

Abstract
Background
Signal Transducer and Activator of Transcription – 3 (STAT-3) is a downstream component of the Epidermal Growth Factor Receptor (EGFr) signaling process that may facilitate the resistance of tumor cells to conventional cancer treatments. Studies were performed to determine if inhibition of this downstream protein may produce radiosensitization.
Methods/Results
A431 cells (human squamous cell carcinoma cells with EGFr overexpression) were found to be sensitized to radiation after treatment with STAT-3 small interfering RNA (siRNA). Therefore, a short hairpin RNA (shRNA) against STAT-3 was designed and cloned into a pBABE vector system modified for shRNA expression. Following transfection, clone 2.1 was selected for further study as it showed a dramatic reduction of STAT-3 protein (and mRNA) when compared to A431 parental cells or a negative control shRNA cell line (transfected with STAT-3 shRNA with 2 base pairs mutated). A431 2.1 showed doubling times of 25-31 h as compared to 18-24 h for the parental cell line. The A431 shRNA knockdown STAT-3 cells A431 were more sensitive to radiation than A431 parental or negative STAT-3 control cells.
Conclusion
A431 cells stably transfected with shRNA against STAT-3 resulted in enhanced radiosensitivity. Further work will be necessary to determine whether inhibition of STAT-3 phosphorylation is a necessary step for the radiosensitization that is induced by inhibition of EGFr.
Introduction
The signal transducer and activator of transcription-three protein (STAT-3) is a cytoplasmic protein that can become activated through tyrosine kinase signaling events. Activation of STAT-3 leads to dimerization and translocation to the nucleus where STAT-3 induces transcriptional events [1-3]. These events are associated with the promotion of tumor proliferation and enhanced survival of tumor cells [1, 2, 4]. These effects are partially mediated by an induced resistance to apoptosis which is mediated through increased transcriptional activity of anti-apoptotic proteins such as BcL-XL and survivin [1, 5-9]. STAT-3 also promotes tumor growth and stability through upregulation of cell cycle regulators (cyclin D1 and C–Myc), and vascular endothelial growth factor (VEGF) [1, 8].
It is known that the activation of STAT-3 is mediated by several signaling pathways including the epidermal growth factor receptor (EGFr) pathway and the Src pathway [7-11]. The inhibition of EGFr signaling or Src signaling leads to decreased potential for cellular growth and it is believed that the downstream inhibition of STAT-3 is an important factor involved in this process. Since STAT-3 is downstream of several promoters of tumor growth, it has become the focus of investigation to determine whether STAT-3 is an important and feasible target for cancer therapy [1, 10].
It is also known that the inhibition of EGFr leads to radiosensitization through the use of monoclonal antibodies that block the ligand binding domain of EGFr or small molecule EGFr tyrosine kinase inhibitors (TKIs) [12-15]. Cetuximab, an anti-EGFr monoclonal antibody, has been shown to enhance the effects of radiotherapy in human squamous cell carcinoma cancer cells in-vitro and in-vivo [10, 13]. Following these pre-clinical studies, a phase III clinical trial demonstrated enhanced locoregional control and survival for patients with squamous cell carcinomas of the head and neck treated with cetuximab and radiation compared to radiation alone [16]. Since in-vitro studies have demonstrated that cetuximab also inhibits activation of STAT-3 through EGFr signaling, we investigated whether the inhibition of STAT-3 leads to radiosensitization. In fact, the inhibition of STAT-3 did enhance the cytotoxic effects of radiation in human A431 squamous cell cancer cells.
Materials and Methods
Cell culture
A431 human epidermoid cancer cells were obtained from American Type Culture Collection (Manassas, VA) and processed for cell culture as described in previous studies from our laboratory [14]. Briefly, the cell lines were maintained in Dulbeco's modified Eagle's medium: F12 (50:50) containing 7% fetal bovine serum supplemented with L-glutamine and incubated at 37° C in 5% CO2.
Cell proliferation
This assay of daily growth was performed as previously described [10, 12]. Following the plating of A431 cells, they were allowed to adhere to the surface of plates. Subsequently, they were assessed for cell number on a daily basis. At the appropriate times following the initiation of the study, cells were removed from plates with trypsin and counted with a cell counter (Beckman Coulter, Fullerton, CA).
Radiation cell survival
The assessment of potential radiosensitization was performed as previously described [12] with minor modifications. Briefly, exponentially growing cells were seeded, in triplicate, into culture dishes to yield between 20 to 200 colonies. The cells were allowed to adhere to the surface of plates, then irradiated with various dose of radiation using a 60Co therapy unit at a dose rate of 80 cGy/minute. After the appropriate radiation dose, the cells were returned to the incubators. After 10-14 days of growth, cultures were fixed with methanol-acetic acid, stained with crystal violet and scored for colonies containing more than 50 cells. The standard errors were contained in the symbols unless otherwise stated. The linear-quadratic model of Fertil et al [17] was utilized to fit curves to the following equation: In [surviving fraction (SF)[ = -αD – βD2 where D represents radiation dose (Gy) and α/β were fit to the specific findings. The values of α (Gy -1), β (Gy -2) and mean inactivation dose were calculated for each curve. Subsequently the surviving fraction at 2 Gy (SF 2Gy) and the dose that provided 37% surviving fraction (D37) were calculated. The survival curves were compared, for various conditions, utilizing a standard paired t-test [18].
Transfection of small interfering RNA (siRNA)
A431 cells were plated for radiation cell survival assays, then transfected with 100nM of STAT3 SMARTPOOL siRNA (Dharmacon, Lafayette, CO) using Oligofectamine (Invitrogen, Carlsbad, CA) for 4h before medium with serum was added. Control cells were mock-transfected with Oligofectamine only. Radiation treatment was given 1h post-serum addition. The cells were grown for 10-14 days, then fixed and stained. Colonies with >50 cells were counted and analyzed for surviving fraction as previously described [12].
Immunoblots
Immunoblots were performed as previously described [19] with minor modifications. A431 cells, and transfected A431 cells, were assessed for STAT-3 and GAPDH. Cells were lysed [19] and subsequently equal amounts of protein were loaded in each gel lane. Separation was performed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Immobilon – P membrane (Millipore Corp, Bedford, MA). The immunoblots were blocked in 10% milk – Tris - buffered saline with Tween-20 (TBS – T) (20 mmol/L Tris HCL [pH 7.5], NaCl 137 mmol/L, and 0.05% Tween–20) for 1 hour at room temperature. The primary antibodies of anti - STAT-3 (Cell Signaling Technology, Beverly, MA), anti phosphorylated STAT-3 (Cell Signaling Technology, Beverly, MA) and anti-GAPDH (Santa Cruz Technologies, Inc., Santa Cruz, CA) were incubated overnight at 4°C with 2% milk – Tris – buffered saline with Tween – 20. After washing in TBS-T, the blots were probed with the secondary antibody, anti-mouse – IgG – horseradish peroxidase antibody (Sigma Chemical Company, St. Louis, MO) at room temperature for 1 hour. The blots were developed by chemiluminescence (Amersham Life Sciences, Inc., Arlington Heights, IL).
Cloning and transfection of STAT-3 short hairpin RNA (shRNA)
A 20b siRNA was designed to anneal to STAT-3 variant1, variant 2 and variant 3 mRNA at nucleotides; 1062-1081, 1040-1059, 954-973, respectively. See Table 1. A STAT-3 shRNA was synthesized by linking the antisense siRNA and sense siRNA with a 8 nucleotide loop, along with adding XhoI and BamHI overhangs for insertion into the shRNA expression vector. A negative control, STAT-3 shRNA with 2 base pairs mutated, was also synthesized. The shRNA and the respective complementary shRNA were synthesized by Qiagen (Valencia, CA). The STAT3 and negative control shRNA oligonucleotides were annealed in a Thermal Cycler (Perkin Elmer) using the protocol described by BD Knockout RNAi (BD Biosciences, Palo Alto, CA). Briefly, 100μM of each oligonucleotide was mixed together in Tris-EDTA buffer, then heated to 95°C for 30sec., followed by 2min at 72°C, 2min at 37°C and 2min at 25°C. The annealed shRNA was ligated into pBABE-U6 vector at the XhoI and BamHI cut sites [20]. Ligated plasmid DNA was amplified in DH5-α cells. A431 cells were transfected with plasmid DNA using Lipofectamine LTX Reagent (Invitrogen) as previously described with minor modifications [19]. Individual colonies were isolated using cloning rings in the presence of puromycin and screened for STAT3 knockdown by immunoblot and Quantitative Real-Time PCR (qRT-PCR).
Table 1. Sequence for STAT-3 shRNA and negative control shRNA.
| shRNA | Sense Sequence (5 ‘→3 ’) |
Genes with 100% homology |
|---|---|---|
| STAT-3 | ACTTCAGACCCGTCAACAAA | STAT-3 v1, v2, v3 |
| Negative STAT-3 (mutated bases highlighted) | ACTTCAGACACGGCAACAAA | none |
RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
RNA was isolated from the parental A431 cells, two stable STAT3 knockdown clones, 2.1 and 6.4, and one stable negative control STAT3 clone, NEG-4.1, using TRIzol Reagent (Invitrogen). cDNA was synthesized using SuperScript III First-Strand Synthesis Systems for RT-PCR (Invitrogen). 50ng cDNA was amplified and quantified by qRT-PCR with STAT-3 and GAPDH TaqMan Gene Expression Primer sets (Applied Biosystems).
Results
Since activated STAT-3 is an important regulator of tumor growth and portends resistance to apoptosis in tumor cells, it was hypothesized that the inhibition of STAT-3, through the use of small interfering RNA (siRNA), may potentiate the effects of radiation in A431 human squamous cell carcinoma cells. In fact, when cells were transfected with 100 nM siRNA, using Oligofectamine, for as little as 4 hours prior to various doses of radiation, the cells were significantly more responsive to the cytotoxic effects of radiation compared to cells treated with Oligofectamine alone (p=0.01, Figure 1). It is noteworthy that the time-course of siRNA-induced inhibition of STAT-3 revealed that STAT-3 levels were not reduced until 48-72 hours after transfection with siRNA (Figure 2), suggesting that the inhibition of STAT-3 after radiation was important for radiosensitization.
Figure 1.
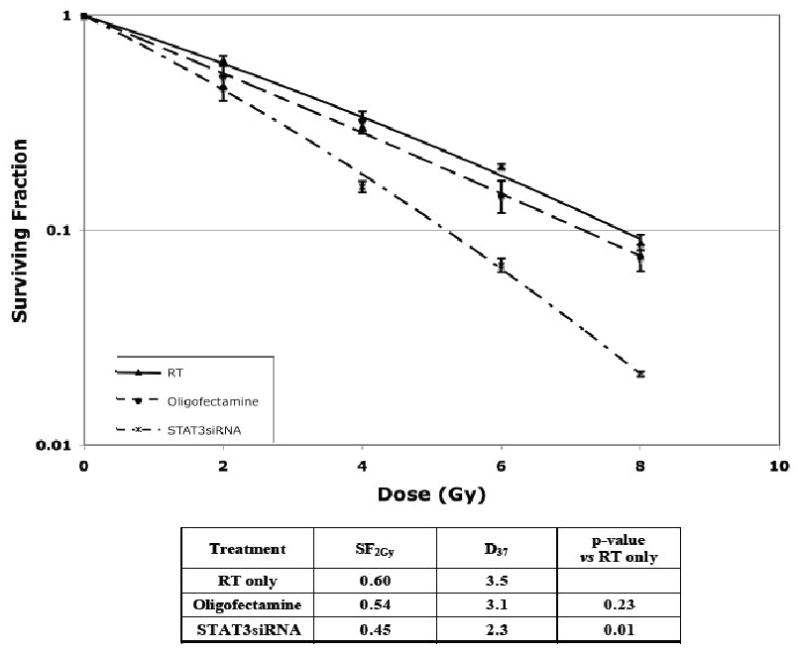
Transfection with anti-STAT-3 siRNA resulted in enhanced sensitivity to radiation. A431 cells were transfected with 100 nM of STAT-3 siRNA with oligofectamine for 4 hours prior to serum exposure for 1 hour and radiation treatment as described in the Materials and Methods section. Control cells were mock transfected with oligofectamine. Plating efficiencies were as follows: Control: 23%, Oligofectamine alone 14%, and STAT-3 siRNA 35%.
Figure 2.
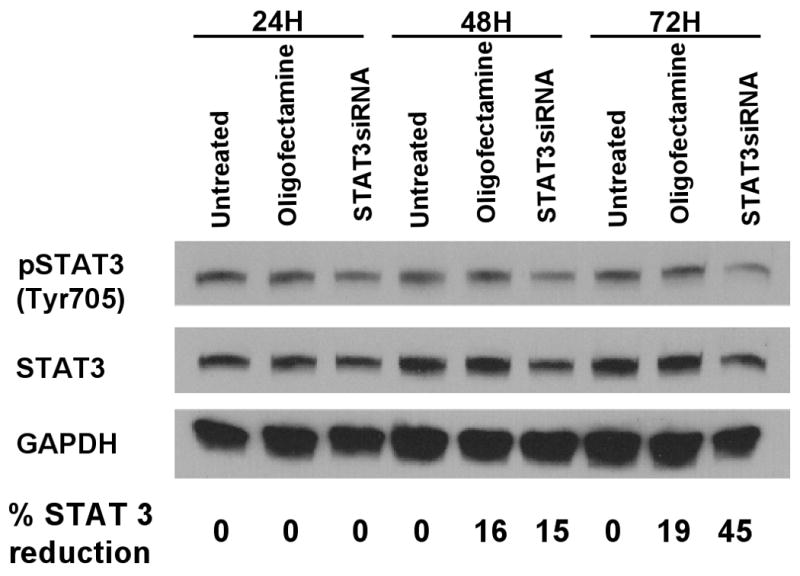
Total STAT-3 and phosphorylated STAT-3 were reduced at 48 and 72 hours following transfection with anti-STAT-3 siRNA. Immunoblot assessments were utilized to assess relative protein levels at various times after transfection (100 nM of STAT-3 siRNA). Cells were treated with 60 ng/ml EGF for 5 minutes prior to being washed and lysed. The percentage of STAT-3 reduction (by densitometry) refers to total STAT-3 relative to control A431 cells.
In order to confirm this radiosensitizing effect, short hairpin RNA (shRNA) oligonucleotides (against STAT-3) were transfected into A431 cells (Table 1). Likewise, a negative control shRNA, identical to STAT-3 shRNA except for 2 base pair mutations, was transfected into cells (STAT-3 neg 4.1). Individual clones were isolated and screened for STAT-3 expression. Using quantitative real time polymerase chain reactions (PCR), two clones (STAT-3 – 2.1, STAT-3 – 6.4) showed a dramatic reduction in STAT-3 mRNA compared to parental control cells or the transfected control cells. The two clones with shRNA against STAT-3 demonstrated a greater than 50% reduction in STAT-3 mRNA (Figure 3). Next, one clone was selected for further analysis (clone 2.1). Immunoblot analysis demonstrated the reduction in total STAT-3 and phosphorylated STAT-3 for this experimental clone with GAPDH immunoblot for loading control (Figure 4). Following brief stimulation with EGF, clone 2.1 demonstrated approximately a 30% reduction in STAT-3 (relative to the parent line) but almost no phosphorylated STAT-3 was detected.
Figure 3.
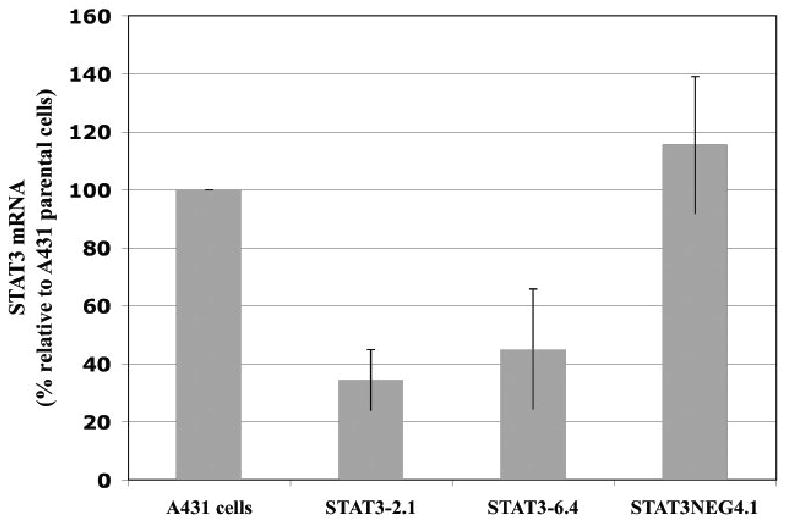
Quantification of mRNA revealed markedly reduced mRNA in A431 cells 2.1 and 6.4 following stable transfection with anti-STAT-3 shRNA compared to parental cells or cells transfected with mutated shRNA (4.1). Quantitative PCR was performed as described in the Materials and Methods section. Means and standard errors are shown.
Figure 4.
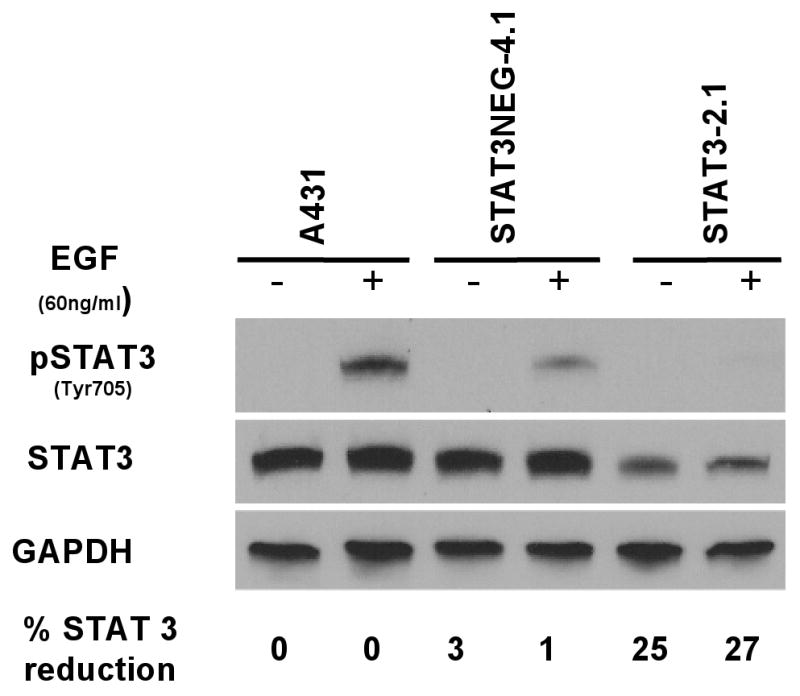
Phosphorylated STAT-3 and overall STAT-3 was reduced in A431 cells 2.1 following stable transfection with anti-STAT-3 shRNA compared to parental cells or cells transfected with mutated shRNA (4.1). Various cellular clones were treated with 60 ng/ml EGF for 5 minutes before cells were washed and lysed. Stable transfectants were produced as described in the Materials and Methods section. The percentage of STAT-3 reduction (by densitometry) refers to total STAT-3 relative to control A431 cells.
Since STAT-3 plays an important role in tumor growth, we examined the proliferative capacity of the various stably transfected clones. A standard proliferation assay revealed that the two cell lines with shRNA knockdown STAT-3 demonstrated marked reduction in the capacity to proliferate over a 3 day period of time. Untreated cells or cells that were stably transfected with the negative control shRNA (STAT-3 neg 4.1) showed more than a 10 fold increase in cell number over four days. However, the shRNA STAT-3 knockdown cell line (2.1) showed a 40-50% lower proliferation during the same 3 day time period (Figure 5). However, the radiation-induced decrement in cell proliferation was not enhanced in the STAT-3 knockdown cell line (Figure 5).
Figure 5.
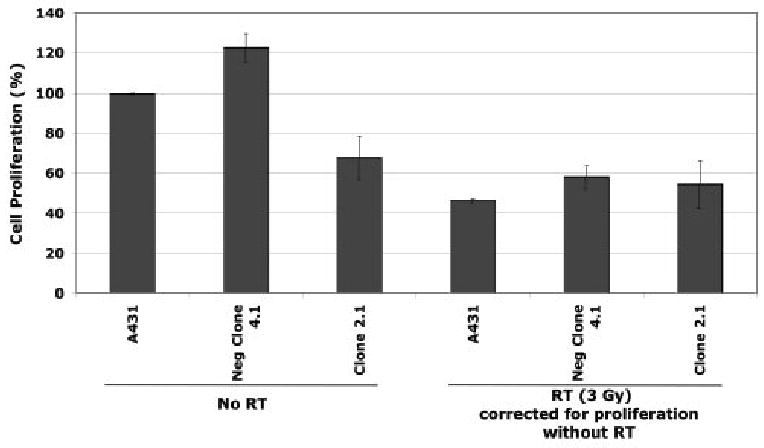
Left with no radiation (No RT): Cellular proliferation was markedly reduced in the stable transfectants with anti-STAT-3 shRNA (2.1) compared to controls (parental and mutant shRNA transfectants Neg clone 4.1). Right with 3 Gy of Radiation: This decrease in proliferation (due to STAT-3 inhibition) did not enhance the radiation-induced anti-proliferative effect. Cellular proliferation was assessed as described in the Materials and Methods section. Means and standard errors are shown.
Having demonstrated STAT-3 knockdown decreases cancer cell growth, we proceeded to examine the effect on radiosensitization. Similar to our transient siRNA knockdown experiments (Figure 1), we found markedly increased radiosensitivity in the STAT-3 knockdown cells compared to controls. A representative example is shown in Figure 6, (p=0.006).
Figure 6.
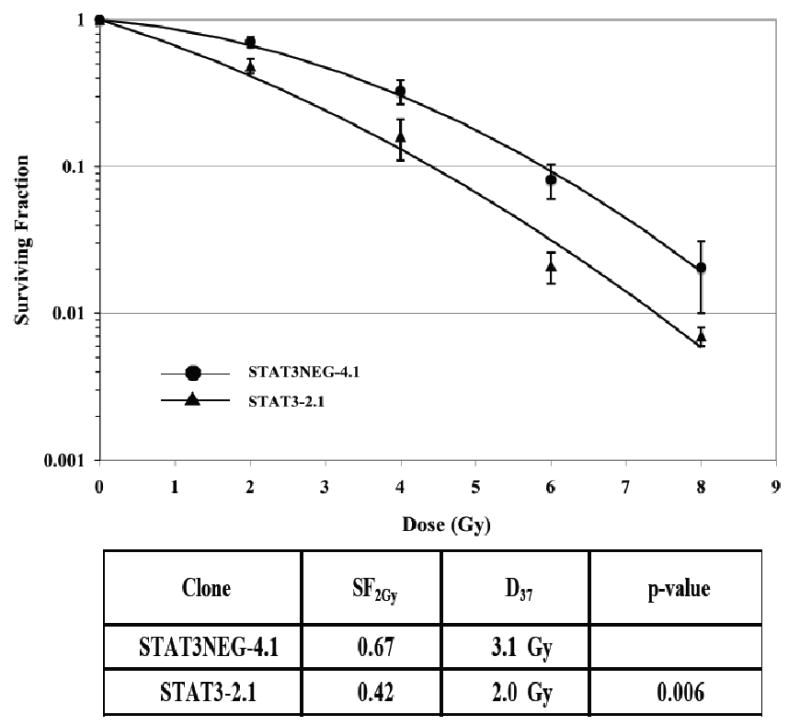
The radiosensitivity of cells stably transfected with STAT-3 shRNA (2.1) revealed enhanced radiosensitivity compared to cells transfected with mutated shRNA control. A representative study is shown. Cells were irradiated and assessed for colony formation as described in the Materials and Methods section. Plating efficiencies were as follows: STAT-3 Neg – 4.1: 55%, and STAT-3.2.1: 14%.
Discussion
Although constitutive activation of STAT-3 has been shown to be characteristic of many tumors [6, 7], additional studies have shown that STAT-3 is greatly influenced by several upstream regulators of tumor growth [8, 11]. For instance, STAT-3 plays an important role in transmitting the growth signals that are initiated at the level of EGFr [3-5, 8]. EGFr is also known to be overexpressed in many tumors and inhibition of EGFr has been shown to enhance the effects of conventional treatments of radiotherapy and chemotherapy in squamous cell malignancies, as well as, many other malignancies [8, 10]. Indeed, recent studies have shown that overexpression of the EGFr family member ErbB2 in breast cancer confers taxol resistance, which can be reversed by inhibiting STAT-3 [21]. The studies reported herein reveal that inhibition of STAT-3 leads to the enhancement of the effects of radiotherapy in a human squamous cell carcinoma cell line (A431 cells). Therefore, STAT-3 may be an important target when considering the development of new agents that will lead to improvements in conventional treatments.
Many studies have examined possible methods to inhibit STAT-3 due to the importance of this protein in tumor progression and growth. STAT-3 is a critical transmitter of several signaling processes (e.g., EGFr, Src, Interleukin 4 and 6, VEGF) that regulate tumor stability and lead to growth [1, 2, 5, 11]. Enhanced activity or overexpression of STAT-3 is associated with increased proliferation and malignant potential of tumor cells [1, 7, 9]. These enhanced growth characteristics are linked to the capacity of activated STAT-3 to increase the resistance of tumor cells to apoptosis [5]. This anti-apoptotic protective effect leads to improved viability of tumors and may increase the likelihood for these cells to survive the cytotoxic effects of many conventional treatments. Also, this protection from apoptosis may have far reaching implications, as it has also been linked to an improvement in the efficiency with which tumors metastasize [22].
Although single agent therapy directed at STAT-3 as a therapeutic target may be an effective cancer therapeutic, it is more likely that the inhibition of STAT-3 will find a place in the treatment armamentarium for cancer patients as a component of combination therapy, i.e. either in combination with other molecularly targeted agents or conventional treatments such as radiotherapy and chemotherapy. Investigations have been undertaken to target various functional aspects of the STAT-3 protein [2]. These strategies have included targeting components of the STAT-3 protein that are responsible for dimerization, DNA binding or the binding of STAT-3 to other downstream targets [2, 23-26]. It is the opinion of Yu and Turkson that the difficulty and slow translation of STAT-3 inhibitors to the clinic has been related to the focus on targeting protein-protein interactions such as the dimerization of STAT-3 proteins [2]. The complexity and large surface area of these interactions makes it difficult to develop agents that can be delivered in vivo and adequately disrupt interactions of this nature. Yu and Turkson support an increased effort in targeting STAT-3 at the level of DNA binding or through oligonucleotide approaches [2]. The results reported herein support these contentions and suggest that the inhibition of STAT-3 may be an important therapy when combined with conventional treatments such as radiation. Further work will be necessary to determine the mechanism by which the inhibition of STAT-3 leads to radiosensitization. The timing of the siRNA experiments suggest that STAT-3 inhibition may interfere with cell survival processes that are initiated after radiation (Figures 1 and 2).
It will be important to investigate the efficacy of inhibiting STAT-3 in combination with inhibitors of other proteins that are either upstream or downstream of STAT-3 with respect to growth promoting signaling pathways. The potential utility of this approach is supported by the fact that many signaling pathways can not be completely blocked by targeting one protein in the pathway, due to the cross-talk between signaling pathways [3-8]. Therefore, future studies should explore the combination of anti-EGFr agents (e.g., cetuximab, erlotinib and gefitinib), anti-VEGF (e.g., bevacizumab and thalidomide) or anti-Src (e.g. dasatinib and AZD-0530) in combination with new agents that block STAT-3. Also, it will be important to test whether new compounds that inhibit STAT-3 may have potential as chemo-preventive agents since this approach may leave transforming cells more susceptible to apoptosis. The future study of STAT-3 inhibition should provide much insight into potential methods of exploiting aberrant cell growth of tumor cells.
Acknowledgments
Supported in part by NIH R01 CA89297.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Presented at the 11th International Wolfsberg Meeting, 2009
Conflict Of Interest Statement
James A. Bonner, M.D.
Occasional honoraria from Bristol-Myers Squibb and ImClone Systems, Inc.
There are no other conflicts of interest.
References
- 1.Heimberger AB, Priebe W. Small molecular inhibitors of p-STAT3: Novel agents for treatment of primary and Metastatic CNS cancers. Recent Patients on CNS Drug Discovery. 2008;3:179–188. doi: 10.2174/157488908786242489. [DOI] [PubMed] [Google Scholar]
- 2.Yue P, Turkson J. Targeting STAT3 in cancer: How successful are we? Expert Opin Investig Drugs. 2009;18(1):45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willey CD, Palanisamy AP, Johnston RK, et al. STAT3 activation in pressure-overloaded feline myocardium: Role for integrins and the tyrosine kinase BMX. Int J Biol Sci. 2008;4(3):184–199. doi: 10.7150/ijbs.4.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haura EB, Zheng Z, Song L, et al. Activated epidermal growth factor receptor – STAT-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res. 2005;11(23):8288–8294. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- 5.Grandis JR, Drenning SD, Zeng Q, et al. Constitutive activation of STAT-3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc National Academy Sciences (PNAS) 2000;97(8):4227–4232. doi: 10.1073/pnas.97.8.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of STAT-3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 7.Boehm AL, Sen M, Seethala R, et al. Combined targeting of epidermal growth factor receptor, signal transducer and activator of transcription-3, and Bcl-Xl enhances antitumor effects in squamous cell carcinoma of the head and neck. Mol Pharmacol. 2008;73(6):1632–1642. doi: 10.1124/mol.107.044636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: A model for targeted therapy. Clin Cancer Res. 2006;12(18):5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 9.Aoki Y, Feldman GM, Tosato G. Inhibition of STAT-3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood. 2003;101(4):1535–1542. doi: 10.1182/blood-2002-07-2130. [DOI] [PubMed] [Google Scholar]
- 10.Bonner JA, Raisch KP, Trummell HQ, et al. Enhanced apoptosis with combination C225/radiation treatment serves as the impetus for clinical investigation in head and neck cancers. J Clin Oncol. 2000;18:47s–53s. [PubMed] [Google Scholar]
- 11.Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12(5):1398–1401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- 12.Bonner JA, Maihle NJ, Folven BR, et al. The interaction of epidermal growth factor and radiation in human head and neck squamous cell carcinoma cell lines with vastly different radiosensitivities. Int J Radiation Oncology Biol Phys. 1994;29(2):243–247. doi: 10.1016/0360-3016(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 13.Huang SM, Bock JM, Harari PM. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Research. 1999;59:1935–1940. [PubMed] [Google Scholar]
- 14.Saleh MN, Raisch KP, Stackhouse MA, et al. Combined modality therapy of A431 human epidermoid cancer using anti-EGFr antibody C225 and radiation. Cancer Biother & Radiopharm. 1999;14(6):451–463. doi: 10.1089/cbr.1999.14.451. [DOI] [PubMed] [Google Scholar]
- 15.Dobelbower MC, Russo SM, Raisch KP, et al. Epidermal growth factor receptor tyrosine kinase inhibitor, erlotnib, and concurrent 5-fluorouracil, cisplatin and radiotherapy for patients with esophageal cancer: a phase I study. Anti-Cancer Drugs. 2006;17:95–102. doi: 10.1097/01.cad.0000185178.26862.4c. [DOI] [PubMed] [Google Scholar]
- 16.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 17.Fertil B, Dertinger H, Courdi A, et al. Mean inactivation dose: A useful concept for intercomparison of human cell survival curves. Radiat Res. 1984;99:73–84. [PubMed] [Google Scholar]
- 18.Colton T. Statistics in Medicine. Boston, MA: Little, brown; 1974. pp. 131–136. [Google Scholar]
- 19.Bonner JA, Buchsbaum DJ, Russo SM, et al. Anti-EGFR – mediated radiosensitization as a result of augmented EGFR expression. Int J Radiation Oncology Biol Phys. 2004;59(2-Supp):2–10. doi: 10.1016/j.ijrobp.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Wang H, Li M, et al. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J Biol Chem. 2004;279(16):16000–16006. doi: 10.1074/jbc.M312264200. [DOI] [PubMed] [Google Scholar]
- 21.Hawthorne VS, Huang WC, Neal CL, et al. ErbB2-mediated Src and signal transducer and activator of transcription 3 activation leads to transcriptional up-regulation of p21Cip1 and chemoresistance in breast cancer cells. Mol Cancer Res. 2009;7(4):592–600. doi: 10.1158/1541-7786.MCR-08-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onishi A, Chen Q, Humtsoe JO, et al. STAT3 signaling is induced by intercellular adhesion in squamous cell carcinoma cells. Experimental Cell Research. 2008;314:377–386. doi: 10.1016/j.yexcr.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turkson J, Ryan D, Kim JS, et al. Phosphotyrosyl peptides block STAT3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276(48):45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 24.Turkson J, Kim JS, Zhang S, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261–269. [PubMed] [Google Scholar]
- 25.Turkson J, Zhang S, Mora LB, et al. A novel platinum compound inhibits constitutive STAT3 signaling and induces cell cycle arrest and apoptosis of malignant Cells. J Biol Chem. 2005;280(38):32979–32988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- 26.Timofeeva OA, Gaponenko V, Lockett SJ, et al. Rationally designed inhibitors identify STAT3 N-domain as a promising anticancer drug target. ACS Chem Biol. 2007;2(12):799–809. doi: 10.1021/cb700186x. [DOI] [PubMed] [Google Scholar]