Compared with nonsmoker group, p<0.01; #Compared with COPD group, p<0.05.
Abbreviations: SEM, standard error of the mean; SR-A, class A scavenger receptor.
Confocal analysis of SR-A in AMs of COPD patients and control subjects
Immunofluorescence showed that SR-A protein was predominantly expressed by AMs. However, the SR-A receptors were not located in the membrane of AMs in patients with COPD and in smokers without COPD. Orr et al has reported that SR-A could recognize engineered nanoparticles as a cell membrane receptor, which leads to internalization of both the nanoparticles and SR-A into the cells.14 In this case, it could be the result of internalization after inflammatory stimulation. From Figure 3, we can see that COPD patients had a higher expression level of SR-A in AMs when compared with control subjects (both smokers and nonsmokers). Also, the level of SR-A was higher in smokers than in nonsmokers. These results were consistent with those illustrated in Figure 1, which indicated that SR-A expression was significantly increased in both smokers and patients with COPD.
Figure 3.

Confocal analyses of SR-A in AMs of COPD patients.
Notes: Representative images of confocal results in AMs for SR-A in nonsmoker control subjects (A), COPD patients (B), and smokers without COPD subjects (C). SR-A (green) was mainly observed in macrophages and was much higher in COPD and smoker groups, especially COPD group. Nucleus is shown in blue. Magnification ×1000.
Abbreviation: AMs, alveolar macrophages.
Cytokine secretion in BAL
As determined by ELISA, concentrations of IL-8 and TNF-α were much higher in the BAL of COPD patients and smokers than in nonsmokers which can be seen in Figure 4. These results were consistent with previous reports and indicated a persistent inflammation in the airways of COPD patients. As a result, we could draw the conclusion that the level of SR-A was elevated in lungs of COPD patients and smokers with long-time inflammation.
Figure 4.

Cytokine secretion in BALF.
Notes: BALF was assayed by ELISA. COPD patients and smoker groups showed significantly higher levels of IL-8 (A) and TNF-α (B) compared with the nonsmoker group, especially COPD group. Data are expressed as mean ± SEM (n=30; Compared with nonsmoker group, *p<0.05, **p<0.01,****p<0.0001; Compared with COPD group, ##p<0.01, ####p<0.0001).
Abbreviations: BALF, bronchoalveolar lavage fluid; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean; TNF-α, tumor necrosis factor-alpha.
Relationships between SR-A mRNA, and cytokine expression levels and FEV1% predicted
In this study, we found a negative correlation between SR-A mRNA levels in AMs and FEV1% predicted in the BAL in this cohort of COPD patients (Figure 5A). Moreover, there were significant positive correlations between SR-A mRNA levels in AMs and IL-8 and TNF-α concentrations (Figure 5B and C).
Figure 5.

Relationships between SR-A mRNA, and cytokine expression levels and FEV1% predicted.
Notes: Correlations between FEV1% predicted, IL-8 expression level, TNF-α expression level and SR-A mRNA level in AMs. There were significantly negative correlations between SR-A mRNA levels in AMs and FEV1% predicted in COPD patients (A). In addition, there were significantly positive correlations between SR-A mRNA levels and IL-8 expression levels, the same as SR-A mRNA levels and TNF-α expression levels (B, C). Data are expressed as mean ± SEM.
Abbreviations: AMs, alveolar macrophages; SEM, standard error of the mean; SR-A, class A scavenger receptor.
Western blots and RT-qPCR results of RAW264.7 cells
RAW264.7 cells that overexpress SR-A were constructed by lentivirus transduction. To confirm whether SR-A level was significantly higher than control cells, we detected both protein and mRNA levels of SR-A by Western blots and real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR). As the result shows, the construction of overexpression cell models was successful (Figure 6).
Figure 6.

Western blots and RT-qPCR results of RAW-SR-A and RAW-control cells.
Notes: (A, B) Results of Western blots and quantified analysis. (C) Result of mRNA expression level by RT-qPCR. Both the protein and mRNA levels were significantly higher in RAW-SR-A cells. (*p<0.05 vs the control group, **p<0.01 vs the control group).
Abbreviations: RT-qPCR, real-time reverse transcription quantitative polymerase chain reaction.
Inflammation induced by CSE, LPS, and Poly(I:C)
Cigarette smoke impairs the immune defense of a host, and it contributes to recurrent infections of respiratory airways, which have a critical role in the progression of COPD.15 Bacterial and viral infections lead to exacerbation of COPD.16 In the present study, we treated RAW cells with CSE, LPS from E. coli (1 μg/mL), and poly(I:C) (50 μg/mL) to simulate environment in patients with COPD. In Figure 7, the expression level of SR-A was obviously increased compared with cells treated with PBS, and the mRNA and protein expression levels of TNF-α, IL-6, and IL-1β in RAW-SR-A cells were significantly higher than in the control ones.
Figure 7.

RT-qPCR and ELISA results of RAW-SR-A and RAW-control cells stimulated by LPS, poly(I:C), and CSE.
Notes: RAW264.7 cells were plated as 106 cells/well in a six-well plate and cultured to 70%–80% confluence. After overnight incubation, cells were treated with PBS, LPS (1 μg/mL), poly(I:C) (50 μg/mL), and CSE for 24 h. Cells were collected for RT-qPCR test, and cytokine secretion in supernatant was measured by ELISA. (A) Expression levels of SR-A increased obviously compared with cells treated with PBS. (B–G) Both protein and mRNA levels of TNF-α, IL-6, and IL-1β significantly increased in RAW-SR-A cells when compared with RAW-control cells. Inflammation induced by LPS was strongest. Although inflammation produced by CSE was low, there is still significant difference between these two kinds of cells. (*p<0.05 vs the control group, **p<0.01 vs the control group, ***p<0.001 vs the control group).
Abbreviations: CSE, cigarette smoke extract; ELISA, enzyme-linked immunosorbent assay; LPS, lipopolysaccharides; ns, nonsignificant; RT-qPCR, real-time reverse transcription quantitative polymerase chain reaction.
Inflammation induced by H1N1 influenza
Previous results showed that inflammation was much higher in RAW-SR-A cells after stimulation with LPS, poly(I:C), and CSE. To simulate the status of infection in COPD patients, we treated RAW cells with PBS and H1N1 influenza virus separated from patients infected with flu at an MOI of 5, 10, and 20. Cells were collected for RT-qPCR test, and cytokine secretion in supernatant was measured by ELISA. Inflammation increased as cells were treated with higher virus concentrations. As shown in Figure 8, the mRNA level of SR-A was significantly higher in RAW-SR-A cells, and so were the mRNA and protein levels of TNF-α, IL-6, and IL-1β.
Figure 8.

RT-qPCR and ELISA results of RAW-SR-A and RAW-control cells stimulated by H1N1 influenza.
Notes: RAW264.7 cells were plated as 106 cells/well in a six-well plate and cultured to 70%–80% confluence. After overnight incubation, the culture medium was replaced with serum-free DMEM containing H1N1 influenza virus at an MOI of 5, 10, and 20 for 24 h with 2 μg/mL Trypsin-TPCK and 25 mM HEPES buffer. The supernatant was collected for ELISA analysis and the adherent cells were collected for real-time PCR. (A) Level of SR-A was significantly higher in RAW-SR-A cells, and it increased as cells were treated with higher concentration of H1N1 influenza virus. (B–G) Similar to previous results, mRNA and protein levels of TNF-α, IL-6, and IL-1β were much higher in RAW-SR-A cells. (*p<0.05 vs the control group).
Abbreviations: ELISA, enzyme-linked immunosorbent assay; ns, nonsignificant; RT-qPCR, real-time reverse transcription quantitative polymerase chain reaction.
Discussion
COPD is associated with chronic inflammation, which is localized to peripheral airways and lung parenchym, while the long-term inflammation could result in progressive and irreversible airflow limitation.3 Hence, it is important to learn the exact mechanisms and relieve inflammation in the disease progression. The present study elucidates a critical role of SR-A in the regulation of inflammation.
SR-A participates in a number of functions in metabolic processes, such as recognizing and removing modified lipoproteins. Also, it has the ability to partner with various co-receptors which further enable the broad range of functions. Recently, more attention has been paid to its role in innate immunity. SR-A has been reported in pathogen clearance; yet, the studies of SR-A in the regulation of inflammation are still limited.
In the present study, we found out that the upregulation of SR-A could increase inflammation induced by CSEs, bacteria, and virus, and that it could be closely related to choric inflammation in COPD. Compared with nonsmoker subjects without COPD, the expression level of SR-A is increased in lung tissues as well as in AMs in smokers and COPD groups. Moreover, the basal level of TNF-α and IL-8 in COPD patients was elevated in the BAL specimens. These results are consistent with previous reports suggesting that cytokines are systematically increased in airways of patients with COPD.17 The present study also illustrated that SR-A mRNA levels in AMs were positively correlated with TNF-α and IL-8, and negatively correlated with FEV1% predicted in this cohort of COPD patients. As the expression level of SR-A was significantly higher than in control subjects, we introduced RAW264.7 cells that overexpress SR-A to investigate its role in the regulation of inflammation in subjects with higher level of SR-A. To our knowledge, this is the first study that investigated the role of SR-A in the regulation of inflammation using RAW cells that overexpress SR-A.
Cigarette smoke is recognized as a risk factor of COPD due to production of chronic oxidative stress within lungs. SR-A is largely responsible for the recognition and engulfment of cigarette smoke.18,19 Kirkham et al have discovered that recognition of cigarette smoke condensates by SR-A leads to macrophage adhesion, which may have important consequences in the development of emphysema in COPD.20 In our study, expression levels of SR-A were elevated in both RAW-SR-A and RAW-control cell groups, which could be caused by better recognition of CSE. Also, the cytokines were significantly higher in RAW-SR-A group, which is consistent with the findings in patients with COPD and control subjects.
Bacterial infection is an important reason for pathogenesis and exacerbation of COPD, which is often associated with impaired respiratory function over prolonged periods of time.16,21,22 LPS is a central component of the outer membrane in Gram-negative bacteria.23 SR-A has been reported as a receptor of both Gram-positive and Gram-negative bacteria in macrophages and is essential for maximal LPS-induced activation of the Toll-like receptor 4 (TLR4), which can lead to inflammatory cascade reaction via NF-κB signaling pathway.24 RAW cells were stimulated with LPS extracted from E. coli at a concentration of 1 μg/mL for 24 h to simulate bacterial infection. SR-A level was elevated in both groups, and inflammation was significantly higher in RAW-SR-A cells compared with control ones. Cumulatively, inflammation led by bacterial infection was elevated when SR-A was overexpressed.
Exacerbation of COPD is often caused by respiratory viral infections. SR-A expression is required for TLR3-mediated responses to viral infection via detection of viral dsRNA.25 Upon activation, TLR3 recruits the adaptor Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF), which further activates NF-κb pathway.26 Stowell et al have also demonstrated that TLR3 activation by dsRNA mimic poly(I:C) could regulate local inflammatory response in the lung, causing lung function impairment and exacerbation.27 To confirm the role of SR-A in viral infection, we treated RAW cells with poly(I:C) at a concentration of 50 μg/mL for 24 h, and we detected protein and mRNA levels of TNF-α, IL-6, and IL-1β. Inflammation increased significantly in RAW-SR-A cells. However, Shimizu et al have declared that TNF-α production is significantly increased in SR-A−/− non-obese diabetic (NOD) bone marrow-derived dendritic cells when compared with their wild-type counterparts.28 The contradictory results could be explained by different experiment models. To our knowledge, this is the first study that investigated the role of SR-A by treating RAW-SR-A cells with poly(I:C). Also, it is possible that there is an innate immune dysfunction in SR-A−/− NOD mice, which might lead to innate immune dysfunction and completely different regulation of inflammation.
Influenza virus is the most common pathogen in severe exacerbations of COPD requiring admission to hospital.29 We extracted H1N1 virus from outpatients infected with flu, which was identified as the most popular strain in Eastern China from December 2015 to March 2016. Viral RNA structures could be recognized through molecules such as SR-A, TLR3, RIG-I, and MDA5.25,30 To confirm whether SR-A has the same role in recognizing and eliminating influenza, we treated RAW-SR-A and RAW-control cells with H1N1 influenza that was separated from patients infected with flu at an MOI of 5, 10, and 20 for 24 h. When RAW cells were treated with H1N1 influenza at an MOI of 20, expression levels of TNF-α, IL-6, and IL-1β were significantly higher in RAW-SR-A cells compared with control ones. Influenza A is a highly contagious single-stranded RNA virus that infects both the upper and lower respiratory tracts of humans. We hypothesize that SR-A could bind ssRNA of H1N1 influenza and induce inflammation after activation of TLR3.
Conclusion
In summary, through interaction with other membrane receptors, SR-A could increase inflammatory responses stimulated by LPS, poly(I:C), CSE, and H1N1 influenza, especially when it is overexpressed. Levels of SR-A were elevated in patients with COPD and could be closely related with long-term inflammation in COPD, which affects the whole respiratory tract from central to peripheral airways. Consequently, our study results may be used for designing new therapies targeting SR-A in the treatment of COPD.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81570029 and 81600004) and Shanghai Key Discipline for Respiratory Diseases (No. 2017ZZ02014).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. doi: 10.1164/rccm.201701-0218PP. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SR, Kalhan R, Mannino DM. Epidemiology of chronic obstructive pulmonary disease: prevalence, morbidity, mortality, and risk factors. Semin Respir Crit Care Med. 2015;36(4):457–469. doi: 10.1055/s-0035-1555607. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi: 10.1016/j.jaci.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 4.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 5.Dunne DW, Resnick D, Greenberg J, Krieger M, Joiner KA. The type I macrophage scavenger receptor binds to gram-positive bacteria and recognizes lipoteichoic acid. Proc Natl Acad Sci U S A. 1994;91(5):1863–1867. doi: 10.1073/pnas.91.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelley JL, Ozment TR, Li C, Schweitzer JB, Williams DL. Scavenger receptor-A (CD204): a two-edged sword in health and disease. Crit Rev Immunol. 2014;34(3):241–261. doi: 10.1615/critrevimmunol.2014010267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki H, Kurihara Y, Takeya M, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386(6622):292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 8.Arredouani MS, Franco F, Imrich A, et al. Scavenger receptors SR-AI/II and MARCO limit pulmonary dendritic cell migration and allergic airway inflammation. J Immunol. 2007;178(9):5912–5920. doi: 10.4049/jimmunol.178.9.5912. [DOI] [PubMed] [Google Scholar]
- 9.Haworth R, Platt N, Keshav S, et al. The macrophage scavenger receptor type A is expressed by activated macrophages and protects the host against lethal endotoxic shock. J Exp Med. 1997;186(9):1431–1439. doi: 10.1084/jem.186.9.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi Y, Miyaji C, Watanabe H, et al. Role of macrophage scavenger receptor in endotoxin shock. J Pathol. 2000;192(2):263–272. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH692>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 11.Drummond R, Cauvi DM, Hawisher D, et al. Deletion of scavenger receptor A gene in mice resulted in protection from septic shock and modulation of TLR4 signaling in isolated peritoneal macrophages. Innate Immun. 2013;19(1):30–41. doi: 10.1177/1753425912449548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu XD, Zhuang Y, Ben JJ, et al. Caveolae-dependent endocytosis is required for class A macrophage scavenger receptor-mediated apoptosis in macrophages. J Biol Chem. 2011;286(10):8231–8239. doi: 10.1074/jbc.M110.145888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat Rev Immunol. 2013;13(9):621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 14.Orr GA, Chrisler WB, Cassens KJ, et al. Cellular recognition and trafficking of amorphous silica nanoparticles by macrophage scavenger receptor A. Nanotoxicology. 2011;5(3):296–311. doi: 10.3109/17435390.2010.513836. [DOI] [PubMed] [Google Scholar]
- 15.Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. doi: 10.1016/S0140-6736(11)60988-4. [DOI] [PubMed] [Google Scholar]
- 16.Sethi S. Molecular diagnosis of respiratory tract infection in acute exacerbations of chronic obstructive pulmonary disease. Clin Infect Dis. 2011;52(Suppl 4):S290–S295. doi: 10.1093/cid/cir044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stankiewicz W, Dabrowski MP, Chcialowski A, Plusa T. Cellular and cytokine immunoregulation in patients with chronic obstructive pulmonary disease and bronchial asthma. Mediators Inflamm. 2002;11(5):307–312. doi: 10.1080/09629350210000015719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87(3):1047–1082. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 19.Guzik K, Skret J, Smagur J, et al. Cigarette smoke-exposed neutrophils die unconventionally but are rapidly phagocytosed by macrophages. Cell Death Dis. 2011;2:e131. doi: 10.1038/cddis.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirkham PA, Spooner G, Ffoulkes-Jones C, Calvez R. Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. 2003;35(7):697–710. doi: 10.1016/s0891-5849(03)00390-3. [DOI] [PubMed] [Google Scholar]
- 21.Svanes C, Sunyer J, Plana E, et al. Early life origins of chronic obstructive pulmonary disease. Thorax. 2010;65(1):14–20. doi: 10.1136/thx.2008.112136. [DOI] [PubMed] [Google Scholar]
- 22.Hogg JC. Childhood viral infection and the pathogenesis of asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S26–S28. doi: 10.1164/ajrccm.160.5.8. [DOI] [PubMed] [Google Scholar]
- 23.Whitfield C, Trent MS. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem. 2014;83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- 24.Yu H, Ha T, Liu L, et al. Scavenger receptor A (SR-A) is required for LPS-induced TLR4 mediated NF-kappaB activation in macrophages. Biochim Biophys Acta. 2012;1823(7):1192–1198. doi: 10.1016/j.bbamcr.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Limmon GV, Arredouani M, McCann KL, Corn Minor RA, Kobzik L, Imani F. Scavenger receptor class-A is a novel cell surface receptor for double-stranded RNA. FASEB J. 2008;22(1):159–167. doi: 10.1096/fj.07-8348com. [DOI] [PubMed] [Google Scholar]
- 26.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442(7098):39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 27.Stowell NC, Seideman J, Raymond HA, et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir Res. 2009;10:43. doi: 10.1186/1465-9921-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu M, Yasuda H, Hara K, Takahashi K, Nagata M, Yokono K. The dual role of scavenger receptor class A in development of diabetes in autoimmune NOD mice. PLoS One. 2014;9(10):e109531. doi: 10.1371/journal.pone.0109531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359(22):2355–2365. doi: 10.1056/NEJMra0800353. [DOI] [PubMed] [Google Scholar]
- 30.Le Goffic R, Pothlichet J, Vitour D, et al. Cutting edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007;178(6):3368–3372. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]