

Abstract
Well-established genetic manipulation procedures along with a fast doubling time, the ability to grow in inexpensive media, and easy scaleup make Escherichia coli (E. coli) a preferred recombinant protein expression platform. Human alpha-1 antitrypsin (AAT) and other serpins are easily expressed in E. coli despite their metastability and complicated topology. Serpins can be produced as soluble proteins or aggregates in inclusion bodies, and both forms can be purified to homogeneity. In this chapter, we describe an ion-exchange chromatography-based protocol that we have developed involving the use of two anion-exchange columns to purify untagged human AAT from E. coli. We also outline methods that can be used to determine the inhibitory activity of both AAT in cell lysates and purified AAT. Our protocol for the purification of bacterially expressed AAT yields pure and active protein at 6–7 mg/l culture.
Keywords: Alpha-1 antitrypsin (AAT), Elastase, Trypsin, Anion-exchange chromatography, Stoichiometry of inhibition, Absorbance spectroscopy
1 Introduction
Serine protease inhibitors (serpins) are a superfamily of proteins with diverse sequences and a common two-domain structure with three β-sheets (A, B, and C), 8–9 α-helices, and a loop called the reactive center loop (RCL) connecting sheets A and C [1]. Most serpins are protease inhibitors, and for these serpins, a solvent-exposed RCL is central to the complex mechanism that is used to trap and inactivate target proteases [2–4]. Mature human alpha-1 antitrypsin (AAT, 394 residues) is a prototypical inhibitory plasma serpin that protects lungs during inflammation by irreversibly inhibiting serine proteases released by leukocytes, particularly human leukocyte elastase [5]. Specific single-point mutations in AAT (and other serpins) are known to result in misfolding-associated genetic disorders in humans, which are referred to as the serpinopathies [6].
Studies on AAT have contributed significantly to the current understanding of serpins. Investigations of the AAT structure, folding, and/or function using a bottom-up approach require significant amounts of the pure protein. AAT has been produced successfully from both prokaryotic and eukaryotic expression systems. Furthermore, plasma AAT is a glycosylated protein with three N-linked glycans, but glycosylation does not affect its structure, native state stability, or function as a protease inhibitor [7, 8]. Therefore, the Gram negative bacterium Escherichia coli (E. coli) remains the most robust and economical host for producing recombinant AAT, even though the protein produced in E. coli is not glycosylated. Previous reports about the use of E. coli for the production and purification of AAT (and its variants) are summarized in Table 1.
Table 1.
Production of AAT protein from E. coli
| Plasmid; Promoter | Expression Strain | Growth condition; Inducer | Induction Temp.; Time; Yield | Ref. |
|---|---|---|---|---|
| AAT expression and purification from soluble cell lysate | ||||
| pOTSα (with N-term deleted AAT); λPL | AR120 | 32 °C; Thermal induction at A650 of 1.0 |
42 °C; 60–90 m; NR |
[9] |
| λPL | TGE7213 | 15 1 culture at 30 °C; Thermal induction at A600 of 10 |
42 °C; 6 h; NR | [10] |
| pTermat; T7 | BL21(DE3) | 101 medium 37 °Ca; IPTG (0.1% w/v) at A600 of 10; Rifampicin at 0.1 mg/ml after 30 min of induction |
37 °C; 3.5 h; 0.8 mg/g wet cell paste |
[11] |
| pEAT8 (M2 AAT); T7 | BL21(DE3) | Semi-defined medium; 0.4 mM IPTG at A600 of 1.4 |
37 °C; 3 h; NR | [12] |
| pMAL-C2x (MBP-AAT); pTac | BL21CPb | 0.3 mM IPTG at A600 of 0.5b | 37 °C; 8 h; 7–9 mg/l |
[13] |
| a. pQE31 (His6-AAT); T5 b. pQE30 (His6-AAT); T5 |
SG13009 (pREP4) |
2xYT medium; a. 1.0 mM IPTG at A600 of 0.8–1 b. 0.5 – 1.0 mM IPTG at A600 of 0.5 – 0.7 |
a. 30 °C; 3 h;NR b. 37 °C; 4 h;NR |
[14] [15] |
| pEAT8–137 (M2 AAT); T7 | BL21(DE3) | 37 ° C, LB medium; 0.4 mM IPTG at A600 of 0.7–0.8 |
30 °C; 5 h; 6–7 mg/1 |
[16]c |
|
| ||||
| AAT expression and purification from inclusion bodies | ||||
| pαBcl (N-terminal deleted AAT); λPL | AR120 | 32 °C until A650 of 1.0; Thermal induction |
42 °C; 60–90 m; NR |
[9] |
| pEAT8 (M2 AAT); T7 | BL21(DE3) | M9ZB medium; 0.4 mM IPTG at A600 of 0.8 |
40 °C; 3 h;NR |
[17] |
| pTermat (Chimeric- AAT); T7 | BL21(DE3) | 37 °C; IPTG (0.1% w/v) at A600 of 10; Rifampicin at 0.1 mg/ml after 30 m of induction |
37 °C; 3.5 h; 10–15 mg/1 |
[18] |
| pQE30(His6-Multi9AAT); T5 | SG-13009 | M9 Minimal medium; 1 mM IPTG at A600 of 0.5 |
4.5 h; NR | [19] |
| pTermat; T7 | BL21(DE3) | 2xYT medium; 0.5–1.0 mM IPTG at A600 of 0.5–0.7; Rifampicin at 0.05 mg/ml after 30 m of induction | 37 °C; 4 h; NR | [15] |
Defined medium for fermentation
Optimized strain and growth condition from [13]
Current Study
NR not reported
Depending on the cell culture growth and protein expression conditions, overexpression of AAT in E. coli is known to result in varying extents of protein partitioning between soluble and insoluble (inclusion bodies) forms. Since inclusion bodies generally contain few E. coli proteins and are relatively pure, AAT can be expressed at elevated temperatures (such as at 40 °C), which favors the inclusion body formation. Active AAT is then purified from the refolded denaturant-solubilized inclusions bodies (Table 1). However, protein purification from inclusion bodies is more expensive and time consuming than purifying soluble AAT from the E. coli cytoplasm, mostly due to the inefficient refolding step. Earlier reports on AAT purification from the soluble cell fraction required either tagged AAT or a purification protocol with more than two steps (Table 1).
Here, we describe a simple two-step anion-exchange chromatography-based protocol for purifying untagged AAT from the cell lysate soluble fraction that can be completed within a day. In order to produce soluble AAT, recombinant AAT expression in E. coli is carried out at 30 °C. The first step in the purification process involves running the crude cell lysate through a manually packed column of Q Sepharose Fast Flow resin at pH 5.92, which yields AAT that is approximately 70% pure. In a subsequent step, more than 95% protein purity is achieved by running the eluted sample from the first column through a MonoQ column at pH 8.0.
The mechanism by which active AAT inhibits proteases is through the distortion of the protease active site and this leads to the formation of a relatively stable acyl-enzyme complex [3, 20]. AAT inhibitory activity can be monitored by using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to detect the AAT-protease covalent complex or spectroscopically by monitoring the residual protease activity. While the SDS-PAGE analysis enables detection of various AAT forms (within a complex, free, or cleaved), the spectroscopy-based measurement allows detection and quantification of the extent of protease inhibition. We, therefore, provide protocols for measuring AAT activity using both methods: (a) the inhibition of porcine pancreatic elastase monitored by SDS-PAGE and (b) bovine pancreatic trypsin inhibition using an absorbance-based assay. The mobility shift of the AAT-protease complex as determined by gel electrophoresis is also the basis of our activity screen to test for functional AAT in soluble cell lysates prior to purification. Such an assay is useful to screen for the effects of mutations on AAT activity as long as some fraction of the variant protein is expressed as soluble protein. This screen can also guide subsequent purification.
2 Materials
2.1 AAT Expression Plasmid
Plasmid pEAT8-137—The parent plasmid, pEAT8, harbors a naturally occurring variant of human AAT, the M2 variant containing the Arg101His, and Glu376Asp mutations relative to the canonical mature protein sequence [21, 22]. M2 was cloned under the T7 promoter in pET8c [17]. A synonymous codon substitution at position 137, TTG(Leu) to CTG(Leu), was introduced in order to eliminate an internal translation initiation site that is downstream of an internal Shine-Dalgarno site. Two additional synonymous substitutions at nucleotide positions 399 and 402 (T399C and T402C) were made to introduce a unique AatII restriction site in order to confirm the mutations through restriction mapping, and the resulting plasmid is pEAT8-137 [23]. The pEAT8-137 plasmid is used as an AAT protein expression vector (see Note1).
2.2 Cell Growth and Protein Expression
E. coli strain BL21(DE3).
Luria Bertaini (LB) broth.
Ampicillin.
Isopropyl β-D-1-thiogalactopyranoside (IPTG).
2.3 Screening for AAT Activity in Cell Lysates Using Western Blots
Lysis buffer: 50 mM HEPES sodium salt, 100 mM NaCl, 10 mM CaCl2, pH 8.0.
Porcine pancreatic elastase.
Bovine pancreatic trypsin.
1 mM and 0.1 M Hydrochloric acid (HCl).
Sodium dodecyl sulfate (SDS) sample buffer with freshly added β-mercaptoethanol.
12% Tris-Glycine SDS-PAGE.
Polyvinylidene difluoride (PVDF) membrane (Biotrace Pall Life Sciences, 0.45 μm).
Transfer buffer: 25 mM Tris, pH 8.3, 192 mM Glycine, 15% (v/v) Methanol, 0.05% (w/v) SDS.
1 × Tris Buffered Saline (TBST): 50 mM Tris, 0.9% (w/v) NaCl pH adjusted to 7.6 with HCl, 0.05% (v/v) Tween-20.
Antibody against AAT (Rabbit Polyclonal AAT antibody, TA590016, Origene, see Note2).
Secondary antibody against the primary antibody (HRP-conjugated-Goat anti-Rabbit secondary antibody, Pierce).
Western blot detection substrate (Clarity, Bio-Rad).
PVDF membrane staining solution: 0.5% (w/v) Naphthol blue black (Amido black, Sigma), 25% (v/v) isopropanol, 10% (v/v) acetic acid.
PVDF membrane destaining solution: 20% (v/v) methanol, 7.5% (v/v) acetic acid.
2.4 AAT Protein Purification
Resuspension buffer: 10 mM Bis-Tris, pH 5.92.
2.5 cm diameter glass column with appropriate flow adaptor.
Q Sepharose Fast Flow resin.
-
Buffers for running sample on Q Sepharose Fast Flow column:
Buffer A: 10 mM Bis-Tris, pH 5.92.
Buffer B: 10 mM Bis-Tris, 1 M NaCl, pH 5.92.
MonoQ 4.6/100 PE (GE Healthcare, see Note3).
-
Buffers for running sample on MonoQ 4.6/100 PE column:
Buffer C: 10 mM HEPES sodium salt, pH 8.0.
Buffer D: 10 mM HEPES sodium salt, 1 M NaCl, pH 8.0.
30 kDa MWCO protein concentrator (Amicon Ultra 15 ml centrifugal filter).
2.5 Gel-Based Stoichiometry of Inhibition (SI) Determination
Purified AAT.
Assay buffer: 50 mM HEPES sodium salt, 100 mM NaCl, 10 mM CaCl2, pH 8.0.
Porcine pancreatic elastase.
Bovine pancreatic trypsin.
1 mM and 0.1 M Hydrochloric acid (HCl).
Sodium dodecyl sulfate (SDS) sample buffer with freshly added β-mercaptoethanol.
12% Tris-Glycine SDS-PAGE.
2.6 Trypsin Active Site Titration
4-nitrophenyl-4-guanidinobenzoate hydrochloride (pNGB).
Dimethylformamide (DMF).
Assay buffer: 50 mM HEPES sodium salt, 100 mM NaCl, 10 mM CaCl2, pH 8.0.
Bovine pancreatic trypsin.
1 mM Hydrochloric acid (HCl).
2.7 Spectroscopy-Based SI Determination
Nα-Benzoyl-L-arginine 4-nitroanilide hydrochloride (BAPNA).
Dimethylformamide (DMF).
Assay buffer: 50 mM HEPES sodium salt, 100 mM NaCl, 10 mM CaCl2, pH 8.0.
Bovine pancreatic trypsin.
Purified AAT.
2.8 Special Equipment
Bacterial cell lysis equipment (Sonicator, Sonics Vibra-cell model VCX 500 or Microfluidizer processor, model M-110L, Microfluidics, Newton MA).
Western blot electroblotting unit (Mini Trans-Blot cell, Bio-Rad).
Chemiluminescence imaging system (GBOX Chemi-XRQ gel documentation unit, Syngene).
FPLC protein purification system (ÅKTA Purifier, GE Healthcare).
UV-Vis spectrophotometer (Varian).
3 Methods
3.1 Cell Growth and AAT Protein Expression
Transform E. coli BL21(DE3) with the pEAT8–137 plasmid.
Inoculate 3–50 ml of LB that contains ampicillin at a 100 μg/ml final concentration with BL21(DE3) cells freshly transformed with the pEAT8-137 plasmid and grow the culture overnight at 30 °C or 37 °C, shaking at 200–250 rpm.
Typically, culture volumes of 10 ml are used to screen variants for AAT activity in cell lysates, and 2 l cultures are grown for protein purification experiments. In particular, 1–2% of the overnight grown culture is used to inoculate fresh LB that contains ampicillin (100 μg/ml), and cells are grown at 37 °C until an OD600 of approximately 0.7–0.8 is reached. For larger overnight cultures (e.g., 50 ml), spin down and resuspend the cells in fresh LB medium that contains ampicillin (100 μg/ml) since these cells will have used up the ampicillin.
IPTG should then be added to a final concentration of 0.4 mM in order to induce AAT protein expression and the bacteria are grown with shaking at 30 °C for 5 h (see Note4).
5. Cells are harvested at 4 °C by centrifugation at 4000 × g for 10 min. The cell pellet from a small scale growth (10 ml culture) is resuspended in 0.5 ml of lysis buffer (see item 1 in Subheading 2.3) and 1 l of culture is resuspended in about 40 ml of resuspension buffer (see item 1 in Subheading 2.4). The resuspended cell suspension should be stored at −80 °C for later use.
3.2 Screening for AAT Activity in Cell Lysates Using Western Blots
The frozen cell pellet suspension is thawed and lysed by sonication on ice using Sonics Vibra-cell model VCX 500 with a 3 mm microtip probe at 30% amplitude for 10 s, with 15 s off time and about 10–11 pulses.
The lysate is centrifuged at 4 °C for 10 min at 12,000 × g and the soluble fraction (supernatant) that is separated from the cell debris is kept on ice or at 4 °C.
A 2 mg/ml stock solution of both porcine pancreatic elastase (in water) and bovine pancreatic trypsin (in 1 mM HCl) should be prepared.
Protease inhibition activity assays are set up in a reaction volume of 15 μl with 1 μl protease (elastase or trypsin), 9 μl of assay buffer, and 5 μl of the cell lysate (supernatant fraction) at 25 °C for 10 min. In order to avoid acidifying the reaction, the protease is added to the assay buffer before the AAT-containing cell lysate is added. The reactions are quenched by adding 1 μl of 0.1 M HCl and SDS-PAGE sample buffer that contains β-mercaptoethanol (1×), and then the samples are boiled for 3–5 min. An assay with purified AAT serves as a positive control (see Note5).
Samples are analyzed on a 12% Tris-Glycine SDS-PAGE gel and the resolved proteins are transferred onto a PVDF membrane using the Bio-Rad Mini Trans-Blot cell. The transfer is carried out for 1 h at a setting of 100 Vand constant 350 mA current.
The membrane is blocked in 5% (w/v) of Bio-Rad blotting grade blocker solution in 1 × TBST for 1 h at room temperature (or overnight at 4 °C).
The membrane is incubated with the diluted primary antibody against AAT for 1 h (with gentle shaking) followed by an incubation with the diluted secondary antibody for 1 h at room temperature. The antibody dilutions are made in 1 × TBST following the manufacturer’s recommendations. The membrane is washed with agitated shaking at least three times for 5 min each with 1 × TBST between the each antibody incubation.
The membrane is probed using the Clarity Western ECL blotting substrate following the manufacturer’s protocol and digitally imaged on the Syngene Chemidoc unit (see Fig. 1a).
After developing the blot, the membrane is irreversibly stained with the amido black PVDF membrane staining solution for less than 5 min followed by extensive destaining in the PVDF membrane destaining solution (see Fig. 1b).
Fig. 1.
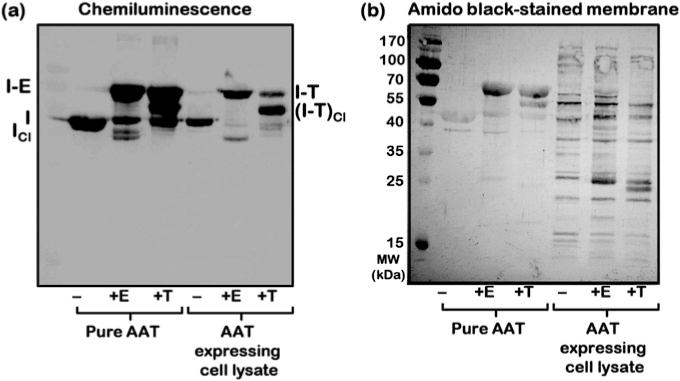
Screening AAT protease inhibition activity in cell lysates. AAT protein in the soluble fraction of cell lysates is tested for function by monitoring the formation of a covalent complex between AAT [I] and proteases, elastase [E] and trypsin [T]. (a) The inhibitor-protease complexes [I-E and I-T] are detected by chemiluminescence imaging of the Western blot by using a primary antibody against AAT. (b) The sample and protein transfer quality are assessed by staining the membrane with Amido black. In the case of the AAT interaction with trypsin, degradation products of the complex, [I-T]Cl, are observed [3]. Cleaved AAT [ICl] is visible in reactions between AAT and elastase
3.3 AAT Protein Purification
3.3.1 Cell Lysis
Thaw the frozen cell pellet under cold water.
Cell lysis is carried out on ice using Sonics Vibra-cell model VCX 500 with a 13 mm diameter probe with a replaceable tip at 30% amplitude for 10 s, with 15 s off time intervals and about 40 pulses. For an 80 ml cell suspension, the total sonication time is 30 min). Alternatively, cell lysis may be carried out on a Microfluidizer processor at a pressure of 16–17 k PSI with 4 passages through the system (see Note6).
The cell lysate is spun at 18,000 × g, 4 °C for 45 min in order to obtain the soluble fraction (supernatant). The supernatant is filtered using a 0.45 μm filter prior to loading it onto the Q Sepharose Fast Flow column. Protein purification is carried out following a 2-step (or column) purification protocol, as described below.
3.3.2 First Chromatographic Step of the AAT Protein Purification—Soluble Cell Extract on a Manually Packed Q Sepharose Fast Flow Column (Column 1)
Pack the Q Sepharose Fast Flow resin in a 2.5 cm diameter glass column in order to achieve a final bed volume of 20 ml for processing the supernatant from a 2 l growth culture.
Filtered supernatant is loaded onto the Q Sepharose column that is equilibrated with buffer A at 4 °C.
Protein is eluted with a linear gradient of 50–140 mM NaCl in buffer A (e.g., 5–14% Buffer B) in 9 column volumes (CV) (see Fig. 2a) or alternatively using a 3-step elution method with a linear gradient of 50–80 mM NaCl in Buffer A (5–8% Buffer B, 3 CV), and a step gradient at 80 mM NaCl in Buffer A (8% Buffer B, 3 CV), followed by a linear gradient of 80–140 mM NaCl in Buffer A (8–14% Buffer B, 3 CV) (see Fig. 2b).
The eluted protein fractions are analyzed on a 12% Tris-Glycine SDS-PAGE gel (see Fig. 2c).
AAT elutes from the column between 70 and 100 mM NaCl (e.g., 7–10% buffer B) in a linear gradient elution (see peak 1 in Fig. 2a), or within the 80 mM NaCl elution step (e.g., 8% Buffer B) in the alternative step gradient elution method (see peak 1 in Fig. 2b).
Fractions containing AAT (peak 1) are pooled and concentrated using a 30 kDa MWCO protein concentrator at 4 °C and buffer exchanged into Buffer C (see Note7).
Fig. 2.
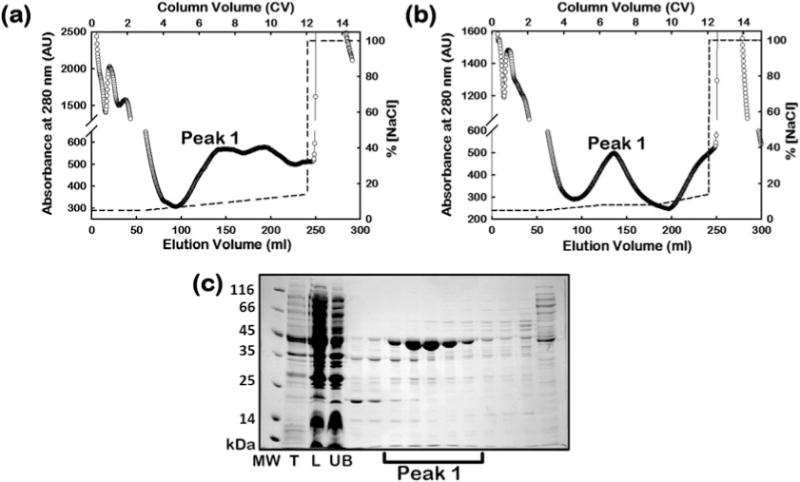
Step 1 of the AAT protein purification process. FPLC chromatogram of AAT in the supernatant (labeled “L”) eluted from the Q Sepharose Fast Flow column using (a) a continuous linear salt gradient method, or (b) a 3-step gradient method. SDS-PAGE of fractions eluted from column 1 in (b) is shown in panel (c). The fractions within peak 1 contain AAT. The dashed line shows the salt gradient used for elution. The labels, T and UB, denote the total cell lysate and flow through fraction from the column respectively
3.3.3 Second Chromatographic Step of the AAT Protein Purification—Final Purification of AAT Protein Eluted from the Q Sepharose Fast Flow Column Using a MonoQ Column (Column 2)
The concentrated protein sample from column 1 is filtered through a 0.2 μm filter to remove any aggregated protein.
The filtered sample is applied to the MonoQ column that is equilibrated in 70 mM NaCl in Buffer C (e.g., in 7% Buffer D).
Protein elution is carried out using a linear gradient of 70–200 mM NaCl in buffer C (e.g., 7–20% buffer D) in 50 CV and 1 ml fractions are collected (Fig. 3a).
Fractions are analyzed on a 12% Tris-Glycine SDS-PAGE gel (see Fig. 3b).
Fractions containing AAT are eluted between 120 and 160 mM NaCl as a broad peak (see peak 2 in Fig. 3a, b), and then pooled and concentrated using a 30 kDa MWCO protein concentrator at 4 °C. The fraction is then buffer exchanged into the desired final buffer (Buffer C).
Pure protein is flash frozen in liquid nitrogen and stored at −80 °C.
The final protein yield is about 6–7 mg/l culture for wild-type AAT, with the protein concentration measured using an extinction coefficient at 280 nm (E1cm, 280nm(1%)) of 5.3 [24].
The extent of purification achieved after each purification step described above is estimated from sample analyses on a 12% Tris-Glycine SDS-PAGE gel (see Fig. 3c).
Fig. 3.
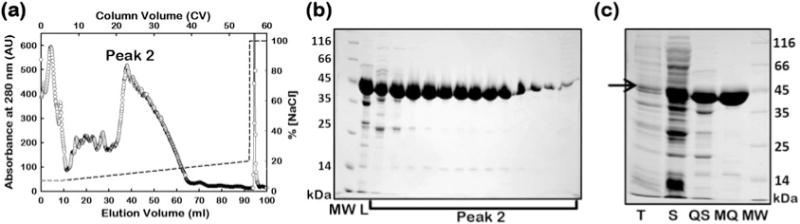
Step 2 of the AAT protein purification process. FPLC chromatogram for the MonoQ column following application of peak 1 (labeled “L”) from the Q Sepharose purification step (a). Fractions eluted from the MonoQ column are analyzed using SDS-PAGE (b). Peak 2 fractions contain the final pure protein. The salt gradient for the elution is indicated by the dashed line. An overall view of protein purity achieved in the individual purification steps can be assessed from panel (c). Samples from different steps of the purification protocol; total cell lysate [T], soluble fraction of the cell lysate [S], eluted protein from the Q Sepharose column [QS], and final pure AAT from the MonoQ column [MQ], are analyzed on a 12% SDS-PAGE gel. The protein is >95% pure after the final column. The arrow in panel C marks AAT in the cell lysate
3.4 Gel-Based Stoichiometry of Inhibition (SI) Determination
Prepare stock solution of porcine pancreatic elastase (2 mg/ml in water).
The AAT activity assay is set up in a reaction volume of 15 μl that contains purified AAT protein and elastase. Molar ratios of AAT to elastase from 0 to 2.0 are used by keeping one of the protein concentrations fixed and increasing the concentration of the other protein (see Fig. 4). The samples are incubated at 25 °C for 10 min.
The reactions are quenched by adding 1 μl of 0.1 M HCl and SDS-PAGE sample buffer that contains β-mercaptoethanol (1×), and then boiling the samples for 3 min.
Samples are analyzed on a 12% Tris-Glycine SDS-PAGE gel (see Fig. 4). The molar ratio at which all of the elastase is consumed and is present only in covalent AAT-elastase (I-E, see Fig. 4) complexes (see in Fig. 4a) or all of the AAT is present as I-E (see in Fig. 4b) represents the SI of elastase by AAT. A similar gel-based assay may be performed to determine the SI of trypsin by AAT (data not shown).
Fig. 4.
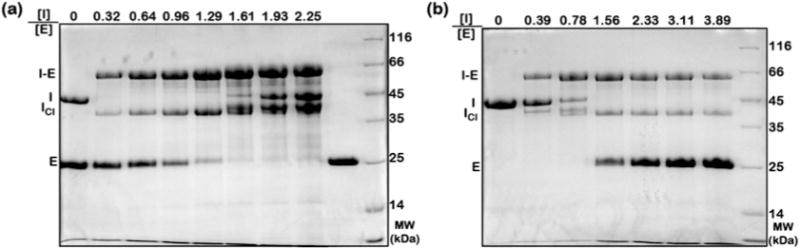
Stoichiometry of inhibition (SI) of elastase by AAT. SDS-PAGE analyses of AAT-elastase covalent complex [I-E] formation in reaction mixtures containing: (a) an increasing concentration of AAT [I] with a fixed concentration of elastase [E], and (b) an increasing concentration of elastase [E] with a fixed concentration of AAT [I]. The SI of elastase by AAT is estimated as 1.6 in this measurement, which is in agreement with the previous reports [25–27]. Icl indicates cleaved AAT
3.5 Trypsin Active Site Titration
The active site titration is used to measure the concentration of active protease (see Note8). One molecule of active trypsin hydrolyses one molecule of p-nitrophenyl-p’-guanidino benzoate (pNGB) resulting in the stoichiometric release of p-nitrophenol and an inactive trypsin molecule [28]. The p-nitrophenol molecule absorbs light at 412 nm. The concentration of the active enzyme is determined by measuring the burst of absorbance at 412 nm when this reaction takes place.
Stock solutions of 0.1 M pNGB in DMF and approximately 2 mg/ml of trypsin in 1 mM HCl should be prepared.
Two μl of pNGB is added to 1 ml of assay buffer and the baseline is monitored at 412 nm for about 1 min. The cuvette is removed without stopping the spectrophotometer and 5–20 μl of the enzyme is added to the solution. The sample is quickly mixed and the cuvette is placed back in the spectrophotometer to measure the absorbance burst by continuing the absorbance measurement at 412 nm (A412) for one additional minute (see Fig. 5a).
The above measurement is repeated at least three times and an average of the three determinations is used to calculate the concentration of active trypsin.
The change in A412 before and after adding trypsin is recorded as the ∆A412. The active site concentration, i.e., active trypsin concentration, is calculated as [Trypsin]Active = ∆A412/ε412, where ε412 = 17,000 M−1 cm−1 at pH 8.0 for the p-nitrophenol released from pNGB.
Fig. 5.
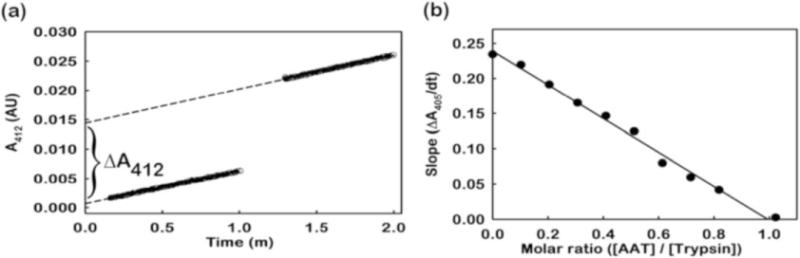
Inhibition of trypsin by AAT. (a) Trypsin active site titration using pNGB. Hydrolysis of pNGB by trypsin releases p-nitrophenol and an increase in the absorbance is observed at 412 nm (A412). The change in absorbance (ΔA412) is used to calculate the concentration of active trypsin. The dotted line represents a linear fit to the data. (b) The stoichiometric inhibition of trypsin by AAT is measured by monitoring the residual trypsin activity in reactions that contain increasing molar ratios of AAT to trypsin and by using BAPNA, a trypsin substrate. The line in the plot is a linear fit to the data (filled circles). The x-intercept obtained from the fitted line (0.99 in this data figure) is the SI for AAT-trypsin interaction
3.6 Spectroscopy-Based SI Determination
BAPNA is a chromogenic trypsin substrate [29]. Trypsin cleaves BAPNA, thereby releasing p-nitroaniline that absorbs around 400 nm. In a reaction with trypsin and BAPNA, the increase in absorbance at 405 nm in relation to the time is proportional to trypsin activity. The incubation of trypsin with AAT leads to the formation of an AAT-trypsin complex, thereby decreasing the concentration of free, active trypsin. Therefore, measuring the residual active trypsin concentration in reaction mixtures that contain a preformed AAT-trypsin complex using BAPNA provides an estimation of the stoichiometric inhibition of trypsin by AAT.
A 100 μl stock solution of 0.1 M BAPNA in DMF should be prepared (see Note9).
The trypsin stock solution with the active protease concentration predetermined (section 3.5) should be used for the SI determination.
Mix the AAT and trypsin at molar ratios ranging from 0 to 1.2 (or 2.0 if assaying new variants) in 1.5 ml centrifuge tubes, making sure to keep the trypsin concentration fixed while increasing the AAT concentration. The total reaction volume is 100 μl. To avoid acidifying the AAT, trypsin is added to the assay buffer and mixed before the addition of AAT. The samples are incubated at 25 °C for 15 min.
After 15 min, add assay buffer to bring the volume to 1 ml.
Monitor the residual trypsin activity by adding 5 μl of BAPNA and measuring the absorbance at 405 nm (A405) for about 2 min.
The plots of A405 versus time should be fitted to a line and the slope (∆A405/dt) should be recorded.
Next, a plot of ∆A405/dt versus molar ratio of AAT to trypsin ([AAT]/[Trypsin]Active) should be made. The x-intercept obtained from the linear fit of the data yields the SI of trypsin by AAT (see Fig. 5b). An SI value of 1 indicates that one mole of AAT inhibits one mole of trypsin and that the purified AAT is 100 percent active.
Acknowledgments
We thank Prof. C. L. Cooney (Massachusetts Institute of Technology) for providing the pEAT8-137 plasmid, and Prof. M. H. Yu (Korea Institute of Science and Technology), who provided the original source of the pEAT8 plasmid. This work was supported by grants from the Alpha-1 Foundation and DBT-Ramalingaswami Re-entry Fellowship (to B.K.), the US National Institutes of Health, OD-00045 (to L.M.G.), and R01 GM094848 (to A.G., D.N.H. & L.M.G.). B.K. thanks CSIR-IMTECH for the protein instrumentation facility.
Footnotes
The protocols described here have also been used successfully for AAT variant genes cloned in pET29b(+) and pET16b plasmids. For AAT variants that contain exposed cysteine residues, 1 mM β-mercaptoethanol is added to all of the buffers that are used for purification just before use. All buffers should be filtered through a 0.2 μm membrane.
The monoclonal antibody against AAT from Abcam (ab9399) also detects AAT-protease complexes in cell lysates [16]. The primary antibody against the protease may also be used in this assay, and we have tested an anti-elastase antibody in our previous study [16].
A smaller MonoQ column, MonoQ 5/50 Global (GE Healthcare), has also been used for the second purification step.
Studies of temperature-dependent wild-type AAT expression at 30 °C, 37 °C and 40 °C indicate that the maximal amount of soluble protein is produced at 30 °C (post-IPTG addition). For purification from inclusion bodies, 40 °C may be used.
Unlike trypsin, excess elastase does not digest the AAT-elastase complex. Therefore, an excess of elastase can be used in the assay.
Lysozyme from chicken egg white (Sigma) and DNaseI from bovine pancreas, grade II (Roche) are added to the thawed cell suspension to final concentrations of 6.25 μg/ml and 2.5 μg/ml, respectively, and incubated on ice for 30 min prior to cell lysis by sonication.
Alternately, the pooled fractions from the Q-Sepharose column may be dialyzed against 10 mM HEPES sodium salt, pH 8 overnight at 4 °C, and then concentrated before loading it on the MonoQ column.
The fraction of active trypsin can be determined from the ratio of the concentration of active trypsin ([Trypsin]Active)) to that of the concentration of prepared trypsin stock solution: ([Trypsin]Total), [Trypsin]Active/[Trypsin]Total.
The BAPNA stock solution must be fresh. BAPNA is very sensitive to hydrolysis and should be kept dry. When making BAPNA stock solutions, take the BAPNA out of the freezer and do not open the bottle until it is at room temperature. Once the BAPNA is at room temperature, open the bottle and weigh out the appropriate amount. Immediately return the bottle to the freezer. If the absorbance at 405 nm versus the time plots are curved (not linear), then the BAPNA must be replaced.
References
- 1.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 2.Huntington JA, Carrell RW. The serpins: nature’s molecular mousetraps. Sci Prog. 2001;84:125–136. doi: 10.3184/003685001783239032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 4.Stratikos E, Gettins PG. Formation of the covalents erpin-proteinase complex involves translocation of the proteinase by more than 70 Å and full insertion of the reactive center loop into beta-sheet A. Proc Natl Acad Sci U S A. 1999;96:4808–4813. doi: 10.1073/pnas.96.9.4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stockley RA. Alpha1-antitrypsin review. Clin Chest Med. 2014;35:39–50. doi: 10.1016/j.ccm.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Gooptu B, Lomas DA. Conformational pathology of the serpins: themes, variations, and therapeutic strategies. Annu Rev Biochem. 2009;78:147–176. doi: 10.1146/annurev.biochem.78.082107.133320. [DOI] [PubMed] [Google Scholar]
- 7.Powell LM, Pain RH. Effects of glycosylation on the folding and stability of human, recombinant and cleaved α1-antitrypsin. J Mol Biol. 1992;224:241–252. doi: 10.1016/0022-2836(92)90587-a. [DOI] [PubMed] [Google Scholar]
- 8.Sarkar A, Wintrode PL. Effects of glycosylation on the stability and flexibility of a metastable protein: the human serpin α1-antitrypsin. Int J Mass Spectrom. 2011;302:69–75. doi: 10.1016/j.ijms.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johansen H, Sutiphong J, Sathe G, Jacobs P, Cravado A, Bollen A, Rosenberg M, Shatzman A. High-level production of fully active human α1-antitrypsin in Escherichia coli. Mol Biol & Med. 1987;4:291–305. [PubMed] [Google Scholar]
- 10.Bischoff R, Speck D, Lepage P, Delatre L, Ledoux C, Brown SW, Roitsch C. Purification and biochemical characterization of recombinant α1-antitrypsin variants expressed in Escherichia coli. Biochemistry. 1991;30:3464–3472. doi: 10.1021/bi00228a017. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins PC, Carrell RW, Stone SR. Effects of mutations in the hinge region of serpins. Biochemistry. 1993;32:7650–7657. doi: 10.1021/bi00081a008. [DOI] [PubMed] [Google Scholar]
- 12.Griffiths SW, Cooney CL. Development of a peptide mapping procedure to identify and quantify methionine oxidation in recombinant human α1-antitrypsin. J Chromat A. 2002;942:133–143. doi: 10.1016/s0021-9673(01)01350-4. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal S, Jha S, Sanyal I, Amla DV. Expression and purification of recombinant human α1-proteinase inhibitor and its single amino acid substituted variants in Escherichia coli for enhanced stability and biological activity. J Biotech. 2010;147:64–72. doi: 10.1016/j.jbiotec.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Zhou A, Carrell RW, Huntington JA. The serpin inhibitory mechanism is critically dependent on the length of the reactive center loop. J Biol Chem. 2001;276:27541–27547. doi: 10.1074/jbc.M102594200. [DOI] [PubMed] [Google Scholar]
- 15.Pearce MC, Cabrita LD. Production of recombinant serpins in Escherichia coli. Methods Enzymol. 2011;501:13–28. doi: 10.1016/B978-0-12-385950-1.00002-X. [DOI] [PubMed] [Google Scholar]
- 16.Krishnan B, Gierasch LM. Dynamic local unfolding in the serpin α-1 antitrypsin provides a mechanism for loop insertion and polymerization. Nat Struct Mol Biol. 2011;18:222–226. doi: 10.1038/nsmb.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwon KS, Lee S, Yu MH. Refolding of α1-antitrypsin expressed as inclusion bodies in Escherichia coli: characterization of aggregation. Biochim Biophys Acta. 1995;1247:179–184. doi: 10.1016/0167-4838(94)00224-5. [DOI] [PubMed] [Google Scholar]
- 18.Bottomley SP, Stone SR. Protein engineering of chimeric Serpins: an investigation into effects of the serpin scaffold and reactive centre loop length. Protein Eng. 1998;11:1243–1247. doi: 10.1093/protein/11.12.1243. [DOI] [PubMed] [Google Scholar]
- 19.Peterson FC, Gordon NC, Gettins PG. Formation of a noncovalent serpin-proteinase complex involves no conformational change in the serpin. Use of 1H-15NHSQC NMR as a sensitive nonperturbing monitor of conformation. Biochemistry. 2000;39:11884–11892. doi: 10.1021/bi001152+. [DOI] [PubMed] [Google Scholar]
- 20.Dementiev A, Dobo J, Gettins PG. Active site distortion is sufficient for proteinase inhibition by serpins: structure of the covalent complex of α1-proteinase inhibitor with porcine pancreatic elastase. J Biol Chem. 2006;281:3452–3457. doi: 10.1074/jbc.M510564200. [DOI] [PubMed] [Google Scholar]
- 21.Huber R, Carrell RW. Implications of the three-dimensional structure of α1-anti-trypsin for structure and function of serpins. Biochemistry. 1989;28:8951–8966. doi: 10.1021/bi00449a001. [DOI] [PubMed] [Google Scholar]
- 22.UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laska ME. Thesis. Massachusetts Institute of Technology; Cambridge, MA: 2001. The effect of dissolved oxygen on recombinant protein degradation in Escherichia coli. [Google Scholar]
- 24.Pannell R, Johnson D, Travis J. Isolation and properties of human plasma α-1-proteinase inhibitor. Biochemistry. 1974;13:5439–5445. doi: 10.1021/bi00723a031. [DOI] [PubMed] [Google Scholar]
- 25.James HL, Cohen AB. Mechanism of inhibition of porcine elastase by human alpha-1-antitrypsin. J Clin Invest. 1978;62:1344–1353. doi: 10.1172/JCI109255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo EJ, Im H, Maeng JS, Kim KE, Yu MH. Distribution of the native strain in human α1-antitrypsin and its association with protease inhibitor function. J Biol Chem. 2000;275:16904–16909. doi: 10.1074/jbc.M001006200. [DOI] [PubMed] [Google Scholar]
- 27.Dolmer K, Gettins PG. How the serpin α1-proteinase inhibitor folds. J Biol Chem. 2012;287:12425–12432. doi: 10.1074/jbc.M111.315465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chase T, Jr, Shaw E. P-Nitrophenyl-p’-guanidinobenzoate HCl: a new active site titrant for trypsin. Biochem Biophys Res Comm. 1967;29:508–514. doi: 10.1016/0006-291x(67)90513-x. [DOI] [PubMed] [Google Scholar]
- 29.Erlanger BF, Kokowsky N, Cohen W. The preparation and properties of two new chromogenic substrates of trypsin. Arch Biochem Biophys. 1961;95:271–278. doi: 10.1016/0003-9861(61)90145-x. [DOI] [PubMed] [Google Scholar]